
Abstract
A method was developed to separate, identify, and quantify 28 fatty acids of potential health relevance using liquid chromatography/electrospray ionization-mass spectrometry (LC/ESI-MS). Optimization of the experimental factors enabled baseline separation of the fatty acids including three pairs of closely related fatty acid isomers (C18:3n-3 and C18:3n-6; C18:1 and C18:1 t; and C20:3n-3 and C20:3n-6) that are challenging to separate. The limits of detection ranged from 0.01 to 0.26 mg L−1 for the 28 fatty acids, and average recovery (mean, n = 4) was found to be 102 ± 12 %. In addition, the proposed method was validated using a quality control standard mix of fatty acids which yielded acceptable precision and accuracy. Fatty acid concentrations in conventional grain-fed and organic grass-fed beef were determined, and the results show that grass-fed beef have a lower omega-6 to omega-3 ratio (1.6–2.8) compared to grain-fed beef (9.3–13.5). Principal component analysis (PCA) was applied on the resulting data to find correlations between significant fatty acid composition and the diet of beef samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dietary fats are a health concern for many reasons including their impacts on cardiovascular health, diabetes, obesity, growth, and development (Ratnayake and Galli 2009). Some dietary fatty acids are essential, while others have known detrimental effects. Research suggests that higher omega-3 fatty acid intake may be associated with lower risk of colorectal and breast cancer (Gerber 2009). High saturated fatty acid consumption has been found to have some relationship to cardiovascular disease (Daley et al. 2010).
The composition of the fat in beef varies, but there are some consistent generalizations about the levels of fatty acids. Saturated fatty acids (SFA) and monounsaturated fatty acids (MUFA) account for the largest part of the lipid content of beef (Daley et al. 2010; Aldai et al. 2009). Aldai and coworkers report that SFA and MUFA account for over 95 % of the fatty acids in Canadian beef. The majority of the minor fats are classified as polyunsaturated fatty acids (PUFAs), composed mainly of omega-3 (n-3) and omega-6 (n-6) fatty acids. Alpha-linolenic acid (18:3 n-3), an omega-3 fatty acid, and linoleic acid (18:2 n-6), an omega-6 fatty acid, are classified as essential fatty acids because humans cannot synthesize them. The n-6/n-3 ratio in food is an important health index (Simopoulos 2008) with recommended levels at or below 4. However, research has revealed that the Western diet tends to have ratios over 15 and some estimates as high as 30:1 (Gomez et al. 2011). The Canadian food industry allows that omega-3 fatty acid claims to be made on food packaging and there is research indicating that a healthy n-6/n-3 ratio is attainable in beef (Canadian Food Inspection 2014). This underlies the importance of having accurate information about what fatty acids are present in food products and at what levels (Aldai et al. 2009). The aim of this work is to quantify SFAs, MUFcbnAs, and other trace level fatty acids that are present in beef.
Gas chromatography (GC) is the current standard technique used for fatty acid profiling (Ratnayake and Galli 2009; Ruiz-Rodriguez et al. 2010). Determination of fatty acid levels in food involves solvent extraction of the crude lipids, hydrolysis, purification, and derivatization to more volatile methyl esters for GC analysis (Bielawska et al. 2010; Ruiz-Rodriguez et al. 2010; Wei and Zeng 2011). Base-catalyzed and acid-catalyzed methylations have been utilized in preparing methyl ester derivatives; however, both techniques have drawbacks: (1) incomplete base-catalyzed methylation of N-acyl lipids and free fatty acids and (2) possible isomerization in acid-catalyzed reactions (Czauderna et al. 2007). Derivatization might still be necessary if detection sensitivity is limited by UV/visible absorbance, conductivity, or fluorescence (Marini and Marini 2012). The advent of mass spectrometry (MS) has eliminated this requirement, and we can now analyze fatty acids in food with much superior specificity and sensitivity (Lacaze et al. 2007; Persson et al. 2010).
High-performance liquid chromatography (HPLC or LC) and capillary electrophoresis (CE) have also been used successfully to separate fatty acids and other types of lipids (Marini and Marini 2012; Soliman et al. 2013). HPLC and CE are liquid-phase separation techniques and do not require analytes to be volatile as required for GC. In addition, high temperatures used in GC may increase the chance of degradation of long-chain highly unsaturated PUFAs, which is not an issue in HPLC and CE. As a consequence, we developed an alternative liquid-phase method using LC/MS to determine fatty acids. The fatty acid profile of beef can be altered by changing the finishing diet of the cattle (Daley et al. 2010). It has been shown that feeding cattle diets with higher n-3 content by increasing grass or adding flax improves the n-3 levels in the meat product (Juarez et al. 2011). Furthermore, this work looked at two different feed regimes for cattle to determine if there is a significant difference in the fatty acid profiles across conventional grain-fed and grass-fed beef.
Material and Methods
Reagents and Chemicals
Analytical grades of acetonitrile (ACN), chloroform, and tetrahydrofuran (THF) and LC/MS grade water were purchased from VWR Canada (ON, Canada). Reagent grade methanol and chloroform were used for the lipid extractions and were obtained from VWR (ON, Canada). Anhydrous ethanol (>90 %), hydrochloric acid (37 %), ammonium acetate (>98 %), ammonium hydroxide (28 % NH3 in H2O), and potassium hydroxide (>85 %) were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). The fatty acid standard mix (GLC 490; Nucheck Prep (Elysian, MN, USA)) used for the analyses contains 26 fatty acids: myristic acid (C14:0), palmitic acid (C16:0), palmitoleic acid (C16:1), heptadecanoic acid (C17:0), stearic acid (C18:0), oleic acid (C18:1), trans-vaccenic acid (C18:1t), α-linoleic acid (C18:2n-6), linolenic acid (C18:3n-3), γ-linolenic acid (C18:3n-6), arachidic acid (C20:0), 11-eicosenoic acid (C20:1), 11,14-eicosadienoic acid (C20:2n-6), 11,14,17-eicosatrienoic acid (C20:3n-3), homo-γ-linolenic acid (C20:3n-6), behenic acid (C22:0), erucic acid (C22:1), arachidonic acid (C20:4n-6), eicosapentaenoic acid (C20:5n-3), 13,16-docosadienoic acid (C22:2n-6), 13,16,19-docosatrienoic acid (C22:3n-3), docosatetraenoic acid (C22:4n-6), docosapentaenoic acid (C22:5n-3), docosahexaenoic acid (C22:6n-6), lignoceric acid (C24:0), and nervonic acid (C24:1). Individual fatty acid standards, tricosanoic acid (C23:0), nonadecanoic acid (C19:0), C18:1, C18:1t, C18:3n-3, C18:3n-6, C19:2n-6, C20:3n-3, and C20:3n-3 were also purchased from Nuchek Prep (Elysian, MN, USA). A representative standard, GLC20A, from animal fat (lard, beef tallow, mutton tallow) and a vegetable oil, palm oil, was obtained from the same supplier to serve as a quality control standard between analyses. All fatty acid standards used were of high purity (>99 %).
A standard reference mixture was prepared from GLC 490 by dissolving the entire contents of the vial in 10 mL of HPLC grade chloroform. The exact weight of the standard was recorded, and the individual concentration of each fatty acid was calculated to be 588 mg L−1. Calibration standards were prepared from this stock by diluting the appropriate volume in chloroform and adding a constant amount of internal standard (C23:0). Individual fatty acid standards were used to spike the standard mixture to confirm the elution order of any isomers.
Extraction and Saponification
Twelve individually wrapped beef ribeye steaks (six from conventional grain-fed and six from organic grass-fed beef) were purchased from different meat retailers in British Columbia, Canada in fall of 2013. Fat was manually trimmed prior to extractions. The detailed procedure for extracting crude lipid from beef muscle tissue has already been described previously (Soliman et al. 2013). After extraction, lipid extracts were dissolved in 1 mL chloroform and stored in a −20 °C freezer until further analysis.
For saponification, an aliquot of 10-μL lipid extract in chloroform was evaporated, and the dried extract was weighed to obtain the exact mass (~1–1.5 mg). Immediately after weighing, 0.5 mL of 1 M KOH in 95 % ethanol and 20 μL of recovery standard (1000 mg L−1 C19:2n-6) were added to the lipid. The vial was mixed thoroughly using a Vortex-Genie® mixer (Scientific Industries, New York, USA). The sample was then heated between 70 and 75 °C. After 30 min, the saponified extract was cooled to room temperature before adding 250 μL water and 100 μL concentrated HCl (12.1 M). The sample was shaken and extracted three times with 500 μL hexane. The extracts were combined, and the solvent was evaporated under a gentle flush of nitrogen. The sample was reconstituted in 190 μL chloroform and 10 μL of 500 mg L−1 C23:0, then filtered through Spin-X centrifuge tube Nylon® filters (Corning, NY, USA). The filtered sample was transferred to borosilicate glass vials containing 150-μL inserts for LC/MS analysis.
LC/MS Parameters and Method
All analyses were carried out on an Agilent 1200 series HPLC system (Agilent Technologies, Mississauga, ON, Canada) coupled to an Agilent 6530 Accurate-Mass Quadrupole Time-of-Flight (Q-TOF) spectrometer equipped with electrospray ionization (ESI) source (gas temperature, 300 °C; drying gas, 8 L/min; nebulizer, 8 psig; sheath gas temperature, 350 °C; sheath gas flow, 10 L/min; Vcap, −3000 V). Fatty acids were analyzed in negative ion mode, and mass spectra were collected between 200 and 700 m/z. A sample volume of 0.5 μL was injected to the LC, and the flow rate was set to 0.35 mL/min. Separation was achieved on a Zorbax Extend-C18 column (100 mm × 2.1 mm; 1.8-μm particle size; Agilent, Canada) kept at a constant temperature of 45 ± 0.2 °C. Mobile phase (A) was composed of 43 % H2O, 6 % THF, 51 % ACN, 2 mM ammonium acetate, and 0.02 % NH4OH; mobile phase (B) was composed of 5 % H2O, 10 % THF, 85 % ACN, 2 mM ammonium acetate, and 0.02 % NH4OH. Gradient elution was programmed as follows: 0 % B for 10 min, ramped to 30 % B in 1 min, 30–35 % B at 11–20 min, 35–100 % B at 20–55 min, held at 100 % B at 55–60 min, ramped down to 0 % B in 1 min and equilibrated at 0 % B at 61–68 min. Diluted samples were analyzed at a modified gradient for shorter analysis time: 0–100 % (B) at 0–45 min, held for 5 min, returned to 0 % (B) in 1 min, and re-equilibrated at 0 % for 10 min.
Principal Component Analysis
To elucidate the correlations between types of beef samples and significant fatty acids, principal component analysis (PCA) was performed. PCA of detected polyunsaturated fatty acids (C18:2n-6, C18:3n-3, C18:3n-6, C20:3n-3, C20:3n-6, C20:4n-6, C22:4n-6, C20:5n-3, C22:5n-3) and C18:1t concentrations of grain-fed and grass-fed samples was performed using Minitab® 16.1.1.0 (PA, USA). Because some samples contained an overall higher fat percentage compared to leaner cuts of steak, the absolute amounts of each fatty acid (mg g−1 of tissue) were normalized per weight of sample before applying PCA.
Results and Discussion
Selection of LC/MS Parameters and Gradient Conditions
A reversed phase C18 column is the traditional choice for fatty acid separation in LC/MS due the nonpolar nature of the analytes. The majority of medium- to long-chain fatty acids have a pKa of 5 for the carboxylic group (Kanicky and Shah 2002). Below the pKa, the fatty acids are neutral, and above the pKa, the carboxylic group deprotonates giving the fatty acid a negative charge. As the chemistry behind ESI relies on the generation of ions in solution prior to reaching the MS detector, higher signal sensitivity was expected by adjusting the pH of the mobile phase 1 pH unit above the pKa and detecting the analytes using negative ion mode. For this reason, a C18 column with an extended stability over higher pH range was selected to avoid dissolution of silica in the column as well as minimize peak tailing due to interactions of ionized carboxylic groups and free silanol sites (Lacaze et al. 2007).
The advantage of an MS detector over other types of detectors for LC (e.g., UV, fluorescence) is that the MS provides an extra dimension for separation based on m/z ratio and thus enables coeluting peaks to be resolved by mass selectivity. This is why a mass spectrometer coupled to LC is far more sensitive and specific compared to using other detectors with LC. However, out of the 28 fatty acids of interest, there are three pairs of isomers that could not be distinguished by their apparent mass-to-charge ratio (m/z), and these are as follows: C18:3n-3 and C18:3n-6; C18:1 and C18:1 t; and C20:3n-3 and C20:3n-6. Therefore, optimization of the LC/MS parameters and elution gradient was primarily focused on achieving chromatographic separation of these isomers.
The initial solvent composition during the study was 2 mM ammonium acetate in water for A and 95 % ACN/5 % 2 mM ammonium acetate in water for B adjusted to pH 5.5, 7.0, and 7.5 with ammonium hydroxide. The mobile phase solvent at pH 7.5 gave the best results, but the main problem encountered was that the solvent system was not strong enough to elute the longer-chain saturated fatty acids (C>20:0) resulting in very broad peaks or late eluting analytes from the preceding run. The addition of 5 % THF increased the elution strength of the mobile phase improving the peak shapes of the analytes as well as eluting the strongly retained fatty acids completely out of the column.
The optimization of the gradient was performed by first analyzing the fatty acids using isocratic conditions at different %B: 95, 90, 85, 80, 75, 70, 65, and 60 %. Separation between the isomers improved considerably from 70 to 60 % B. However, below 60 %, the peaks became broader, and the analysis time lengthened drastically to more than 90 min. Figure 1 shows the separation of the isomers when B is 70, 65, and 60 %. Based on these results, a new mobile phase A was prepared which was composed of 60 % B (43 % H2O, 6 % THF, 51 % ACN, 2 mM ammonium acetate, and 0.02 % NH4OH) while B was maintained at its original composition. The original plan was to develop a segmented gradient based on these individual isocratic analyses since the medium-chain analytes (C<18) were eluting very close together. The details on the selected gradient program can be found in LC/MS Parameters and Method section. The temperature of the column was also studied from 30 to 50 °C. The best separation was obtained at 45 °C. Figure 2 shows the separation of the fatty acid standard mix including the internal and recovery standards.

Extracted ion chromatograms of three pairs of isomeric fatty acids separated isocratically at different %B: a C18:3n-3 and C18:3n-6, 277.0000–277.5000 m/z; b C18:1t and C18:1, 281.0000–281.5000 m/z; c C20:3n-3 and C20:3n-6, 305.0000–305.5000 m/z. Mobile phase A: 2 mM ammonium acetate/0.02 % NH4OH in H2O; B: 90 % ACN/5 % THF/5 % H2O/2 mM ammonium acetate/0.02 % NH4OH

Extracted ion chromatograms of 28 fatty acid standards (100 mg L−1) in negative-ion mode LC/MS. Instrument parameters and gradient conditions are reported in the LC/MS Parameters and Method section
Fatty Acid Analysis
The fatty acids extracted from conventional grain-fed and organic grass-fed beef were quantified using external calibrations generated for the analytes (Table 1). The peak areas of the fatty acids were normalized to the peak area of the internal standard (C23:0). The signal used for all quantitative analyses was the deprotonated ion, [M-H]−, of the analytes. The limits of detection (LODs) for each fatty acid in GLC 490 were also determined. The LOD was obtained by calculating the concentration of the analyte that has an equivalent signal to noise (S/N) ratio of three. The values reported in Table 1 are all less than 1 mg L−1. A quality control standard (GLC 20A) was also analyzed between every six injections. This standard mix is a representative of animal fat (lard, beef tallow, mutton tallow, and palm, a vegetable oil) that contains 2 % of C14:0, 30 % of C16:0, 3 % of C16:1, 14 % of C18:0, 41 % of C18:1, 7 % of C18:2n-3, and 3 % of C18:3n-3. The purpose of the quality control was to ensure that the experimental concentrations of known reference standard stayed within 20 % of the actual values throughout the duration of the calibration and sample analyses.
Sample Analysis
A modified method by Folch and coworkers (Folch et al. 1957) was used to extract the lipids from trimmed ground beef followed by saponification and another extraction to isolate the fatty acids (Soliman et al. 2013). A recovery standard (C19:2n-6) was also added to each sample to evaluate the efficacy of the procedure. Four replicates were analyzed for each sample, and the average % recovery was found to be 102 ± 12 %. Representative ion chromatograms of grain-fed and grass-fed beef are shown in Fig. 3. The amount of fatty acids found for conventional grain-fed beef and grass-fed beef are summarized in Fig. 4. The three main fatty acids detected in both groups were the monounsaturated fatty acid C18:1, which is also known as oleic acid, and the saturated fatty acids, C18:0 and C16:0.

Representative extracted ion chromatograms for a conventional grain-fed and b grass-fed beef. LC/MS parameters and gradient conditions are reported in the LC/MS Parameters and Method section

Summary of fatty acid concentrations (mg/g of tissue) in conventional grain-fed (a) and organic grass-fed beef (b). Note: outliers denoted by *
Research has shown that grass-fed beef has lower n-6/n-3 ratio than grain-fed beef. Based on literature, the n-6/n-3 ratio ranges of grain-fed and grass-fed beef are 3.00–13.6 and 1.44–1.96, respectively (Daley et al. 2010). Figure 5 exhibits the experimental n-6/n-3 ratios of the samples. Experimental n-6/n-3 ratios of 9.3 to 13.5 were obtained from our grain-fed samples, while our grass-fed samples were found to have an n-6/n-3 range of between 1.6 and 2.8. The importance of PUFAs has already been mentioned earlier. In addition to their health benefits, they also act as precursors to conjugated linoleic acids (CLAs), a group of C18:2n-6 isomers with reported anticarcinogenic properties (Griinari et al. 2000). However, CLAs can also be synthesized by endogenous desaturation of trans-vaccenic acid (C18:1t) (Griinari et al. 2000). In order to look at the relationship between PUFAs and C18:1t with respect to the samples, we used the multivariate analysis technique of PCA.

Comparison of omega-6/omega-3 ratio between conventional grain-fed and grass-fed beef (n = 6)
Principal Component Analysis
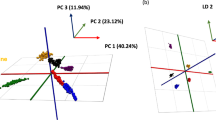
PCA is a multivariate analysis tool that can be used to identify and distinguish the variations among multiple factors. PCA is an unsupervised exploratory technique that generates a set of new orthogonal variables called principal components (PCs). The maximum amount of the variance contained in the original data set is concentrated in the first PCs, reducing the number of variables. Each PC is a linear combination of the original variables, whereby each successive PC explains the maximum amount of variance possible in the data set. PCA was performed on the LC/MS data from the 12 beef samples to find correlation between types of samples and significant fatty acids. The scores and loading plots are shown in Fig. 6. The variance of the first principal component (PC1) and second principal component (PC2) were 51.3 and 28.6 %, respectively. The cumulative proportion from PC1 to PC4 was found to be 95.6 %. Two distinct groups are observed in Fig. 6a, corresponding to conventional grain-fed and grass-fed beef correlations. Interestingly, the loading plot shows that all of the fatty acids contributed to PC1 whereas all except C18:3n-6 contributed to PC2. However, on examining the position of C18:3n-6 on the loading plot, one notices that most of the grain-fed samples (A1, A3, A4, and A5) are found in the same vicinity on the score plot. Furthermore, by inspecting the top and bottom of the score plot, two groups of fatty acids are negatively correlated to each other with respect to PC2. The top group consists of C18:1t and all n-6 fatty acids, while the bottom group consists entirely of n-3 fatty acids with one n-6 fatty acid (C20:3). This can easily be interpreted in conjunction to the score plot. From Fig. 6a, it can be seen that the grass-fed beef samples are located in the bottom quadrants signifying higher n-3 fatty acids. On the other hand, conventional grain-fed beef samples are located in the upper quadrants on the score plot. Surprisingly, we expected C18:1t to be positively correlated with n-3 fatty acids based on review conducted by Daley et al. (2010). This was not the case as described by the loading plot. However, the relationship between C18:1t and C18:2n-6 can be explained by the fact that C18:1t is considered a precursor to CLAs which are isomers of linoleic acid (C18:2). There were other peaks detected in the extracted ion chromatogram of C18:2, in addition to n-3 and n-6, which may be due to other isomers. These peaks were not identified in this work due to lack of reference CLA standards.

Scores plot (a) of the first (PC1) and second principal (PC2) components. Samples labeled from A1 to A6 are conventional grain-fed samples while B1 to B6 are grass-fed samples. The correlation between fatty acids used for PCA is described by the loading plot in b of PC1 and PC2. The variance of the first principal component (PC1) and second principal component (PC2) was 51.3 and 28.6 %, respectively
Conclusion
A sensitive LC/ESI-MS method has been developed for the simultaneous separation and quantification of 28 relevant fatty acids including saturated, monounsaturated, and polyunsaturated fatty acids. The proposed method was used to examine fatty acids present in grain-fed and grass-fed ribeye steaks. Eighteen fatty acids were detected and quantified in the samples, showing overall higher omega-6/omega-3 ratios for grain-fed beef. This work could therefore be used for testing the authenticity of grass versus grain-fed beef commercial products. Furthermore, the developed method allowed the successful separation of three pairs of closely related fatty acid isomers (C18:3n-3 and C18:3n-6; C18:1 and C18:1 t; and C20:3n-3 and C20:3n-6) which are potentially important in beef. Results from the PCA revealed correlation between n-3 fatty acids and grass-fed beef supporting earlier findings in literature. The proposed method can serve as an alternative or complementary technique to GC for fatty acid analysis in various food samples. It also has the potential of analyzing other fatty acid isomers such as CLAs in the future.
References
Aldai N, Dugan MER, Rolland DC, Kramer JKG (2009) Survey of the fatty acid composition of Canadian beef: Backfat and longissimus lumborum muscle. Can J Anim Sci 89:315–329
Bielawska K, Dziakowska I, Roszkowska-Jakimiec W (2010) Chromatographic determination of fatty acids in biological material. Toxicol Mech Methods 20:526–537
Canadian Food Inspection Agency (2014) Guide to food labelling and advertising, Chapter -Nutrient content claims. Section 7.19: Omega-3 and omega-6 polyunsaturated fatty acid claims. http://www.inspection.gc.ca/english/fssa/labeti/guide/ch7be.shtml (02.21.14)
Czauderna M, Kowalczyk J, Korniluk K, Wasowska I (2007) Improved saponification than mild base and acid-catalyzed methylation is a useful method for quantifying fatty acids, with special emphasis on conjugated dienes. Acta Chromatogr 18:59–71
Daley CA, Abbott A, Doyle PS, Nader GA, Larson S (2010) A review of fatty acid profiles and antioxidant content in grass-fed and grain-fed beef. Nutr J 9:1–12
Folch J, Lees M, Stanley GHS (1957) A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem 226:497–509
Gerber M (2009) Background review paper on total fat, fatty acid intake and cancers. Ann Nutr Metabol 55:140–161
Gomez CC, Bermejo LLM, Loria KV (2011) Importance of a balanced omega 6/omega 3 ratio for the maintenance of health.Nutritional recommendations. Nutr Hosp 26:323–329
Griinari JM, Corl BA, Lacy SH, Chouinard PY, Nurmela KVV, Bauman DE (2000) Conjugated linoleic acid is synthesized endogenously in lactating dairy cows by Delta(9)-desaturase. J Nutr 130:2285–2291
Juarez M, Dugan MER, Aalhus JL, Aldai N, Basarab JA, Baron VS, McAllister TA (2011) Effects of vitamin E and flaxseed on rumen-derived fatty acid intermediates in beef intramuscular fat. Meat Sci 88:434–440
Kanicky JR, Shah DO (2002) Effect of degree, type, and position of unsaturation on the pKa of long-chain fatty acids. J Colloid Interface Sci 256:201–207
Lacaze J-PCL, Stobo LA, Turrell EA, Quilliam MA (2007) Solid-phase extraction and liquid chromatography-mass spectrometry for the determination of free fatty acids in shellfish. J Chromatogr A 1145:51–57
Marini D, Marini F (2012) Neutral Lipids. In: Nollet LML, Toldra F (eds) Food analysis by HPLC, 3rd edn. CRC Press, Boca Raton, FL, USA, pp 140–217
Persson XT, Blachnio-Zabielska AU, Jensen MD (2010) Rapid measurement of plasma free fatty acid concentration and isotopic enrichment using LC/MS. J Lipid Res 51:2761–2765
Ratnayake WMN, Galli C (2009) Fat and fatty acid terminology, methods of analysis andfat digestion and metabolism: abackground review paper. Ann Nutr Metabol 55:8–43
Ruiz-Rodriguez A, Reglero G, Ibanez E (2010) Recent trends in the advanced analysis of bioactive fatty acids. J Pharm Biomed Anal 51:305–326
Simopoulos AP (2008) The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med 233:674–688
Soliman LC, Donkor KK, Church JS, Cinel B, Prema D, Dugan MER (2013) Separation of dietary omega-3 and omega-6 fatty acids in food by capillary electrophoresis. J SepSci 36:3440–3448
Wei G, Zeng EY (2011) Gas chromatography-mass spectrometry and high-performance liquid chromatography-tandem mass spectrometry in quantifying fatty acids. Trends Anal Chem 30:1429–1436
Acknowledgments
This work is supported by funding from Investment Agriculture Foundation of British Columbia through the British Columbia’s Ranching Task Force Funding Initiative, with JS Church as principal investigator of the funding. We also acknowledge Thompson Rivers University for the use of their facilities and instrumentations. The LC/MS instrument used was funded by Western Economic Diversification of Canada.
Conflict of Interest
Laiel C. Soliman declares that she has no conflict of interest. Elizabeth M. Andrucson declares that she has no conflict of interest. Kingsley K. Donkor declares that he has no conflict of interest. John S. Church declares that he has no conflict of interest. Bruno Cinel declares that he has no conflict of interest.
Compliance with Ethics Standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Soliman, L.C., Andrucson, E.M., Donkor, K.K. et al. Determination of Fatty Acids in Beef by Liquid Chromatography-Electrospray Ionization Tandem Mass Spectrometry. Food Anal. Methods 9, 630–637 (2016). https://doi.org/10.1007/s12161-015-0229-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-015-0229-0