
Abstract
Growth hormone (GH) is an important driver for somatic growth and increase in height in children. The development of recombinant human GH has greatly increased its availability, and hence the potential for its use and abuse. GH therapy should only be offered to patients with established and approved indications. Common pediatric indications for treatment include growth hormone deficiency, Turner syndrome, Prader–Willi syndrome, small for gestational age, chronic renal insufficiency, and idiopathic short stature. Before initiating treatment, the family should be counseled about the treatment goals, costs, and possible adverse effects from the treatment. It is important for patients to have realistic expectations from the treatment. The dose of GH should be individualized for the indication and will require titration in each patient based on response to the treatment and the adverse effects. Overall, GH has a good safety record. However, GH treatment has many potential and real adverse effects that need to be considered and monitored during treatment. Recently, safety concerns regarding the long-term effect of GH therapy on cardiovascular morbidity have come under scrutiny.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Growth Hormone (GH) is a peptide hormone secreted by the anterior pituitary gland. As its name suggests, it is an important driver of somatic growth in children. Deficiency of GH results in short stature. The use of GH therapy for short stature was introduced in the 1950s with the use of cadaveric GH derived from postmortem human pituitary glands [1]. The source of the hormone greatly limited its availability till the 1980s, when recombinant human GH (rhGH) was approved for clinical use. This availability has increased potential for the use and the abuse of GH. Potential adverse effects and very high cost limit its use.
The authors briefly discuss below the basic principles involved in the use of GH for common pediatric indications (Table 1), including dosing, contraindications, and adverse effects.
Principles of Growth Hormone Therapy
It is believed that tall stature is a socially and cosmetically desirable quality. GH, as its name implies, may seem like an attractive option to enhance statural growth. However, GH has the potential to cause serious adverse effects and should only be used after weighing the pros and cons.
-
1.
The goal of GH therapy is to increase growth velocity to achieve final adult height appropriate for the child’s genetic potential and to a physically and socially acceptable range.
-
2.
The indication to initiate GH therapy should be based on accepted and established clinical and laboratory criteria and not just on demand.
-
3.
The lowest possible dose of GH that can attain optimum growth velocity should be used.
-
4.
The patients and their family should be counseled regarding the adverse effects that may arise during treatment.
Specific Indications for Growth Hormone Therapy
Growth Hormone Deficiency
Growth hormone deficiency (GHD) is the most obvious and accepted indication for GH therapy. GHD is primarily a clinical and auxologic diagnosis and a single isolated nonstimulated low serum GH value should not be used to establish a diagnosis of GHD. It may be difficult to accurately establish a diagnosis of GHD, especially in patients with partial GH deficiency. In all cases, the diagnosis should be established as per appropriate guidelines [2]. It is also important to recognize concomitant deficiencies of other pituitary (in particular, anterior pituitary) hormones. Children with GHD can have deficiency of one or more of the following hormones—adrenocorticotrophic hormone (ACTH) resulting in decreased levels of serum cortisol, thyroid stimulating hormone (TSH) resulting in decreased circulating levels of thyroxine, and deficiency of luteinizing hormone (LH) and follicle stimulating hormone (FSH) resulting in delayed or stalled pubertal development. Whereas deficiency of FSH and LH may only become apparent in the peripubertal age, cortisol and thyroxine deficiency should be investigated before starting GH treatment. If there is cortisol or thyroxine deficiency along with GHD, they should be treated in the following order—cortisol replacement, followed a few days later by thyroxine replacement, and last should be GH replacement after stabilization of cortisol and thyroxine levels.
GH therapy will induce a rapid catch-up increase in the growth velocity in these children during the initial two to three years of treatment. There will be a subsequent decline in the growth velocity. The Pediatric Endocrine Society (PES) recommends a starting at a dose of 22–35 μg/kg/d [2]. For most preparations of rhGH, the conversion formula from units to mg is 3.0 IU = 1 mg (for some, it is 2.5 IU = 1 mg). Subsequent dose schedule should be individualized to maintain growth velocity and IGF1 levels in the normal range. Once growth velocity has decreased to less than 2 cm per year, GH treatment at the pediatric doses should be discontinued and the patient reassessed for adult GHD, if indicated. The thyroid axis should be reassessed after initiating GH therapy for acquired tertiary/secondary hypothyroidism that can develop as a side effect of GH treatment.
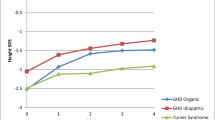
A study from India of patients with GHD has shown that the maximum height gain is during the first year. The response to treatment was similar in both genders. The response to treatment negatively correlated with the chronological age, bone age, and height age at presentation [3]. Long-term follow-up of patients treated for idiopathic GHD has shown that the final height of these children is in the midparental height range but the absolute height outcome remains in the lower part of the normal range [4].
Turner Syndrome
Turner syndrome (TS) has a prevalence of 25 to 50 per 100,000 females and nearly all patients have short stature. A multinational survey reported that the mean adult height of women with TS was 144.3 ± 6.7 cm compared to the population mean height in non-TS women of 160.2 ± 6.3 cm [5]. GH therapy is recommended in children with TS starting around 4 to 6 y of age (or earlier if there is decrease in growth velocity), if the child has evidence of growth failure i.e., the child has short stature, has a short predicted adult height, or the height velocity observed over a 6 mo period is less than the 50th centile [6]. The recommended dose of GH is 45–50 μg/kg/d or 1.3–1.5 mg/m2/d (4.0–4.5 IU/m2/d). The dose may be increased up to 68 μg/kg/d (2.0 mg/m2/d) if the predicted adult height potential is substantially compromised. The average height gain compared to the baseline predicted, or historical controls is 5 to 8 cm when treated for a period exceeding 5–6 y. The outcome is better in children with taller parents, younger age at initiation of treatment, and a longer duration between initiation of GH treatment and puberty. The treatment should be discontinued once bone age is 14 y. Children with TS are at greater risk of adverse effects such as scoliosis, raised intracranial pressure, and slipped capital femoral epiphysis while on treatment with GH than other children [7, 8]. If a late diagnosis is made and the predicted adult height is unsatisfactory, oxandrolone (0.03 mg/kg/d) may be added after 10 y of age. Oxandrolone is an anabolic steroid, and the patient must be monitored for virilizing adverse effects.
Noonan Syndrome
Noonan syndrome (NS) is an autosomal dominant condition occurring in approximately 1:2000 births. The phenotype varies but can include short stature, dysmorphic facies (hypertelorism, down-slanting palpebral fissures, ptosis, and low-set posteriorly rotated ears), neck webbing, increased carrying angle, developmental delay, cardiac abnormalities, cryptorchidism, and/or hypogonadism, and coagulation abnormalities.
In NS there appear to be abnormalities with the GH–IGF1 axis including a degree of GH resistance. Studies indicate that GH can improve final adult height to a variable degree (0.6–2.0 SDS, or approximately 4–13 cm) in these children. As for most indications for GH treatment, the greatest benefits are seen when treatment is started at an early age. GH doses of between 0.23 mg/kg/wk and 0.46 mg/kg/wk have been used for treatment of short stature associated with NS.
Chronic Renal Insufficiency
Children with chronic renal insufficiency (CRI) may exhibit short stature and reduced adult height due to malnutrition, dyselectrolytemia, metabolic disturbances, anemia, bone disorders, and underlying comorbidities. These children also develop GH insensitivity. Hence, GH therapy is recommended for children with CRI stage 3 to 5, if they exhibit growth failure defined as height below the 3rd centile, growth velocity below the 25th centile over a period of 3 mo in infants and 6 mo in children and other treatable causes of growth failure have been excluded or treated [9]. GH therapy should not be initiated if the epiphyses have fused. In post-transplant patients, GH therapy is indicated only if there is no catch-up growth after 1 y of transplant. Contraindications to GH include severe secondary hyperparathyroidism (parathyroid hormone levels > 500 pg/mL), proliferative or severe nonproliferative diabetic retinopathy, acute critical illness, or active malignancy.
Studies have shown that children on conservative treatment respond better to GH therapy than those receiving dialysis [10]. The recommended dose of GH in CRI patients is 0.045–0.05 mg/kg/d. The dose should be adjusted according to body weight at regular intervals. A Cochrane review estimated that 28 IU/m2/wk of GH in children with CRI resulted in an increase in height velocity of 3.8 cm/y as compared to untreated patients [11].
Treatment with GH should be stopped once the epiphyses fuse, at the time of kidney transplantation, if the patient develops severe secondary hyperparathyroidism, slipped capital femoral epiphyses, intracranial hypertension, or in the event of an unexplained decrease in estimated glomerular filtration rate. Before initiating treatment, the cost and the burden of care in these children should be discussed with the family.
Small for Gestational Age
Small for Gestational Age (SGA) is defined as birth weight less than –2 SD below the mean. Most of these children have catch-up growth and reach a height above –2 SD for their age by the time they are 2–3 y old. Children who do not achieve this catch-up growth are at risk of being short adults. Studies have shown that GH therapy is effective in normalizing height in short, non-GH deficient SGA children [12]. In 2001, GH was approved by the FDA for SGA children who failed to exhibit catch up. It is indicated for SGA children who at 2–3 y of age, do not achieve catch-up growth, continue to have a height below –2 SD for their age, and have an average or subnormal growth velocity, provided other causes of short stature have been excluded [13]. The recommended doses are between 0.24 to 0.48 mg/kg/wk (0.1–0.2 IU/kg/d). Studies report that the height gain as adults in SGA children treated with GH is 0.9 SDS more than untreated control patients [14]. These children should be regularly monitored for effects of GH on glucose and lipid metabolism and blood pressure during and after treatment with GH.
Prader–Willi Syndrome
Prader–Willi syndrome (PWS) is a complex genetic syndrome characterized by obesity, hypothalamic-pituitary dysfunction, short stature, intellectual disability, and obstructive or central sleep apnea. GH treatment is approved in PWS after 2 y of age [15]. Testing for GH deficiency is not routinely indicated in children with PWS. GH treatment leads to improvement in lean body mass, motor development, and normalization of body habitus. Some cases of sudden death have been reported in children with PWS on GH treatment, although the role of GH treatment in the mortality is not clear. Before initiating treatment, a basic metabolic profile, radiograph for bone age, Serum IGF1 and IGFBP3 levels, and thyroid function tests should be obtained. Serum IGF1 level should be measured at least twice a year to maintain it in the physiologic range. Polysomnography should be done at the start of the treatment, 6 to 10 wk after initiation of therapy, and repeated on an annual basis or if there are new symptoms. Head circumference should also be monitored at every visit as GH is known to cause abnormal head growth in young children.
Idiopathic Short Stature
Idiopathic short stature (ISS) refers to a condition where a child is short, but no cause can be identified, and the child is not GH deficient. GH therapy may be started in these children with height SDS ≤ − 2.25 on a case-by-case basis after discussing the risks and benefits of GH treatment [2]. The group of children with ISS is very heterogenous and there is marked variation in the response. GH therapy can be started at a dose of 0.24 mg/kg/wk. The dose should not exceed 0.47 mg/kg/wk. A close follow-up is required for ongoing dose optimization.
SHOX Gene Mutations
SHOX gene function is dose-dependent, with haploinsufficiency (loss of function of one allele) resulting in SHOX deficiency and short stature, and loss of both SHOX alleles causing the more extreme phenotype of Langer syndrome osteodysplasia (extreme short stature, mesomelia, and limb deformity). SHOX deletions also cause the Leri–Weill syndrome (LWS). With haploinsufficiency, the phenotype presents a variable spectrum from isolated short stature with no obvious skeletal phenotype, often diagnosed as ISS, to the more classic phenotype seen in LWS or TS.
Children with SHOX mutations have been shown to respond to GH, with very similar outcomes to that seen in TS. Short stature due to SHOX mutation is an FDA approved indication for the use of GH. GH doses similar to that for TS are appropriate, and the outcome is influenced by many of the patient-specific variables listed above for GHD and TS.
GH Initiation and Administration
GH is available only as an injectable preparation. There are no oral formulations. It is available as a solution in prefilled pen injectable devices or as a dry powder form that must be reconstituted prior to administration. The drug must be injected subcutaneously daily. Administration in the evening is recommended. Longer acting (e.g., weekly) preparations are currently under clinical trials. The dose of GH is measured in IU as well as milligrams. For most preparations 3.0 IU = 1 mg.
Prior to initiating GH therapy other potentially treatable causes of GHD should be identified and addressed. The cost–benefit ratio and potential side effects should be discussed with the family. It is strongly recommended to collect accurate anthropometric data over a period of 6 mo before initiating treatment so that response to treatment can be adequately assessed (except in GH-deficient infants presenting with hypoglycemia where treatment must be initiated urgently.)
Safety and Adverse Effects
Recombinant GH therapy in its present form has been in use since 1985 for GHD with several newer indications being added over time. Overall, the drug has a good safety record. However, GH treatment has many potential and real adverse effects that need to be considered and monitored during treatment (Table 2).
Malignancy
GH, primarily via its action of increasing IGF1 levels, promotes mitogenic activity and tissue growth, hence, the concern regarding its ability to increase risk of various malignancies in children treated with GH. Initial reports raised an alarm on the possibility of increased incidence of hematologic malignancies in children treated with GH. However, two reports from large-scale databases have concluded that the risk of leukemia in GH-treated patients is comparable to the general population [16] and increased occurrence of leukemia is seen only in GH-treated patients who have underlying risk factors for malignancy such as Fanconi anemia, previous radiation, or chemotherapy [17]. There is no association between GH therapy and the recurrence or occurrence of a brain tumor [18]. It is recommended that GH treatment be initiated only after a minimum of 1-y malignancy-free period with the recognition that GH therapy may increase chances of a second malignancy [19]. For children with genetic syndromes that can increase risk of hematologic malignancies, appropriate counseling should be instituted [2].
Intracranial Hypertension
Intracranial hypertension (pseudotumor cerebri) is a known complication of GH therapy. It is mostly seen at the initiation of treatment or with an increase in the dose. GH treatment must be stopped immediately as continued treatment can lead to blindness and even death. In a prospective study it was observed that GH-deficient patients with hypothalamic–pituitary anomalies or genetic or chromosomal diseases are at higher risk for pseudotumor cerebri [20]. The KIGS (Pfizer International Growth Study) database has shown that the incidence intracranial hypertension was 27.7 per 100,000 treatment years. The incidence is higher in children with TS, PWS, CRI, and congenital GHD [7].
Slipped Capital Femoral Epiphyses
Slipped Capital Femoral Epiphyses (SCFE) is a condition where there is a displacement of the femoral head epiphysis from its usual alignment. SCFE presents with hip joint pain with a limping gait. SCFE has an incidence of 73.4 per 100,000 treatment years. The median duration of onset of SCFE after initiation of GH therapy was 0.4 to 2.5 y [7]. In all suspected cases radiograph of the hip joint must be done with appropriate orthopedic evaluation and treatment.
Scoliosis
Scoliosis may develop or be accentuated during GH therapy. It is more commonly seen in patients with TS or PWS. Evaluation for presence and progression of scoliosis should be done in children receiving GH therapy [2].
Hypothyroidism
Initiation of GH therapy may lead to secondary/tertiary hypothyroidism which, if left untreated, may lead to a suboptimal response to GH therapy. Hence, thyroid function should be monitored periodically in all children on GH therapy.
Long-Term Deleterious Cardiovascular Effects
Recently, studies predominantly from Europe, have raised concerns about the long-term deleterious cardiovascular effects of GH treatment [21, 22]. However, conclusions of causality are still limited and the absolute risk remains low. Further long-term robust studies will be needed to make definite statements on this matter.
Adjuvants to Growth Hormone Therapy
The use of agents, such as gonadotropin-releasing hormone analogs and aromatase inhibitors, to augment GH-induced growth is discussed in the review article by Saroufim and Eugster in this issue of the Journal [23].
References
Ayyar VS. History of growth hormone therapy. Indian J Endocrinol Metab. 2011;15 Suppl 3:S162–5.
Grimberg A, Divall SA, Polychronakos C, et al. Guidelines for growth hormone and insulin-like growth factor-I treatment in children and adolescents: growth hormone deficiency, idiopathic short stature, and primary insulin-like growth factor-I deficiency. Horm Res Paediatr. 2016;86:361–97.
Garg MK, Pakhetra R, Dutta MK, Gundgurthi A. Response to growth hormone therapy in Indian patients. Indian J Pediatr. 2010;77:639–42.
Reiter EO, Price DA, Wilton P, Albertsson-Wikland K, Ranke MB. Effect of growth hormone (GH) treatment on the near-final height of 1258 patients with idiopathic GH deficiency: analysis of a large international database. J Clin Endocrinol Metab. 2006;91:2047–54.
Ranke MB, Grauer ML. Adult height in Turner syndrome: results of a multinational survey 1993. Horm Res. 1994;42:90–4.
Gravholt CH, Andersen NH, Conway GS, et al. Clinical practice guidelines for the care of girls and women with turner syndrome: proceedings from the 2016 cincinnati international turner syndrome meeting. Eur J Endocrinol. 2017;177:G1–70.
Darendeliler F, Karagiannis G, Wilton P. Headache, idiopathic intracranial hypertension and slipped capital femoral epiphysis during growth hormone treatment: a safety update from the KIGS database. Horm Res. 2007;68 Suppl 5:41–7.
Bell J, Parker KL, Swinford RD, Hoffman AR, Maneatis T, Lippe B. Long-term safety of recombinant human growth hormone in children. J ClinEndocrinolMetab. 2010;95:167–77.
Drube J, Wan M, Bonthuis M, et al. clinical practice recommendations for growth hormone treatment in children with chronic kidney disease. Nat Rev Nephrol. 2019;15:577–89.
Wühl E, Haffner D, Nissel R, Schaefer F, Mehls O. Short dialyzed children respond less to growth hormone than patients prior to dialysis. German study group for growth hormone treatment in chronic renal failure. Pediatr Nephrol. 1996;10:294–8.
Vimalachandra D, Hodson EM, Willis NS, Craig JC, Cowell C, Knight JF. Growth hormone for children with chronic kidney disease. Cochrane Database Syst Rev. 2006;19:CD003264.
de Zegher F, Albertsson-Wikland K, Wollmann HA, et al. Growth hormone treatment of short children born small for gestational age: growth responses with continuous and discontinuous regimens over 6 years. J ClinEndocrinolMetab. 2000;85:2816–21.
Lee PA, Chernausek SD, Hokken-Koelega AC, Czernichow P. International Small for Gestational Age Advisory Board. International small for gestational age advisory board consensus development conference statement: management of short children born small for gestational age, April 24-October 1, 2001. Pediatrics. 2003;111:1253–61.
Maiorana A, Cianfarani S. Impact of growth hormone therapy on adult height of children born small for gestational age. Pediatrics. 2009;124:e519–31.
McCandless SE; Committee on Genetics. Clinical report—health supervision for children with Prader-Willi syndrome. Pediatrics. 2011;127:195–204.
Allen DB, Rundle AC, Graves DA, Blethen SL. Risk of leukemia in children treated with human growth hormone: review and reanalysis. J Pediatr. 1997;131:S32–6.
Nishi Y, Tanaka T, Takano K, et al. Recent status in the occurrence of leukemia in growth hormone-treated patients in Japan.GH treatment study committee of the foundation for growth science, Japan. J Clin Endocrinol Metab. 1999;84:1961–5.
Teramoto A. Growth hormone therapy and brain tumors. Pediatr Endocrinol Rev. 2017;14Suppl 1(Suppl 1):229–34.
Raman S, Grimberg A, Waguespack SG, et al. Risk of neoplasia in pediatric patients receiving growth hormone therapy–a report from the pediatric endocrine society drug and therapeutics committee. J Clin Endocrinol Metab. 2015;100:2192–203.
Martín-Begué N, Mogas E, WolleyDod C, et al. Growth hormone treatment and papilledema: a prospective pilot study. J Clin Res Pediatr Endocrinol. 2021;13:146–51.
Carel JC, Ecosse E, Landier F, et al. Long-term mortality after recombinant growth hormone treatment for isolated growth hormone deficiency or childhood short stature: preliminary report of the French SAGhE study. J Clin Endocrinol Metab. 2012;97:416–25.
Tidblad A, Bottai M, Kieler H, Albertsson-WiklandK, Sävendahl L. Association of childhood growth hormone treatment with long-term cardiovascular morbidity. JAMA Pediatr. 2021;175:e205199.
Saroufim R, Eugster EA. Non-GH Agents and Novel Therapeutics in the Management of Short Stature. Indian J Pediatr. 2021. https://doi.org/10.1007/s12098-021-03824-3.
Author information
Authors and Affiliations
Contributions
VV and RKM have contributed equally to the manuscript and approved the final version of the manuscript. RKM is the guarantor for this paper.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Vyas, V., Menon, R.K. Management of Short Stature: Use of Growth Hormone in GH-Deficient and non-GH–Deficient Conditions. Indian J Pediatr 88, 1203–1208 (2021). https://doi.org/10.1007/s12098-021-03892-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-021-03892-5