
Abstract
Objectives
To screen for variants in the MC4R and LEP genes in 46 patients with clinical suspicion of non-syndromic early onset severe obesity (NEOSO).
Methods
Children with early onset obesity satisfying WHO criteria of obesity were studied. The MC4R and LEP genes were sequenced using a PCR amplicon based NGS on Illumina MiSeq next generation sequencer using an in-house developed protocol.
Results
Of the 46 children tested, four were found to have novel pathogenic/likely-pathogenic variants (one in the MC4R gene and three in the LEP gene). In three out of the 4 families, the presence of the variants was confirmed using standard bidirectional capillary sequencing in the probands.
Conclusions
Four children with novel likely pathogenic variants in the MC4R and LEP genes are reported. Genetic analysis is crucial in children with early onset obesity and should be considered.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Childhood obesity is a serious problem in today’s world and is increasing at an alarming rate [1]. The etiology is multifactorial, resulting from a complex interaction of genetic and environmental factors. Apart from well-known dietary and lifestyle factors, childhood obesity is also influenced by genetic factors [2]. In fact, genetic factors are reported to contribute to 40–70% variation in body mass between individuals [3, 4]. Genetic causes include chromosomal [Prader Willi syndrome (PWS)] and non-chromosomal obesity syndromes (Bardet Beidl syndrome, Cohen syndrome etc.) [5]. Another form of hereditary obesity is non-syndromic early onset severe obesity (NEOSO), resulting from pathogenic/likely-pathogenic variants in the genes involved in the hypothalamic-leptin-melanocortin pathway (LEP, LEPR, MC4R, PC1/ PC3/ PCSK1, POMC) and hypothalamic formation (SIM1, BDNF, NTRK2) which are responsible for controlling food intake and body weight [6]. Genetic variants in the LEP, LEPR and MC4R genes are most frequent cause of monogenic forms of non-syndromic obesity [6].
Leptin is a protein secreted by adipocytes, and its concentration in blood positively correlates with body fat mass and body mass index (BMI). Mutations in LEP gene result in undetectable to low serum leptin levels resulting from improper protein folding, intracellular transport and secretion, or loss of biological activity of secreted protein [7]. Mutations in LEPR often lead to a truncated receptor, preventing leptin binding and signaling through hypothalamic-leptin-melanocortin pathway [8].
MC4R is a seven-transmembrane G protein coupled receptor principally expressed in brain, including hypothalamus. MC4R gene mutations represent some of the most common monogenic causes of NEOSO and MC4R deficiency is the most common monogenic form of obesity [9].
Approximately 7% NEOSO are monogenic and are caused due to gene variants of major effect [10, 11]. The clinical presentation is non-specific. Identifying these genetic variants is important to plan personalized clinical management (for example, leptin replacement therapy for patients with LEP gene mutations) and provide genetic counseling to patient and family (these mutations are autosomal recessive and parents are usually carriers) [12].
Before the advent of Next generation sequencing (NGS), sequential capillary sequencing of LEP, LEPR and MC4R genes was the mainstay of molecular diagnosis, which was both time consuming and labor intensive. NGS has superseded capillary sequencing for molecular typing because of its huge multiplexing capability for genomic targets as well as number of samples, and reduced relative cost. The authors have developed a targeted NGS assay comprising of two NEOSO associated genes, LEP and MC4R. With this panel, authors screened for variants in the MC4R and LEP genes in 46 patients with NEOSO, and identified novel, likely pathogenic frameshift variants in MC4R and LEP genes in four patients.
Material and Methods
Children from a tertiary level care pediatric endocrine unit with NEOSO (age less than 5 y) were included in the study. Obesity was defined for <2 y of age as sex-specific weight for recumbent length > 97.7th percentile and 2–5 y as BMI > 95th percentile for age and sex [12]. A detailed history was recorded, anthropometric measures were noted and converted to Z scores [13] and clinical examination was performed. Children who appeared to have nutritional obesity were excluded from the study. Three milliliters of peripheral blood was collected from patients in K2 – EDTA vacutainers (Becton Dickinson, MD, USA). Blood samples were also collected from parents who were willing to check their carrier status. The institutional ethics committee approved the study and parents gave written informed consent.
Genomic DNA was extracted using DNeasy Blood and Tissue kit (Qiagen, Germany) as per the manufacturers’ instructions. The MC4R and LEP genes were sequenced using a PCR amplicon based NGS on an Illumina MiSeq next generation sequencer. The broad steps involved were: generation of PCR amplicons, tagmenation of amplicons, ligation of Illumina specific index-adapter sequences and quantitation, purification and pooling of amplicons for sequencing. Primer sequences were designed using Primer Express v2.0 (Applied Biosystems, CA, USA) (Table 1). A long range PCR protocol using Q5 Polymerase (NEB) and an in-house master mix was used to amplify the 1.4 kb product of the MC4R gene. For the LEP gene, a multiplex PCR was designed to amplify both exons of the LEP gene using the Qiagen Quantitect (Qiagen) PCR mix (per the manufacturer’s instructions). PCR amplicons were purified and diluted to 0.2 ng/μL before tagmentation. In one step, tagmentation protocol allows fragmentation of the PCR amplicons to about 250 bp length and integrates the Illumina adapters. An index PCR aids in ligation of the indices on the amplicons. These indices act as barcodes that allow processing of large number of samples simultaneously, and were incorporated through a secondary PCR step. Further, the PCR products were quantified using the Qubit fluorometric system. Concentrations were molar normalized for pooling of samples. The pooled library was purified using the Purelink PCR Purification Kit (Invitrogen) and diluted down to 4 nM final concentration using Resuspension Buffer (RSB – Illumina, CA, USA). The library was denatured using 0.2 N NaOH and further neutralized and diluted to a final concentration of 15pM using HT1 Buffer (Illumina, CA, USA). Further, a 5% phiX library spike (Illumina, CA, USA) was added as a control and diversity enhancer. The final library was loaded onto an Illumina MiSeq cartridge (Illumina, CA, USA) and run in 2*300 or 2*250 mode on an Illumina MiSeq (Illumina, CA, USA).
In all samples, the average coverage exceeded 10x with a sequence quality (>Q30) of more than 80% in all the targeted regions. Bioinformatics analysis was carried out using Illumina's BaseSpace cloud analytics platform along with an in-house developed pipeline. Generated .fastq files were trimmed for Q score > 30 and adaptors followed by alignment to release GRCH37/ hg19 of the reference human genome assembly. Identified variants were analyzed using VarSeq (VarSeq™ v2.x, Golden Helix, Inc., Bozeman, MT).) and .bam files were visualized in the GenomeBrowse (GoldenHelix Inc., Bozeman, MT, USA) tool. Mutations were detected and identified manually.
Results
Targeted next generation sequencing was performed using a panel comprising of the MC4R and LEP genes (two exons) on 46 patients with NEOSO. The mean age of the children at the time of diagnosis was 25.2 ± 19 mo (n = 46, 22 girls, 24 boys). Fourteen children were younger than 2 y and 32 were > 2 y of age when tested. Mean length for age Z score in children younger than 2 y was 6.6, weight for age Z score was 7.6, and weight for length Z score was 4.9 (all above +2). For children older than 2 y of age, height Z score was 2.1, weight Z score was 4.5 and BMI Z score was 4.4 (all above +2) as per WHO standard [10].
Of the 46 patients tested, four patients were identified with pathogenic/ likely-pathogenic variants in the MC4R or LEP gene. Out of these 4 variants, one variant in the MC4R gene has previously been reported in heterozygous state in the ExAC database [14]. The three variants in the LEP gene are novel and have, to the best of authors’ knowledge, not been described in published literature. The authors did not detect any clinically relevant small sequence variants in the other cases.
Description of Patients with Novel Mutations in the MC4R and LEP Genes
Patient 1
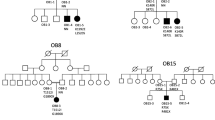
A 2.5-y-old girl weighing 28.7 kg (Z score = 6.9), height 101.2 cm (Z = 2.8SD) and BMI 27.9 kg/m2 (Z = 7.2SD) was referred for severe obesity. The child was born to parents with 3rd degree consanguinity. Parents reported a delay in achieving early milestones. Hearing, vision and speech were normal. She did not have any history of frequent infections or comorbidities. Thyroid function tests and glucose concentrations were within reference range. Serum leptin levels were 3.7 ng/ml. Targeted NGS panel showed presence of homozygous deletion of two nucleotides MC4R:c.63_64delCA. This variant had been reported once in the ExAC database in a heterozygous form, but had not been reported, to the best of authors' knowledge, in the homozygous state prior to the diagnosis of this case [15]. This frameshift variant changes codon for tyrosine (Y) to a stop codon resulting in a premature termination of the amino acid chain at the 21st position (p.Tyr21Ter) (Fig. 1). Using in silico mutation prediction (MutationTaster), this variant was predicted to be disease causing. The parents were subsequently shown to be heterozygous carriers for the same variant. Targeted bidirectional capillary sequencing independently confirmed presence of variant in homozygous state in the child and in heterozygous states in each parent. The predicted loss of function in MC4R gene in the patient correlates with the phenotypic presentation of early onset severe obesity. The family was provided dietary advice, genetic counseling and the option of prenatal diagnosis for future pregnancies.

MC4R gene c.63_64 delCA homozygous frameshift mutation. (a) Schematic representation of location of MC4R frameshift deletion in chromosome 18. (b) NGS read pile up showing MC4R:c.63_64delCA (These NGS data are in reverse complement orientation) in the proband (homozygous) and in the carrier parents (heterozygous) (c) Capillary DNA sequencing showing patients’ c.63_64delCA GCAGTTA//GACTG compared to normal sequence.
Patient 2
An 8-mo-old boy presented with severe obesity with weight of 14.4 kg (Z = 5.1), length of 80 cm (Z = 4.2) and weight for length Z score of 3.7. The child was born to parents with 3rd degree consanguinity. He had a birth weight of 3 kg and showed a rapid weight gain with age. Early developmental milestones were normal. Hearing, vision and speech were apparently normal. Thyroid function tests and glucose concentrations were within reference range. Serum leptin concentration was 1.1 ng/ml.
The NGS assay revealed the presence of a novel frameshift variant NM_000230.2:c.142_143delAC with a deletion of two nucleotides in exon 2 of the LEP gene. This frameshift variant results in a change in codon at the 48th position (of a 167 amino acid protein) from a Threonine residue to a stop (p. The48Ter) (Fig. 2a), causing a premature termination of the protein. In silico prediction software classified this variant as “disease causing” or “deleterious”. The truncated gene was predicted to result in a protein that has limited or no activity, which correlates with clinical observation of reduced leptin levels. The mother was confirmed to be heterozygous carrier. The father’s sample was not available. Further, bidirectional capillary sequencing confirmed the presence of the variant in the child and his mother. The family was advised dietary management and lifestyle changes as leptin therapy is not available in India.

Data of patients with novel likely pathogenic variants in the LEP gene. (a) NGS read pile up in patient 2, showing LEP:c.142_143 homozygous deletion in patient and heterozygous deletion in the parent (mother). (b) Capillary DNA sequencing showing patients’ c.142_143 delCA CATTTCACAC//GGTA compared to normal sequence seen in patient 2. (Note that the 'CA' deletion is shown in different positions due to the NGS data being left aligned and the capillary sequence data being right aligned) (c) NGS read pile up showing LEP: NM_000230:c.453delG single nucleotide deletion in the patient 3 (upper lane) and LEP c.461 T > C / p.Leu154Pro variant in the patient 4 (lower lane)
Patient 3
A 6-mo-old girl presented with severe, early onset obesity with weight of 13.9 kg (Z score 5.7), length of 69 cm (Z score 1.4) and weight for length Z score of 6.3. The child was born to parents with 2nd degree consanguinity. She had a birth weight of 3.3 kg and showed rapid weight gain from 3 mo of age. Early developmental milestones, hearing, vision and speech were normal. Serum leptin concentrations were 0.9 ng/ml.
The NGS assay revealed presence of a single nucleotide deletion NM_000230:c.453delG [NP_000221.1:p.Ser153fs] in a homozygous state in exon 3 of the LEP gene (Fig. 2c). This variant is predicted to result in a prolonged protein; in silico prediction (MutationTaster) classified this variant as “likely pathogenic”. Further, bidirectional capillary sequencing confirmed the presence of the variant. The family was provided genetic counseling; the parents, however, did not consent to testing of their blood. To best of authors’ knowledge, this variant has never been reported in literature.
Patient 4
A 4-mo-old girl presented with severe, early onset obesity with weight of 12 kg (Z score 4.8), length of 68 cm (Z score 1.9) and weight for length Z score of 4.7. The child was born to parents with 2nd degree consanguinity. She had a birth weight of 3.4 kg and showed a rapid weight gain from 3 mo of age. Early developmental milestones including hearing, vision and speech were normal. Serum leptin levels were 0.7 ng/ml.
The NGS assay revealed presence of missense variant NM_000230.2:c.461 T > C [NP_000221.1:p.Leu154Pro, ClinVar submission ID 448905] in homozygous state in exon 3 of LEP gene (Fig. 2c). In silico prediction (Sift, Polyphen and MutationTaster) classified this variant as “disease causing” or “pathogenic”. Further, bidirectional capillary sequencing confirmed the presence of the variant. The parents did not consent to genetic testing. To best of authors’ knowledge, this variant has also never been reported in literature.
The clinical characteristics of cases in which variants were detected are summarized in Table 2.
Discussion
The authors have used a custom in-house developed NGS assay to analyze the LEP and MC4R genes in 46 patients with clinical diagnosis of NEOSO, and identified novel, previously unreported, pathogenic variants in the MC4R and LEP genes in four patients. The variant in the MC4R gene was reported only once in ExAC database in a heterozygous state, but had not been reported in a homozygous state. In all families there was a history of consanguinity and presence of variants was confirmed in heterozygous form in two sets of parents. The mutations in the LEP gene are novel variants that, to best of authors’ knowledge, have not been reported in published literature. The LEPR gene is also one of the common genes associated with monogenic non-syndromic early onset obesity, however, authors were not able to include it in the current panel.
The eight important genes causing NEOSO are LEP, LEPR, MC4R, PC1/ PC3/ PCSK1, POMC, SIM1, BDNF and NTRK2 which are mainly involved in satiety control, energy homeostasis and development of the hypothalamus. The LEP, MC4R and LEPR genes are probably the most significant, contributing to 4–7% of all cases of childhood obesity (70–80% of all cases of NEOSO) [10]. There are very few clinical clues for presence of pathogenic variants in a particular gene. Children with mutations in the LEP gene are known to have endocrine (hypogonadotropic hypogonadism and hypothyroidism) and immunological abnormalities [14]. Identification of the pathogenic/ likely pathogenic variants in the LEP gene helps in management, counseling, prediction of recurrence risk and providing prenatal diagnosis in any subsequent pregnancy. Further, patients with LEP mutations are known to respond to dietary management and leptin therapy [16].
Variations in the MC4R gene (OMIM *155541) have a population prevalence of at least one in 2000 (0.05%), and are found in 0.5–1% of obese adults and are accountable for 6% of all severe cases of disease starting in childhood [9, 17]. Pathogenic variants in the MC4R gene have been reported to be associated with both autosomal dominant and recessive forms of NEOSO with dominant form being much more common [18]. In index case reported (Case 1), patient had a homozygous frame shift mutation and parents were heterozygous.
Variants in the LEP and MC4R were classified as “likely pathogenic” based on available evidence from the published literature and databases and through in silico mutation prediction, according to guidelines laid down by American College of Medical Genetics and Genomics [19]. The clinical features of the four patients were consistent with previous reports of loss of function mutations in the MC4R and LEP genes [20, 21]. Pathogenic variants in the LEP or LEPR genes are inherited in an autosomal recessive form, hence homozygous mutations result in the disorder [7]. In the cases reported above, homozygous mutations were found in patients 2, 3 and 4. However, some reports suggest that even heterozygous mutations in the LEP gene (ΔG133 LEP) lead to low leptin levels and are responsible for obesity [22].
With advent of whole exome sequencing, a large number of genes and genetic variants are being newly identified to be associated with NEOSO. The identification of these newer variants has contributed significantly to pathophysiology of non-syndromic obesity and has potential to develop novel therapeutic options.
In conclusion, authors report 4 children with NEOSO with novel likely pathogenic variants in the MC4R and LEP genes. Genetic analysis is crucial in children with early onset obesity despite not having any syndromic features and can be considered. This will help in prognosis, treatment and genetic counseling and option of prenatal diagnosis.
References
Roberto CA, Swinburn B, Hawkes C, et al. Patchy progress on obesity prevention: emerging examples, entrenched barriers, and new thinking. Lancet. 2015;385:2400–9.
Stunkard AJ, Harris JR, Pedersen NL, McClearn GE. The body-mass index of twins who have been reared apart. N Engl J Med. 1990;322:1483–7.
Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA. 1986;256:51–4.
Carnell WS, Haworth CM, Plomin R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87:398–404.
O'Rahilly S, Farooqi IS. Genetics of obesity. Philos Trans R Soc Lond Ser B Biol Sci. 2006;361:1095–105.
Huvenne H, Dubern B, Clément K, Poitou C. Rare genetic forms of obesity: clinical approach and current treatments in 2016. Obes Facts. 2016;9:158–73.
Paz-Filho G, Mastronardi C, Delibasi T, Wong ML, Licinio J. Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol. 2010;54:690–7.
Farooqi IS, Wangensteen T, Collins S, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007;356:237–47.
Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O'Rahilly S. Clinical spectrum of obesity mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–95.
Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206.
O'Rahilly S. Human genetics illuminates the paths to metabolic disease. Nature. 2009;462:307–14.
Styne DM, Arslanian SA, Connor EL, et al. Pediatric obesity—assessment, treatment, and prevention: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2017;102:709–57.
de Onis M, Onyango AW, Borghi E, Siyam A, Nishida C, Siekmann J. Development of a WHO growth reference for school-aged children and adolescents. Bull World Health Organ. 2007;85:660–7.
Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–5.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Farooqi IS, Jebb SA, Langmack G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–84.
Dubern B, Clément K, Pelloux V, et al. 2001. Mutational analysis of melanocortin-4 receptor, agouti-related protein, and alpha-melanocyte-stimulating hormone genes in severely obese children. J Pediatr. 2001;139:204–9.
Farooqi IS, Yeo GS, Keogh JM, et al. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106:271–9.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Nordang GBN, Busk ØL, Tveten K, et al. Next-generation sequencing of the monogenic obesity genes LEP, LEPR, MC4R, PCSK1 and POMC in a Norwegian cohort of patients with morbid obesity and normal weight controls. Mol Genet Metab. 2017;121:51–6.
Rouskas K, Meyre D, Stutzmann F, et al. Loss-of-function mutations in MC4R are very rare in the Greek severely obese adult population. Obesity (Silver Spring). 2012;20:2278–82.
Farooqi IS, Keogh JM, Kamath S, et al. Partial leptin deficiency and human adiposity. Nature. 2001;414:34–5.
Author information
Authors and Affiliations
Contributions
VK: Concept, patient management and manuscript draft; NP: NGS assay design; NG, PG, RLO: Data collection and manuscript draft; NP, MA, AK, NL: Patient management and manuscript draft. SR, TR, KP, AP, SA, AB: Manuscript draft. KK: Data collection, analysis and manuscript writing. AK is the guarantor for this paper.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Khadilkar, V., Gogate, N., Gangodkar, P. et al. A Targeted Next Generation Sequencing Panel for Non-syndromic Early Onset Severe Obesity and Identification of Novel Likely -Pathogenic Variants in the MC4R and LEP Genes. Indian J Pediatr 87, 105–110 (2020). https://doi.org/10.1007/s12098-019-03129-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-019-03129-6