
Abstract
Increasingly, congenital thoracic malformations (CTMs) are diagnosed on antenatal ultrasound, but we lack the evidence to suggest rational management, not least because descriptive terms are used inconsistently. This review describes a simplified clinical classification of CTMs, and contrasts it with pathological descriptions. The age related presentations of CTM are described, together with the differential diagnoses of cystic masses presenting both antenatally and postnatally. Antenatally diagnosed CTMs rarely require intervention before birth; and urgent treatment is only required postnatally if the baby is symptomatic and does not respond to medical management. The asymptomatic baby with an antenatal diagnosis of a CTM presents a management conundrum. Definitive imaging is with high-resolution computed tomography (HRCT), but the optimal timing of imaging is unclear. Whether surgery should be offered to asymptomatic infants is also unclear; in the medium term, 5 % of asymptomatic babies will require surgery for complications of the disease. The most vexed question is malignant change; the risk in the medium term is probably less than 5 %, but we have no way of delineating a high-risk group. Indeed, malignancy has been described even after complete resection of a CTM. The author’s personal management is to advocate surgery in the second year of life for all except for the most trivial CTMs, but many would differ and advocate conservative management. More data are needed if we are to rationalise our approach to these infants.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The widespread adoption of antenatal anomalies ultrasound scanning has brought a resurgence of interest in congenital lung disease, and a realisation of our lack of an evidence base to manage these conditions. The field is plagued by confusing nomenclature (terms like malinosculation and congenital pulmonary airway malformation) which is not used consistently antenatally and postnatally. Antenatal diagnosis of a congenital thoracic malformation (CTM) leads to management quandaries after delivery—what actually do we do now? This review will discuss the clinical and pathological classification of CTMs, and suggest a sadly non-evidence-based management strategy.
Clinical Classification of Congenital Thoracic Malformations
The spectrum of intrathoracic congenital malformations is large, encompassing simple and complex cysts, large vessel abnormalities, chest wall deformities and thoracic manifestations of complex syndromes, including ciliopathies, reviewed in detail elsewhere [1–3]. The scope of this manuscript is to describe only the management of parenchymal cystic and solid congenital malformations.
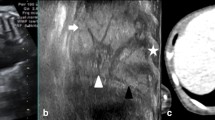
It is important to start by restating some important general principles, most clearly stated by the late, great Richard Asher [4]. The first is to acknowledge that clinicians are seeing only grey scale images and the likelihood of accurately describing the pathology from these is close to zero, in particular since mixed pathology is common within a single lesion [5]. So the first principle, to be applied both on ante-natal and postnatal images, is to describe what you actually see and do not speculate about what is invisible. Thus a congenital thoracic malformation (CTM) may be solid or cystic; and if cystic, single or multiple cysts, thin or thick walled, and with or without a significant solid component [6]. The arterial supply (from aorta or pulmonary artery) and venous drainage (to pulmonary or systemic veins) should also be worked out, to form a complete clinical description of the CTM. The second principle is obvious from the above—do not use Latin or Greek terms, but everyday language that is clear. Finally do not speculate about embryology—you will almost certainly be wrong! A recent example is so-called ‘alveolar-capillary dysplasia with misalignment of the pulmonary veins’—but recently, the so-called misaligned pulmonary veins were in fact shown to be dilated and distended bronchial veins [7]! So the author’s suggestion is to describe the malformation shown in Fig. 1 as a ‘left lower lobe multicystic CTM with aortic blood supply and venous drainage to the hemi-azygous system’ rather than ‘sequestration’—especially since the histopathology, may show any or all of sequestration, congenital cystic adenomatoid malformation (CCAM) and bronchial atresia. So for example, in one study of 24 patients (16 males), median age 3 y (IQR 2.1-9.5) in which imaging and histopathology data were correlated, there were 12 CCAMs (Type 1, n = 5; Type 2, n = 6; Type 4, n = 1); one bronchial atresia; four pleuropulmonary blastomas; and seven sequestrations, of which only one was extralobar. Histological overlaps were common, and could not be detected on imaging; 5 of 7 ‘sequestrations’ had features of CCAM, CCAM Type 4 overlapped with pleuropulmonary blastoma [6]. The issues are different for the pathologist, who can use the microscope to determine histopathology, and in consequence produce more complex classifications (discussed in more detail below [8, 9]). This clinical classification is summarised in Table 1.

HRCT with contrast, which shows what might conventionally be described as an extralobar sequestration, or possibly a CCAM. In the simplified nomenclature this would be described as a left lower lobe multicystic CTM, with arterial supply from aorta and venous drainage to the azygous system (a) HRCT shows the CTM has variable sized cysts (b) The arterial supply is from the aorta (red arrows) (c) The blue arrows show venous drainage running cranially to join the azygous system; the red arrow indicated the aorta
Pathological Classification of Congenital Thoracic Malformations
The complexities of classification of CTMs are given in Table 2. As above, features of more than one lesion may be present in the same CTM, and clinical predications are frequently revised by the pathologist.
Extralobar Sequestration
Sequestrations are localised lesions comprising lung parenchyma, receiving their blood supply via aberrant systemic arteries and lacking continuity with the upper respiratory tract. Extralobar sequestration is less common than intralobar, and has its own pleural investment. Typically the lesion is situated below the left lower lobe, but 15 % are abdominal. Diagnosis is usually antenatal.
Intralobar Sequestration
This lesion is found within the lung, especially the left lower lobe. There is controversy as to whether it is congenital or acquired. Favouring the former is the association with other congenital abnormalities; favouring the latter that it is often a later diagnosis.
Type 0 Congenital Cystic Adenomatoid Malformation
This is also known as acinar dysplasia, and presents with neonatal onset, severe respiratory failure, incompatible with prolonged life; it is often associated with other congenital abnormalities. At autopsy, the lungs are small and firm, with bronchial type airways and abundant mesenchyme.
Type 1 Congenital Cystic Adenomatoid Malformation
This malformation comprises cysts > 2 cm diameter and usually involves only a single lobe; the prognosis is good. Resection specimens show pseudostratified columnar epithelium, sometimes with mucous cell hyperplasia; there is bronchiolar overgrowth with alveolar underdevelopment. There is rare overlap with bronchoalveolar cell carcinoma; the relationship between CTMs and malignant disease is discussed in more detail below.
Type 2 Congenital Cystic Adenomatoid Malformation
This lesion comprises multiple small cysts and may be associated with renal agenesis, cardiovascular defects, diaphragmatic hernia and syringomyelia. Histologically, there is bronchiolar epithelium with overgrowth, separated by alveolar tissue which was underdeveloped.
Type 3 Congenital Cystic Adenomatoid Malformation
This CTM is mainly seen in males; typically a whole lobe is involved, the others being compressed. Macroscopically, the lesion appears solid and not cystic, and microscopically, there is an excess of bronchiolar structures separated by air spaces that resemble late fetal lung, with a virtual absence of small, medium and large pulmonary arteries within the lesion.
Type 4 Congenital Cystic Adenomatoid Malformation
This lesion is characterized by peripheral thin-walled cysts that are often multiloculated. The cystic spaces are typically lined by alveolar type I or type II cells with the intervening stroma being thin and comprising loose mesenchymal tissue. There may be a spectrum of disease between these lesions and type 1 pleuropulmonary blastomas.
Other Congenital Cystic Thoracic Malformations
Bronchogenic cysts are defined pathologically by having a wall containing cartilage. Enterogenous cysts are divided into esophageal, which are lined by squamous or respiratory epithelium, and gastroenteric, lined by gastric or enteric mucosa.
Bronchial Atresia
This abnormality may be commoner than was thought previously. It is often misdiagnosed as CCAM prior to resection, because the atretic airway is often not seen on HRCT. There is overlap with CCAMs and sequestrations, both of which may also show atretic airways.
Congenital Lobar Emphysema
The author’s preferred term is congenital large hyperlucent lobe. Pathologically, there may be very few primitive alveoli, or actually a greater number than normal (polyalveolar lobe).
Presentation of Congenital Thoracic Malformations
Usually the diagnosis is made on a routine 20 wk fetal anomaly scan. A mass lesion may be seen, which is either cystic or solid; there may be mediastinal shift in consequence. The second presentation is as polyhydramnios, due to compression of the fetal esophagus and failure of normal fetal swallowing. An abnormal vascular supply may be identified. The differential diagnosis is given in Table 3. Recently fetal MRI has been used to delineate these abnormalities, but although the images are easier for the non-radiologist to interpret, in author’s view they have yet to be shown to improve outcomes.
What is important to acknowledge is the extreme anxiety felt by the parents. The uncertainty, and the inability of physicians to give a definitive opinion, leads to more anxiety during the pregnancy than in couples with a definite bad prognosis lesion like diaphragmatic hernia [10]. The author likes to see couples soon after the diagnosis, and go through the usually benign antenatal prognosis (below), touching on antenatal options as well as the fact that likely only an elective procedure, if any, is going to be needed.
Although increasingly diagnosis of CTM is antenatal, the diagnosis may be missed and late presentations have been described (Table 4).
Management of Antenatally Presenting Congenital Thoracic Malformations
Usually, observation with repeated scans is all that is required. Generally, the mass can be expected to enlarge in the 20th-30th wk of pregnancy, and then regress, and indeed it may be invisible at term. However, this does not mean they have totally regressed. Only if the mass is progressively enlarging with no sign of regression in the last 10 wk of the pregnancy, or if there is fetal hydrops, should any intervention be contemplated. If intervention is undertaken [11–16], the options are given in Table 5; the evidence base for any of these is limited, and all should be considered as a last resort.
Immediate Postnatal Management of a Baby with Antenatal Presentation of Congenital Thoracic Malformations
The majority of these babies are delivered normally and are well in the postnatal period, and these babies present a therapeutic dilemma discussed in detail below. If the baby is symptomatic with respiratory distress, poor feeding and failure to gain weight, then intervention is required. Symptoms may be from any of high output cardiac failure due to flow through aortopulmonary collaterals, the mass effect of a large CTM, or airway compression. The baby should first be stabilised as far as possible. The conventional option is surgery. A few CTMs have been submitted to embolic procedures, often via the umbilical vein [17]. The results are variable—unsurprisingly, the solid components regress more after the procedure than cystic elements.
Also to be considered under this category is the symptomatic baby with a cystic malformation, whose antenatal ultrasound scans were apparently normal. One unlikely but not impossible explanation is that an abnormality was missed. More likely is that the cystic abnormality is acquired. The differential diagnosis is wide (Table 6). However, pulmonary interstitial emphysema in a non-ventilated, term baby should be highlighted, not least because identification may avoid unnecessary surgery. Typically, the baby is born in good condition, and develops unexpected respiratory distress in the first few days of life. Imaging reveals a cystic lobe or lung (Fig. 2). The abnormalities resolve with conservative management, but if the significance of the findings is not appreciated, the baby may undergo unnecessary surgery [18].

CT scan of a term baby who had normal antenatal ultrasound scans and was initially completely well, but later developed severe respiratory distress. The CT scan shows over-expansion of the left upper lobe by a multi-cystic mass. This was localised pulmonary interstitial emphysema in a term baby
Management of An Asymptomatic Baby with Antenatal Diagnosis of Congenital Thoracic Malformations
This is a difficult situation for all concerned. The obvious (and unanswerable) questions are:
-
1.
What imaging is indicated and when should it be performed?
-
2.
How many babies with a CTM have lived out their natural life with no complications from it?
-
3.
For those who develop a complication, when is this likely to happen, and is the outcome worse than if prophylactic surgery had been performed?
Imaging Options
Most babies will have a chest radiograph (CXR) done shortly after the birth, and in many it will be normal; CXR is only 61 % sensitive to the presence of a CTM [19]. The author’s own policy is to recommend an early HRCT scan without contrast to confirm that the antenatally diagnosed CTM is still present; if it is not, or of trivial size, the author would perform no further imaging. Since there are documented cases of postnatal recession of CTMs, he would observe the baby for around 12–18 mo, at which point he would perform HRCT with contrast (a) to confirm that the malformation is still present, and (b) to delineate the arterial supply and venous drainage prior to planning surgery. There is no evidence base for this approach, and some would advocate doing only a single HRCT, and that only if the parents are contemplating surgery. However, the argument for at least one CT with contrast is that, if the CTM has a very large aortic blood supply, this should probably be embolised, or at least the opinion of an interventional radiologist or pediatric cardiologist sought. In the future, MRI may be a good option, but the present requirement for a general anaesthetic currently limits its usefulness.
A Special Situation: Congenital Large Hyperlucent Lobe (Congenital Lobar Emphysema)
Treatment options are straightforward. Unless the baby is symptomatic in the newborn period, and does not respond to medical management, in which case surgery is required, then the baby will remain well. Although the appearances of the large lobe may be dramatic, with growth they regress, and there is no evidence that growth of the underlying lung is improved by surgery.
Potential Complications of Untreated Congenital Thoracic Malformations
These include infection of the CTM (including fungal and mycobacterial), bleeding including hemothorax, air embolism, high output cardiac failure from shunting through large systemic collaterals and malignant change. If a complication has developed, it is relatively non-controversial to recommend surgery. If the child has developed an infection within a cystic CTM, it is so unlikely to clear that resection is necessary. Perhaps the most difficult prospect to deal with is the risk of malignancy; this is rare, but there is no easy answer as to what to do.
Risk of Complications
There is no hard and fast evidence to guide the clinician. A meta-analysis gives the best evidence, but the follow-up was short (less than 20 y). There were 41 series (published between 1996 and 2008), that included 1070 patients of whom 79 % were diagnosed antenatally [20]. Of these, 505 were not operated on neonatally, of whom sixteen (3.2 %) subsequently became symptomatic and required an operation. The median age of complications was 6.9 mo (range 2.5–10 mo), and the median age of surgery for symptoms was 56 mo (range 2 mo–8.5 y). There were more complications with emergency as against elective surgery, but this likely relates to most of the emergency work being performed in the neonatal period, whereas most of the elective operations were performed in older children.
It is important to note that, although some complications of a CTM (such as infection) will be prevented by prophylactic surgery, this is not the case for malignancy. A recent single centre study [21] reported on 74 fetuses, of whom 72 were live births (n = 1 each intra-uterine death and termination for fetal hydrops), in the period 2001–11. A conservative policy was adopted unless the parents requested otherwise. One baby underwent an emergency lobectomy as a neonate, and three others subsequently underwent elective lobectomy, two at parental request and one because of a family history of synovial sarcoma. Three others underwent treatment—two emergency lobectomies for infection in the CTM, and one embolization of a feeding vessel, thus 3/68 (4.5 %) underwent emergency treatment; most remained asymptomatic during an average 6 y follow up. It has to be said that there is no uniform policy across pediatric centres, with some advocating surgery for all, some observation for all, and some operating a variable policy [22].
An argument advanced in one paper is that surgery should be performed because in fact there may be occult inflammation in the malformation, and that symptoms from inflammation are only appreciated once the malformation has been removed. Certainly occult inflammation can be found if sought; in one study, 24 CTMs were excised (18 operated within the first two weeks of life, 6 at 3 mo of age) [23]. Of these, there was evidence of inflammation in 19, especially those with CCAM type 2 (8/8). All cultures were negative, and this group did not report any change in symptoms after surgery. The author has seen one case who, although thought to be asymptomatic before elective surgery, was in fact ‘a new child’ after resection had been performed.
Malignancy and Congenital Thoracic Malformations
This is a difficult area. We know that primary intrathoracic malignancy in childhood is rare, and that for most but not all CTMs there is no evidence of increased risk. We also know that there are reports of co-existence of CTMs with a wide range of malignancies, such as rhabdomyosarcoma, pleuropulmonary blastoma (PPB) and bronchioloalveolar carcinoma (BAC). PPB should be considered a risk especially if other tumors, such as cystic nephroma, childhood cancers, stromal sex-chord ovarian tumors, seminomas or dysgerminomas, intestinal polyps, thyroid hyperplasias, and hamartomas are present [24]. Nearly 10 % of PPB patients also had renal tumors. Finally, surgery does not prevent malignancy developing elsewhere in the lung; there are well documented cases with malignant disease after complete resection of a CTM [25].
The relationship between cavitation, CTMs and tumors is complex. Cavitation may be caused by tumor necrosis, or carcinoma may develop from cysts (BAC in CCAM). Tumors may cause cystic changes and may be part of a spectrum of cystic disease (type 4 CCAM and type 1 PPB). Finally, tumors may be unrelated to the cysts. There is great pathological debate about whether type 4 CCAM is in fact a regressed PPB, or the relationship goes the other way, but this is of academic interest to the clinician puzzling over pulmonary images.
The biggest single series which gives an idea of prevalence comes from Canada [26]. They resected 74/129 CTMs, of whom three were found to be PPB; worryingly, there was absolutely no distinguishing feature of these three CTMs. Two further CTMs were diagnosed in the same time period, giving a prevalence of 1 in 250,000 with a mortality rate of 20 %. The prevalence of CTMs was 1 in 12,000, and the prevalence of PPB in ‘benign’ resected CTM was 4 %.
It may also be that intracystic mucinous proliferations in type 1 CCAM may be precursors to BAC [27]. There are worrying pathological commonalities between carcinomas and some CTMs. Genomic imbalances most frequently found in chromosomes 2 and 4 in CTMs have also been found in adenocarcinomas arising in non-smokers [28]. Both intracystic and intra-alveolar areas of mucinous proliferation show K-ras mutations and microsatellite instability at the p16 locus, similar to the molecular abnormalities and differentiation profiles in mucinous BAC ‘de novo’ [29].
Conclusions: Elective Surgery or Not?
The author’s own approach (Fig. 3) is to go through the risks of complications as set above, and recommend elective surgery, while recognising that equally respected respiratory pediatricians would recommend the opposite. If the family declines, he will follow up the child, but he does not have a set protocol for routine re-imaging. If surgery is contemplated, he advises operation in the second year of life, to give an opportunity for any regression of the malformation. Increasingly surgery is by video-assisted thoracic surgery (VATS). Complications are rare. He will continue to follow these children up, but again, do not routinely re-image them.

The author’s suggested, but non-evidence based, management plan
Conclusions
The rational management of CTMs is hampered by the lack of the sort of evidence that pediatricians and families need if they are to make rational decisions. We know that most babies diagnosed with a CTM will do well in the medium term. However, a very small number will develop a serious complication such as a major infection, and a few will develop a largely treatment-resistant malignancy, and at the moment we cannot clinically detect the high-risk group. We all need to be gathering data prospectively to try to address this situation.
References
Bush A. Prenatal presentation and postnatal management of congenital thoracic malformations. Early Hum Dev. 2009;85:679–84.
Abel RM, Bush A, Chitty LS, Harcourt J, Nicholson AG. Congenital lung disease. In: Wilmott R, Boat T, Bush A, Chernick V, Deterding R, Ratjen F, editors. Kendig and Chernick’s Disorders of the Respiratory Tract in Children. 8th ed. 2012. p. 317–57.
Bush A, Hogg C. Primary ciliary dyskinesia: recent advances in epidemiology, diagnosis, management and relationship with the expanding spectrum of ciliopathy. Expert Rev Respir Med. 2012;6:663–82.
Asher R. Talking sense. London: Publ Pitman Medical; 1972.
Griffin N, Devaraj A, Goldstraw P, Bush A, Nicholson AG, Padley S. CT and histopathological correlation of congenital cystic pulmonary lesions: a common pathogenesis? Clin Radiol. 2008;63:995–1005.
Bush A. Congenital lung disease: a plea for clear thinking and clear nomenclature. Pediatr Pulmonol. 2001;32:328–37.
Galambos C, Sims-Lucas S, Ali N, Gien J, Dishop MK, Abman SH. Intrapulmonary vascular shunt pathways in alveolar capillary dysplasia with misalignment of pulmonary veins. Thorax. 2015;70:84–5.
Stocker JT. Congenital pulmonary airway malformation: a new name for and an expanded classification of congenital cystic adenomatoid malformation of the lung. Histopathology. 2002;41:424–31.
Langston C. New concepts in pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12:17–37.
Aite L, Zaccara A, Trucchi A, et al. When uncertainty generates more anxiety than severity: the prenatal experience with cystic adenomatoid malformation of the lung. J Perinal Med. 2009;37:539–42.
Tsao K, Hawgood S, Vu L, et al. Resolution of hydrops fetalis in congenital cystic adenomatoid malformation after prenatal steroid therapy. J Pediatr Surg. 2003;38:508–10.
Knox EM, Kilby MD, Martin WL, Khan KS. In-utero pulmonary drainage in the management of primary hydrothorax and congenital cystic lung lesion: a systematic review. Ultrasound Obstet Gynecol. 2006;28:726–34.
Vu L, Tsao K, Lee H, et al. Characteristics of congenital cystic adenomatoid malformations associated with nonimmune hydrops and outcome. J Pediatr Surg. 2007;42:1351–6.
Peranteau WH, Wilson RD, Liechty KW, et al. Effect of maternal betamethasone administration on prenatal congenital cystic adenomatoid malformation growth and fetal survival. Fetal Diagn Ther. 2007;22:365–71.
Morris LM, Lim FY, Livingston JC, Polzin WJ, Crombleholme TM. High-risk fetal congenital pulmonary airway malformations have a variable response to steroids. J Pediatr Surg. 2009;44:60–5.
Leung WC, Ngai C, Lam TP, Chan KL, Lao TT, Tang MH. Unexpected intrauterine death following resolution of hydrops fetalis after betamethasone treatment in a fetus with a large cystic adenomatoid malformation of the lung. Ultrasound Obstet Gynecol. 2005;25:610–2.
Lee BS, Kim JT, Kim EA, et al. Neonatal pulmonary sequestration: clinical experience with transumbilical arterial embolization. Pediatr Pulmonol. 2008;43:404–13.
Freysdottir D, Olutoye O, Langston C, Fernandes CJ, Tatevian N. Spontaneous pulmonary interstitial emphysema in a term unventilated infant. Pediatr Pulmonol. 2006;41:374–8.
Calvert JK, Lakhoo K. Antenatally suspected congenital cystic adenomatoid malformation of the lung: postnatal investigation and timing of surgery. J Pediatr Surg. 2007;42:411–4.
Stanton M, Njere I, Ade-Ajayi N, Patel S, Davenport M. Systematic review and meta-analysis of the postnatal management of congenital cystic lung lesions. J Pediatr Surg. 2009;44:1027–33.
Ng C, Stanwell J, Burge DM, Stanton MP. Conservative management of antenatally diagnosed cystic lung malformations. Arch Dis Child. 2014;99:432–7.
Lo AY, Jones S. Lack of consensus among Canadian pediatric surgeons regarding the management of congenital cystic adenomatoid malformation of the lung. J Pediatr Surg. 2008;43:797–9.
Pelizzo G, Barbi E, Codrich D, et al. Chronic inflammation in congenital cystic adenomatoid malformations. An underestimated risk factor? J Pediatr Surg. 2009;44:616–9.
Priest JR, Williams GM, Hill DA, Dehner LP, Jaffé A. Pulmonary cysts in early childhood and the risk of malignancy. Pediatr Pulmonol. 2009;44:14–30.
Papagiannopoulos KA, Sheppard M, Bush A, Goldstraw P. Pleuropulmonary blastoma: is prophylactic resection of congenital lung cysts effective? Ann Thorac Surg. 2001;72:604–5.
Nasr A, Himidan S, Pastor AC, Taylor G, Kim PCW. Is congenital cystic adenomatoid malformation a premalignant lesion for pleuropulmonary blastoma? J Pediatr Surg. 2010;45:1086–9.
MacSweeney F, Papagiannopoulos K, Goldstraw P, Sheppard MN, Corrin B, Nicholson AG. An assessment of the expanded classification of congenital cystic adenomatoid malformations, and their relationship to malignant transformation. Am J Surg Pathol. 2003;27:1139–46.
Stacher E, Ullmann R, Halbwedl I, et al. Atypical goblet cell hyperplasia in congenital cystic adenomatoid malformation as a possible preneoplasia for pulmonary adenocarcinoma in childhood: a genetic analysis. Hum Pathol. 2004;35:565–70.
Lantuejoul S, Nicholson AG, Sartori G, et al. Mucinous cells in type 1 pulmonary congenital cystic adenomatoid malformation as mucinous bronchioloalveolar carcinoma precursors. Am J Surg Pathol. 2007;31:961–9.
Conflict of Interest
None.
Source of Funding
AB was supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bush, A. Rare Lung Diseases: Congenital Malformations. Indian J Pediatr 82, 833–840 (2015). https://doi.org/10.1007/s12098-015-1800-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-015-1800-9