
Abstract
Li-Fraumeni syndrome is caused by heterozygous germline pathogenic variants in the TP53 gene. It involves a high risk of a variety of malignant tumors in childhood and adulthood, the main ones being premenopausal breast cancer, soft tissue sarcomas and osteosarcomas, central nervous system tumors, and adrenocortical carcinomas. The variability of the associated clinical manifestations, which do not always fit the classic criteria of Li-Fraumeni syndrome, has led the concept of SLF to extend to a more overarching cancer predisposition syndrome, termed hereditable TP53-related cancer syndrome (hTP53rc). However, prospective studies are needed to assess genotype–phenotype characteristics, as well as to evaluate and validate risk-adjusted recommendations. This guideline aims to establish the basis for interpreting pathogenic variants in the TP53 gene and provide recommendations for effective screening and prevention of associated cancers in carrier individuals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Li-Fraumeni syndrome (LFS) is characterized by a high risk of developing a wide variety of malignant tumors in childhood and adulthood, caused by heterozygous germline pathogenic variants in the TP53 gene. Based on penetrance data from familial presentation cases, the highest cumulative incidences in females are 56% and 15% for breast cancer (BC) and soft tissue sarcoma (STS) respectively, while in males, they said incidences are 20% both for STS and for brain cancer [1]. The predominant cancers in people with LFS are osteosarcoma and STS, premenopausal BC, brain tumors, and adrenocortical carcinoma (ACC); similarly, rare tumors, such as choroid plexus carcinoma, hypodiploid acute lymphoblastic leukemia, anaplastic embryonal rhabdomyosarcoma, subtype sonic hedgehog-driven medulloblastoma, and jaw osteosarcoma are highly suggestive of LFS [2, 3].
In recent years, the development of multigene panels for cancer has resulted in increased germline TP53 testing in oncology patients. Therefore, more tumors potentially linked to germline alterations of TP53 have been reported. The heterogeneity of clinical presentations associated with germline TP53 alterations justifies the extending the LFS concept to a wider cancer predisposition syndrome designated heritable TP53-related cancer (hTP53rc) syndrome. Moreover, cancer risk and cancer surveillance recommendations are evolving as new genotype–phenotype relationships are being described. Here, we aim to provide an updated clinical guideline for the identification of TP53 germline pathogenic variants, cancer risk estimation, and surveillance recommendations.
TP53 gene testing
Criteria for germline TP53 variant testing have evolved since Birch´s first definition in 1994. Patients with cancer who meet the latest modified ´Chompret Criteria´ should be tested for germline TP53 variants (Table 1). Likewise, individuals should be tested who develop a second primary tumor within the radiotherapy field of a first core TP53-tumor that occurred before 46 years. Cascade genetic testing of the germline disease-causing TP53 variant should be offered to adult family members.
Child and adolescent cancer patients should also be tested for germline TP53 variants if presenting with hypodiploid acute lymphoblastic leukemia (ALL), unexplained sonic hedgehog-driven medulloblastoma, or osteosarcoma of the jaw. Healthy children who are first-degree relatives of individuals with a germline disease-causing TP53 variant should be offered predictive testing if the genetic variant confers a high cancer risk in childhood (reported in families with childhood cancers carrying the same pathogenic variant, childhood cancers have been observed within the family, or it is a dominant-negative missense variant) [4]. When there is insufficient evidence to determine the childhood cancer risk, the decision to perform genetic testing in children will be made on a case-by-case basis.
Interpretation of TP53 variants
Constitutional TP53 variants are classified as per TP53-specific ACMG/AMP guidelines, which are based on the integration of multiple lines of evidence, including variant frequency in general population, information on phenotype, bioinformatic predictions, and functional data [5]. Unlike loss-of-function variants (nonsense, frameshift, splicing, gross rearrangements), the interpretation of the most common TP53 missense variants is not always obvious. Furthermore, a subclass of missense variants exert a dominant-negative effect resulting in mutant proteins that form tetramers with wild-type TP53, inhibiting the transcriptional activity of the protein complex and causing larger defects in response to DNA damage [6,7,8,9,10].
The penetrance of TP53 variants is incomplete and varies depending on the type of variant and modifying factors [4]. Dominant-negative missense TP53 variants have been reported generally as highly penetrant and detected in families with childhood cancers [11]. In contrast, debate continues regarding phenotype-genotype correlation associated with loss of function and non-dominant negative variants [1, 11, 12]. Also, the different penetrance among carriers from the same family suggests the coexistence of genetic and environmental modifiers [13, 14].
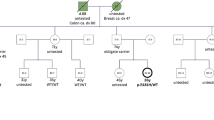
The detection of a TP53 pathogenic variant in the tumor or plasma of patients with early onset diagnosis (< 30 years of age) of TP53 core tumors (breast cancer, soft-tissue sarcoma, osteosarcoma, central nervous system tumor, adrenocortical carcinoma), or other tumors exhibiting an enriched germline conversion rate, such as non-small cell lung cancer or colon cancer, requires further assessment at a Hereditary Cancer Unit (Fig. 1) [15]. In individuals who meet Chompret’s criteria, the identification of a TP53 (likely) pathogenic variant with a variant allele frequency (VAF) of 40–50% in lymphocyte blood testing can be presumed to be germline and associated with a TP53-related cancer syndrome. Additionally, the detection of a TP53 variant at lower VAFs (10–40%) in blood DNA (suggesting germline mosaicism) should be confirmed with supplementary tests that assess the presence of the TP53 variant in unaffected tissues with no lymphocyte content (e.g., skin biopsy, follicle bulbs), to rule out other possibilities, such as TP53 clonal hematopoiesis of indeterminate potential (CHIP) or circulating tumor DNA [16,17,18] (Fig. 1). For interpretation purposes, offspring testing might be also useful and informative after identifying additional carriers.

Algorithm proposal for the interpretation of TP53 pathogenic variants
When a TP53 gene (likely) pathogenic variant is found in the setting of multigene panel testing in a patient not fulfilling Chompret criteria, causes other than those of germline origin must be ruled out and results should be discussed among a multidisciplinary board.
Surveillance and management recommendations
Surveillance
Previous debate about the possible lack of benefit of clinical surveillance in the heritable TP53-related cancer syndrome has been resolved after reporting the clinical impact of the Toronto protocol on patient outcomes. This protocol has demonstrated improved 5-year survival rates among individuals undergoing surveillance compared to the non-surveillance group (88% vs 59.6%), with reported psychological benefits [19,20,21,22]. Patient surveillance also proved to be cost-effective [20, 23].
In 2017, an international consortium established a list of cancer screening recommendations for individuals with germline disease-causing TP53 variants [24]. More recently, two additional consensus surveillance guidelines have been published, the first by the United Kingdom Cancer Genetics Group (UKCGG) Consensus Group [25] and the second by the European Reference Network (ERN) on Genetic Tumour Risk Syndromes (GENTURIS) [4].
Female breast cancer is the most frequent LFS-associated cancer [26], especially in premenopausal women [2, 11, 27]. Breast magnetic resonance imaging (MRI) has been demonstrated to be more sensitive than mammography in women with familial and genetic predisposition, including TP53 carriers, in first and subsequent rounds for detection of early breast cancer [28, 29]. Annual breast MRI can be alternated with whole-body MRI (WBMRI) at 6 months. Mammography is not recommended so as to avoid radiation [4, 24].
In TP53 carriers there is high lifetime risk of developing a sarcoma, with osteosarcomas usually diagnosed in children [2, 11]. Annual WBMRI with diffusion-weighted imaging, vertex to feet, using fast sequences, without gadolinium contrast is recommended. This procedure is effective for the early detection of solid tumors in addition to sarcomas. A meta-analysis of baseline WBMRI in TP53 variant carriers yielded an overall estimated detection rate of new tumors of 7%, with a false-positive rate of 42.5% [30]. Absence of clinical correlation and targeted imaging will help to prevent unnecessary biopsies. Radiologists should follow the guidelines for WBMRI acquisition, interpretation, and reporting for cancer screening [31]. Another important issue is the need for sedation to perform WBMRI in children, with the risk that this entails [30, 32, 33]. Most tumors are detected in early stages and are curable with surgery. With decreased therapeutic intensity, comes decreased therapy-associated complications and can lead to improved quality of life [30].
The risk of brain tumors lasts a lifetime [2, 11]. In various studies, baseline brain MRI has a sensitivity of 60% and specificity of 80%. The baseline cancer detection rate ranges from 1.7% [34] to 8.6% [35]; the cumulative cancer detection rate is 13.6% [20]. The recommendation is for yearly brain MRIs (first, with gadolinium-based contrast and, if normal, subsequent MRIs may be done without contrast).
The risk of ACC in children is approximately 4% and decreases after the first decade of life. Surveillance consisting of clinical examination for signs of virilization, early puberty, Cushing-like features, determination of 17-OH-progesterone, total testosterone, dehydroepiandrosterone sulfate and androstenedione, while abdominal and pelvic ultrasound enables early diagnosis to be made, mostly in stage I, with better chances of cure and survival [20, 36].
For hematopoietic malignancies, there is no evidence that screening procedures lead to a presymptomatic diagnosis and improved survival in healthy individuals [37]. In oncology patients who have received leukemogenic drugs (alkylating agents, topoisomerase inhibitors), annual complete blood count is recommended [4, 24, 38].
If the TP53 carrier patient has received abdominal radiotherapy for the treatment of a previous cancer or if there is a familial history of colorectal tumor suggestive of increased risk, colonoscopy is recommended every five years from 18 years of age onward.
Of note, the spectrum of tumors depends based on the age of the proband; consequently, screening measures should be adapted throughout life.
Who should be offered surveillance?
The full screening protocol should be offered to patients harboring TP53 likely pathogenic or pathogenic variant, whether germline or constitutional mosaicism. As previously mentioned for genetic screening, we emphasize that surveillance measures in children should only be offered when the variant confers a high cancer risk in childhood. Additionally, those patients affected with cancer satisfying classic LFS criteria without a pathogenic TP53 variant identified should be offered surveillance [24, 25].
When to begin surveillance?
In general, it is recommended that surveillance commence as soon as the carrier status is known and be maintained throughout life (Table 2). Whenever possible, screening should continue even after the diagnosis of a primary malignant tumor, adapted to the disease stage or situation. Said surveillance is integrated into the specific clinical follow-up of the cancer diagnosed.
Who should coordinate screening?
Screening in children should be managed by a pediatric oncologist or a trained specialist. In the case of adults, it should preferably be coordinated by a specialist trained in genetics and knowledgeable about the syndrome. For healthy carriers, this high-risk surveillance program should be performed in a non-oncological setting. Ideally, patients should be referred to specialized units and screening findings be discussed in specific multidisciplinary boards.
Individuals harboring a germline pathogenic variant should be encouraged to lead a healthy lifestyle by avoiding smoking, exposure to known carcinogens, and sunlight, as well as by using high protection factor sunscreen, eating a healthy diet, and exercising.
Management
Typically, LFS tumors are treated according to standard protocols, except for surgical treatment for breast cancer. Bilateral mastectomy is recommended instead of breast-conserving surgery to avoid the need for radiotherapy and reduce the risk of a second primary breast cancer [27].
There is clinical and in vivo evidence of increased sensitivity to ionizing radiation and genotoxic chemotherapies [39, 40]. However, until more evidence is gathered, standard chemotherapy regimens should be administered. Ideally, treatment should be personalized using non-leukemogenic drugs.
Despite recent studies demonstrating that the risk of radio-induced cancers is lower than previously reported [41, 42], exposure to radiation should be avoided whenever possible [43].
Risk-reducing bilateral mastectomy should be discussed with female healthy carriers and breast cancer patients [4, 24, 25].
Future directions
Different consensus groups advise against a surveillance protocol based on genotype and modifiers, inasmuch as they have not been validated prospectively [24, 25]. Clinical and genetic registries are necessary to assess genotype–phenotype features. The reported outcomes of surveillance of TP53 pathogenic variant carriers will enable us to evaluate and validate screening recommendations that might be tailored on the type of variant, genetic modifiers, and family history.
In the meantime, expert multidisciplinary boards should be set up for consultation of clinical interpretation of TP53 variants and to establish the best surveillance recommendations for the individual and the family. We propose the creation of referral centers for individuals with heritable TP53-related cancer syndrome.
References
de Andrade KC, Khincha PP, Hatton JN, Frone MN, Wegman-Ostrosky T, Mai PL, et al. Cancer incidence, patterns, and genotype–phenotype associations in individuals with pathogenic or likely pathogenic germline TP53 variants: an observational cohort study. Lancet Oncol. 2021;22:1787–98. https://doi.org/10.1016/S1470-2045(21)00580-5.
Mai PL, Best AF, Peters JA, DeCastro RM, Khincha PP, Loud JT, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the national cancer institute Li-Fraumeni syndrome cohort: cancer risk in TP53 mutation carriers. Cancer. 2016;122:3673–81. https://doi.org/10.1002/cncr.30248.
Guha T, Malkin D. Inherited TP53 mutations and the Li–Fraumeni syndrome. Cold Spring Harb Perspect Med. 2017;7:a026187. https://doi.org/10.1101/cshperspect.a026187.
The European Reference Network GENTURIS, Frebourg T, BajalicaLagercrantz S, Oliveira C, Magenheim R, Evans DG. Guidelines for the Li–Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet. 2020;28:1379–86. https://doi.org/10.1038/s41431-020-0638-4.
Fortuno C, Lee K, Olivier M, Pesaran T, Mai PL, Andrade KC, et al. Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants. Hum Mutat. 2021;42:223–36. https://doi.org/10.1002/humu.24152.
Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50:1381–7. https://doi.org/10.1038/s41588-018-0204-y.
Kato S, Han S-Y, Liu W, Otsuka K, Shibata H, Kanamaru R, et al. Understanding the function–structure and function–mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci. 2003;100:8424–9. https://doi.org/10.1073/pnas.1431692100.
Kotler E, Shani O, Goldfeld G, Lotan-Pompan M, Tarcic O, Gershoni A, et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell. 2018;71:178-190.e8. https://doi.org/10.1016/j.molcel.2018.08.013.
Raad S, Rolain M, Coutant S, Derambure C, Lanos R, Charbonnier F, et al. Blood functional assay for rapid clinical interpretation of germline TP53 variants. J Med Genet. 2021;58:796–805. https://doi.org/10.1136/jmedgenet-2020-107059.
Zerdoumi Y, Lanos R, Raad S, Flaman J-M, Bougeard G, Frebourg T, et al. Germline TP53 mutations result into a constitutive defect of p53 DNA binding and transcriptional response to DNA damage. Hum Mol Genet. 2017;26:2812–2812. https://doi.org/10.1093/hmg/ddx165.
Bougeard G, Renaux-Petel M, Flaman J-M, Charbonnier C, Fermey P, Belotti M, et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol Off J Am Soc Clin Oncol. 2015;33:2345–52. https://doi.org/10.1200/JCO.2014.59.5728.
Rana HQ, Gelman R, LaDuca H, McFarland R, Dalton E, Thompson J, et al. Differences in TP53 mutation carrier phenotypes emerge from panel-based testing. JNCI J Natl Cancer Inst. 2018;110:863–70. https://doi.org/10.1093/jnci/djy001.
Chan CS, Sun Y, Ke H, Zhao Y, Belete M, Zhang C, et al. Genetic and stochastic influences upon tumor formation and tumor types in Li-Fraumeni mouse models. Life Sci Alliance. 2021;4:e202000952. https://doi.org/10.26508/lsa.202000952.
Majhi PD, Griner NB, Mayfield JA, Compton S, Kane JJ, Baptiste TA, et al. Genetic modifiers regulating DNA replication and double-strand break repair are associated with differences in mammary tumors in mouse models of Li-Fraumeni syndrome. Oncogene. 2021;40:5026–37. https://doi.org/10.1038/s41388-021-01892-5.
Mandelker D, Donoghue M, Talukdar S, Bandlamudi C, Srinivasan P, Vivek M, et al. Germline-focussed analysis of tumour-only sequencing: recommendations from the ESMO precision medicine working group. Ann Oncol. 2019;30:1221–31. https://doi.org/10.1093/annonc/mdz136.
Gonzalez KD, Buzin CH, Noltner KA, Gu D, Li W, Malkin D, et al. High frequency of de novo mutations in Li-Fraumeni syndrome. J Med Genet. 2009;46:689–93. https://doi.org/10.1136/jmg.2008.058958.
Renaux-Petel M, Charbonnier F, Théry J-C, Fermey P, Lienard G, Bou J, et al. Contribution of de novo and mosaic TP53 mutations to Li-Fraumeni syndrome. J Med Genet. 2018;55:173–80. https://doi.org/10.1136/jmedgenet-2017-104976.
Weitzel JN, Chao EC, Nehoray B, Van Tongeren LR, LaDuca H, Blazer KR, et al. Somatic TP53 variants frequently confound germ-line testing results. Genet Med. 2018;20:809–16. https://doi.org/10.1038/gim.2017.196.
Villani A, Tabori U, Schiffman J, Shlien A, Beyene J, Druker H, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study. Lancet Oncol. 2011;12:559–67. https://doi.org/10.1016/S1470-2045(11)70119-X.
Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, Druker H, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol. 2016;17:1295–305. https://doi.org/10.1016/S1470-2045(16)30249-2.
Lammens CRM, Bleiker EMA, Aaronson NK, Wagner A, Sijmons RH, Ausems MGEM, et al. Regular surveillance for Li-Fraumeni syndrome: advice, adherence and perceived benefits. Fam Cancer. 2010;9:647–54. https://doi.org/10.1007/s10689-010-9368-z.
McBride KA, Ballinger ML, Schlub TE, Young M-A, Tattersall MHN, Kirk J, et al. Psychosocial morbidity in TP53 mutation carriers: is whole-body cancer screening beneficial? Fam Cancer Netherlands. 2017;16:423–32. https://doi.org/10.1007/s10689-016-9964-7.
Tak CR, Biltaji E, Kohlmann W, Maese L, Hainaut P, Villani A, et al. Cost-effectiveness of early cancer surveillance for patients with Li–Fraumeni syndrome. Pediatr Blood Cancer. 2019;66:e27629. https://doi.org/10.1002/pbc.27629.
Kratz CP, Achatz MI, Brugières L, Frebourg T, Garber JE, Greer M-LC, et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin Cancer Res. 2017;23:e38–45. https://doi.org/10.1158/1078-0432.CCR-17-0408.
Hanson H, Brady AF, Crawford G, Eeles RA, Gibson S, Jorgensen M, et al. UKCGG consensus group guidelines for the management of patients with constitutional TP53 pathogenic variants. J Med Genet. 2021;58:135–9. https://doi.org/10.1136/jmedgenet-2020-106876.
Schneider K, Zelley K, Nichols KE, Garber J. Li-Fraumeni Syndrome. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993 [cited 2022 Sep 15]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1311/ PMID: 20301488
Schon K, Tischkowitz M. Clinical implications of germline mutations in breast cancer: TP53. Breast Cancer Res Treat. 2018;167:417–23. https://doi.org/10.1007/s10549-017-4531-y.
Leach MO, Boggis CRM, Dixon AK, Easton DF, Eeles RA, Evans DGR, et al. Screening with magnetic resonance imaging and mammography of a UK population at high familial risk of breast cancer: a prospective multicentre cohort study (MARIBS). Lancet Lond Engl England. 2005;365:1769–78. https://doi.org/10.1016/S0140-6736(05)66481-1.
Kriege M, Brekelmans CTM, Boetes C, Muller SH, Zonderland HM, Obdeijn IM, et al. Differences between first and subsequent rounds of the MRISC breast cancer screening program for women with a familial or genetic predisposition. Cancer United States. 2006;106:2318–26. https://doi.org/10.1002/cncr.21863.
Ballinger ML, Best A, Mai PL, Khincha PP, Loud JT, Peters JA, et al. Baseline surveillance in Li-Fraumeni syndrome using whole-body magnetic resonance imaging: a meta-analysis. JAMA Oncol. 2017;3:1634. https://doi.org/10.1001/jamaoncol.2017.1968.
Petralia G, Koh DM, Attariwala R, Busch JJ, Eeles R, Karow D, et al. Oncologically relevant findings reporting and data system (ONCO-RADS): guidelines for the acquisition, interpretation, and reporting of whole-body MRI for cancer screening. Radiol Radiol Soc N Am. 2021;299:494–507. https://doi.org/10.1148/radiol.2021201740.
Greer M-LC, Voss SD, States LJ. Pediatric cancer predisposition imaging: focus on whole-body MRI. Clin Cancer Res. 2017;23:e6-13. https://doi.org/10.1158/1078-0432.CCR-17-0515.
Summers P, Saia G, Colombo A, Pricolo P, Zugni F, Alessi S, et al. Whole-body magnetic resonance imaging: technique, guidelines and key applications. ecancermedicalscience [Internet]. 2021 [cited 2022 Mar 30];15. Available from: https://ecancer.org/en/journal/article/1164-whole-body-magnetic-resonance-imaging-technique-guidelines-and-key-applications doi: https://doi.org/10.3332/ecancer.2021.1164. eCollection 2021.
Mai PL, Khincha PP, Loud JT, DeCastro RM, Bremer RC, Peters JA, et al. Prevalence of cancer at baseline screening in the national cancer institute Li-Fraumeni syndrome cohort. JAMA Oncol. 2017;3:1640–5. https://doi.org/10.1001/jamaoncol.2017.1350.
Bojadzieva J, Amini B, Day SF, Jackson TL, Thomas PS, Willis BJ, et al. Whole body magnetic resonance imaging (WB-MRI) and brain MRI baseline surveillance in TP53 germline mutation carriers: experience from the Li-Fraumeni syndrome education and early detection (LEAD) clinic. Fam Cancer. 2018;17:287–94. https://doi.org/10.1007/s10689-017-0034-6.
Tosin KCF, Legal EF, Pianovski MAD, Ibañez HC, Custódio G, Carvalho DS, et al. Newborn screening for the detection of the TP53 R337H variant and surveillance for early diagnosis of pediatric adrenocortical tumors: lessons learned and way forward. Cancers. 2021;13:6111. https://doi.org/10.3390/cancers13236111.
Porter CC, Druley TE, Erez A, Kuiper RP, Onel K, Schiffman JD, et al. Recommendations for surveillance for children with leukemia-predisposing conditions. Clin Cancer Res. 2017;23:e14-22. https://doi.org/10.1158/1078-0432.CCR-17-0428.
Swaminathan M, Bannon SA, Routbort M, Naqvi K, Kadia TM, Takahashi K, et al. Hematologic malignancies and Li–Fraumeni syndrome. Mol Case Stud. 2019;5:a003210. https://doi.org/10.1101/mcs.a003210.
Heymann S, Delaloge S, Rahal A, Caron O, Frebourg T, Barreau L, et al. Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat Oncol. 2010;5:104. https://doi.org/10.1186/1748-717X-5-104.
Kasper E, Angot E, Colasse E, Nicol L, Sabourin J-C, Adriouch S, et al. Contribution of genotoxic anticancer treatments to the development of multiple primary tumours in the context of germline TP53 mutations. Eur J Cancer. 2018;101:254–62. https://doi.org/10.1016/j.ejca.2018.06.011.
Le AN, Harton J, Desai H, Powers J, Zelley K, Bradbury AR, et al. Frequency of radiation-induced malignancies post-adjuvant radiotherapy for breast cancer in patients with Li-Fraumeni syndrome. Breast Cancer Res Treat. 2020;181:181–8. https://doi.org/10.1007/s10549-020-05612-7.
Petry V, Bonadio RC, Cagnacci AQC, Senna LAL, Campos RNG, Cotti GC, et al. Radiotherapy-induced malignancies in breast cancer patients with TP53 pathogenic germline variants (Li-Fraumeni syndrome). Fam Cancer. 2020;19:47–53. https://doi.org/10.1007/s10689-019-00153-5.
Thariat J, Chevalier F, Orbach D, Ollivier L, Marcy P-Y, Corradini N, et al. Avoidance or adaptation of radiotherapy in patients with cancer with Li-Fraumeni and heritable TP53-related cancer syndromes. Lancet Oncol England. 2021;22:e562–74. https://doi.org/10.1016/S1470-2045(21)00425-3.
Acknowledgements
We thank Héctor Salvador and Juan Cadiñanos for their careful review of this manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declared no conflicts of interest with respect to the authorship, and/or publication of this article.
Ethical approval
Ethics approval and Informed consent is not necessary for this clinical guideline. The research has been conducted in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sánchez-Heras, A.B., Ramon y Cajal, T., Pineda, M. et al. SEOM clinical guideline on heritable TP53-related cancer syndrome (2022). Clin Transl Oncol 25, 2627–2633 (2023). https://doi.org/10.1007/s12094-023-03202-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-023-03202-9