
Abstract
Purpose
Panitumumab is extensively used for RAS-WT metastatic colorectal cancer. This study assessed the efficacy and safety of panitumumab plus first-line chemotherapy [docetaxel (DOC) and cisplatin (CIS)] in treatment-naïve advanced gastric or gastro-oesophageal junction (GEJ) adenocarcinoma (ADC) patients.
Methods
Phase II, open-label, single-arm study includes treatment-naïve advanced gastric or GEJ-ADC patients from ten Spanish centres. Patients received panitumumab (6 mg/kg) plus DOC and CIS (50 mg/m2 both) every 2 weeks until disease progression, unacceptable toxicity, or patient withdrawal. Primary endpoint: objective response rate (ORR); main secondary endpoints: disease control rate (DCR), duration of response (DoR), time to progressive disease (TTP), progression-free-survival (PFS), overall survival (OS), and safety.
Results
Forty-four patients were included; median age: 67.8 (range 43.3–82.7) years, 68.2% male. The ORR was 27.3% (95% CI 15.0, 42.8); median PFS and OS: 5.0 (95% CI 3.6, 6.9) and 7.2 (5.5, 9.0) months, respectively. Median TTP, DCR and DoR: 5.3 (range 3.8–7.0) months, 70.5% (95% CI 54.8, 83.2%), and 4.8 (1.8, NE) months. Median panitumumab treatment duration: 11.9 (range 0.1–34.9) weeks; 25.0% patients had a dose reduction and 40.9% discontinued treatment. Grade 3–4 adverse events (AEs): 68.2%/22.2% patients. Most common AEs: asthenia (75.0%) and mucosal inflammation (54.5%). Serious AEs were experienced by 54.6% patients; 9.1%, 13.6%, and 15.9% related to panitumumab, DOC, and CIS, respectively. Three (6.8%) patients died due to AEs not related to study treatment.
Conclusions
The addition of panitumumab to standard chemotherapy as the first-line treatment in advanced gastric or GEJ-ADC does not appear to improve the efficacy outcomes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastric cancer ranks fifth for cancer incidence and second for cancer deaths in the world, with approximately 1 million new cases diagnosed per year [1, 2]. Despite considerable advances in surgical techniques, clinical diagnostics, and new chemotherapy regimens, the clinical outcome for advanced gastric or gastro-oesophageal junction (GEJ) adenocarcinoma (ADC) patients is generally poor, with survival rates of 5 years between 20 and 30% [3]. Numerous strategies have aimed to improve treatment results by adding adjuvant or neoadjuvant systemic therapy to surgery, sometimes, combined with radiotherapy [3]. Until recently, the standard treatment for gastric cancer was based in regimens of 5-fluorouracil (5-FU) and cisplatin (CIS), with the later addition of docetaxel (DOC), a regimen which improved response rate and survival, becoming one of the standard first-line treatments in treatment-naïve patients [4,5,6]. The Galician Oncology Research Group (GGIO, Grupo Gallego de Investigaciones Oncológicas) conducted a phase II study to evaluate efficacy and safety of a biweekly DOC and CIS regimen in advanced gastric cancer treatment-naïve patients. This study reported an objective response rate (ORR) of 42.3% (95% CI 28.9–52.7), with a median time to progression (TTP) of 5.5 months (95% CI 4.0–7.0) showing promising antitumor activity as the first-line treatment in advanced gastric cancer [7].
Many cell surface growth factor receptor pathways have been implicated in the pathogenesis of gastric and GEJ cancer including the receptor tyrosine kinases and their targeted therapies: epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGR), and hepatocyte growth factor receptor (MET) [8]. The EGFR pathway is an established target in colorectal cancer (CRC), and is expressed in 35–81% of all gastric cancer cases and is associated with depth of invasion in resected samples and may also be associated with poorer survival [9]. Monoclonal antibodies’ (mAtb) anti-EGFR such as cetuximab, panitumumab, and nimotuzumab targets the extracellular domain of EGFR. Both cetuximab and panitumumab have been tested extensively in metastatic gastric cancer which encouraged two large phase III trials: REAL3 and EXPAND [10, 11]. The REAL3 was designed to assess the addition of panitumumab to EOC regimen (epirubicin/oxaliplatin/capecitabine) in patients with advanced esophagogastric adenocarcinoma. Median overall survival (OS) in EOC and EOC plus panitumumab groups was 11.3 months (95% CI 9.6–13.0) and 8.8 months (95% CI 7.7–9.8), respectively (HR [95% CI] 1.37 [1.07–1.76]; P = 0.013). There were increased rates of Grade 3–4 diarrhea, rash, mucositis, and hypomagnesaemia adverse events (AEs) but a reduced incidence of hematological toxicity observed in the EOC plus panitumumab group [11]. The EXPAND trial showed a median progression-free survival (PFS) of 4.4 months in the patients receiving cetuximab plus chemotherapy vs 5.6 in the chemotherapy alone group; the OS was 9.4 and 10.7 months, respectively (PFS, P = 0.3158; OS, P = 0.9547) [10]. Both these trials showed no benefit in the addition of cetuximab/panitumumab to chemotherapy [10]. Another phase II study (ATTAX3) combined DOC, CIS, and fluoropyrimidines with or without panitumumab found similar results in both study arms, showing no additional benefit in panitumumab addition [12]. However, in another phase II trial (DOCETUX), the addition of cetuximab to the CIS/DOC regimen as a first-line treatment improved the ORR, but did not prolong the TTP and OS. The toxicity of CIS/DOC chemotherapy was not affected by the addition of cetuximab [13].
At the time of the initiation of this study, the benefit of the addition of mAtbs such as cetuximab and panitumumab was still unclear, as many phase II studies showed promise for the addition of mAtb to chemotherapy for gastric cancer adenocarcinoma with a median OS of 9–11 months [14, 15]. This study aimed to assess the efficacy and safety of the addition of panitumumab as a first-line treatment combination with DOC and CIS every 2 weeks in treatment-naïve advanced gastric or GEJ-ADC patients.
Materials and methods
Study design
This was a phase II, prospective, open-label, single-arm study conducted in ten Spanish hospitals. The Institutional Review Board at each site and the Spanish Medicine Agency approved the study protocol (Study GGIO-2010-01; clinicaltrials.gov identifier NCT01379807; EudraCT No.: 2010-021,192-87).
Patients
The inclusion criteria were: patients ≥ 18 years of age; histologically or cytologically confirmed advanced or unresectable stomach adenocarcinoma or GEJ disease; measurable disease according to the modified Response Evaluation Criteria in Solid Tumour (RECIST) criteria version 1.1; Eastern Cooperative Oncology Group (ECOG) status ≤ 2; seven days prior to the start of treatment: neutrophil count ≥ 1.5 × 109 cells/L; platelet count ≥ 100 × 109 cells/L; hemoglobin ≥ 9 g/dL; bilirubin levels ≤ 1.5 × upper limit of normal (ULN); creatinine clearance ≥ 50 mL/min and AST or ALT levels ≤ 2.5 × ULN ( ≤ 5xULN if liver metastasis is present); magnesium and calcium levels equal or greater than the lower limit of normal (LLN).
Main exclusion criteria were: prior systemic therapy for advanced unresectable or metastatic disease or treatment with monoclonal anti-EGFR antibodies or EGFR inhibitor therapy, < 12 months since end of prior chemotherapy treatment or adjuvant/neoadjuvant chemo-radiotherapy; HER2-positive tumour (IHC 2 + or 3 + ); past or current history ( < 5 years prior to treatment start) of other malignancies except gastric cancer (patients with curatively treated basal and squamous cell carcinoma of the skin or in situ carcinoma of the cervix were eligible); current or prior history of central nervous system metastases; treatment with other investigational drug (s) or participation in another clinical trial within 30 days prior to enrolment; known hypersensitivity to any of the study drugs; clinically significant cardiovascular disease (including myocardial infarction, unstable angina, symptomatic congestive heart failure and severe uncontrolled cardiac arrhythmia) ≤ 1 year before inclusion; history of interstitial lung disease, like pneumonitis and pulmonary fibrosis, or evidence of interstitial lung disease on the baseline chest computed tomography (CT) scan; pregnant or breastfeeding women, or planning to become pregnant within 6 months after the end of treatment; patients (male or female) who do not want to use highly effective contraception during treatment and 6 months after the end of treatment.
Study treatment
Panitumumab (Vectibix®, Amgen Europe B.V.) was administered every 14 days at a dose of 6 mg/kg, over a 60 ( ± 15)-min intravenous infusion using a low protein binding apyrogenic filter with a 0.20–0.22 μm pore size. Docetaxel 50 mg/m2 and cisplatin 50 mg/m2 consisted of a 60-min intravenous infusion after the administration of panitumumab.
Panitumumab does not require systematic prophylactic premedication from the first infusion, while docetaxel and cisplatin were administered with standard prophylactic premedication: dexamethasone 16 mg and antiemetics (5-HT3 antagonist or aprepitant). Administration of this investigational regimen was planned until progression, unacceptable toxicity, or patient withdrawal. If, during or after panitumumab infusion, a reaction occurred, premedication with acetaminophen/paracetamol and/or histamine H1 blockers such as diphenhydramine was administered for subsequent cycles.
Panitumumab administration was withheld if a skin- or nail-related serious AE occurred, or in the case of infusion reactions or Grade 3–4 toxicity, with the following exceptions: symptomatic hypomagnesaemia or hypocalcaemia, Grade 3–4 nausea, vomiting or diarrhea, and Grade ≥ 3 anemia or Grade 4 thrombocytopenia. Panitumumab was restarted once skin- or nail-related toxicity improved to Grade ≤ 2, or other toxicities improved to Grade ≤ 1, according to label specifications. Dose modifications of panitumumab included up to two steps for decreasing doses to 4.8 mg/kg (80% of the initial dose) and to 3.6 mg/kg (60%) when recovered from a Grade 3–4 skin toxicity to Grade ≤ 2.
Study procedures
Pre-study evaluations included complete medical history, HER-2 status, physical examination (including weight and height), ECOG performance status, electrocardiogram, and radiological imaging of the chest, abdomen, pelvis, and all other sites of disease by CT or magnetic resonance imaging (MRI) if clinically indicated. Seven days prior to initiating treatment ECOG performance status, hematology, and biochemistry tests were completed. EGFR expression and KRAS mutation status were not determined.
Tumour response was assessed by the RECIST version 1.1 criteria every 8 ± 2 weeks until disease progression was determined or patient withdrawal occurred.
Adverse events and concomitant medication were collected throughout the study until 30 ± 7 days after the last dose of panitumumab AEs were graded based on the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTC-AE) version 3.0.
Sample size
The sample size (49 patients) was calculated to produce a 95% confidence interval (CI) of 33–61% for the ORR, assuming an estimated rate of 47% (10% higher than the 37% rate observed the previous study with CIS and DOC treatment [7].
Statistical analysis
The primary endpoint was ORR [complete response (CP) plus partial response (PR)], reported using descriptive statistics and 95% CI. Secondary endpoints included: disease control rate (DCR) (CR, PR, and stable disease [SD]), duration of response (DoR), time to response (TTR), time to progressive disease (TTP), time to treatment failure (TTF), duration of SD, PFS, and OS. Safety and tolerability was evaluated by incidence, severity, and outcomes of AEs.
The analysis set included all patients who received at least one complete cycle of panitumumab, DOC, and CIS [intention to treat (ITT)]. All efficacy variables were reported using descriptive statistics that include point estimates. Time-to-event variables were estimated by the Kaplan–Meier method. PFS and OS were calculated as the time from the start of the treatment to first evidence of clinical progression or death by any cause. Data analysis was performed using the SAS® statistical package for Windows (v.9.4, SAS Institute Inc., Cary, U.S.).
Results
Patients’ characteristics
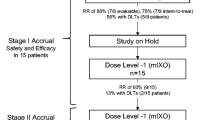
A total of 44 patients were enrolled between April 2011 and October 2013. All were included in both the ITT and safety populations (Fig. 1). The overall patient population had a median age of 67.8 (range 43.3–82.7) years, 68.2% male with minimal-to-moderate symptoms (75% ECOG = 1) at baseline. The most common tumour site was the stomach (84.1%) and most tumours were metastatic (93.2%) and poorly differentiated (43.2%). Only 22.7% of the patients had prior surgery of the primary tumour (Table 1).

Patient flow through the study
Study treatment
The median (range) duration of treatment with panitumumab, DOC, and CIS was 11.9 (0.1–34.9), 13.6 (0.1–34.9), and 12.7 (0.1–34.9) weeks, respectively. A total of 25.0%, of the patients had at least one panitumumab dose reduction, in most cases due to acneiform rash, asthenia, mucosal inflammation, or hypomagnesemia (4.6% each). The number of patients with at least one dose reduction of DOC (29.0%) and CIS (25.0%) was similar to that of panitumumab, most commonly due to asthenia (9.1%), neutropenia (6.8%), and diarrhea (6.8%).
Almost half the patients (40.9%) discontinued panitumumab treatment, with pneumonia, respiratory tract infection, nausea, vomiting, asthenia, and rash (4.6% each) being the most common causes. A total of 36.4% discontinued DOC and CIS treatment, most prevalent causes being nausea, vomiting, pneumonia, asthenia, and decreased appetite (4.6% each). The second-line treatments were only recorded for 22 patients. The most common schemes used in these patients were FOLFIRI (n = 7), followed by capecitabine (n = 4) and irinotecan (n = 4).
Efficacy
The median follow-up time was 7.6 (range 1.8–36.6) months. Treatment efficacy results are summarized in Table 2. The primary endpoint of this study, and the ORR was of 27.3% (95% CI 15.0, 42.8). Three patients (6.8%) presented a CR and 20.5% (n = 9) presented a PR. However, most patients (43.2% [n = 19]) showed an SD and 25.0% (n = 11) of the patients showed PD. The median duration was of 5.3 (3.8–7.0) months, and the median TTF and TTP was of 3.2 (2.3–3.8) and 5.3 (3.8–7.0) months, respectively (Table 2). The DCR observed was of 70.5% (95% CI 54.8, 83.2), with a median DoR of 4.8 (1.8-NE) months.
The median PFS and OS was of 5.0 (95% CI 3.6, 6.9) and 7.2 (5.5, 9.0) months, respectively (Table 2 and Fig. 2).

Kaplan–Meier curves used to estimate a PFS and b OS distribution
Safety
Table 3 summarizes the incidence of AEs during the study. All patients suffered at least one AE and 90.9% suffered a Grade 3–4 AEs (Grade 3/Grade 4: 68.2%/22.2%). The most frequently reported AEs were: asthenia (75.0%), mucosal inflammation (54.5%), diarrhea (45.5%), rash (43.2%) and anemia (40.9%). Asthenia (29.5%) and neutropenia (25.0%) were the most common severe AEs (Grade ≥ 3), followed by vomiting, pneumonia, pulmonary embolism, and rash (all 9.1%). A total of 22.7% patients presented a Grade 4 AE, with neutropenia being the most common (15.9%). Other Grade 4 AEs included anemia, febrile neutropenia, and duodenal obstruction (all 2.3%) (Table 3).
Overall, 90.1% presented at least one panitumumab-related AE, most being Grade 2 (38.3%). Nevertheless, 34.1% and 9.1% of the patients reported at least one Grade 3 and Grade 4 treatment-related AE, respectively. Asthenia (11.4%), rash (9.1%), and mucosal inflammation (6.8%) were the most common Grade 3 panitumumab-related AEs, and neutropenia (N = 3, 6.8%) and rash (2.3%) were the only panitumumab-related Grade 4 AEs (Table 3).
At least one DOC- and CIS-related AE was experienced by 95.5% and 90.9% of patients, respectively. A total of 72.7% of the patients presented a Grade 3–4 AE related to DOC or CIS, the most common being asthenia (both 29.5%), neutropenia (both 25.0%), vomiting (both 9.1%), anemia (DOC/CIS 4.5%/6.8%), and mucosal inflammation (both 6.8%). The only Grade 4 AE related to DOC or CIS was neutropenia (both 15.9%) (Table 3).
At least one serious AE (SAEs) was reported in 54.6% patients; 9.1%, 13.6% and 15.9% were related to panitumumab, DOC and CIS, respectively. The most common SAE (pneumonia) was not related to any study treatment. SAEs such as nausea were only related to DOC and CIS and anemia was only related to panitumumab and cisplatin (Table 3).
Discussion
The present open-label, multicenter phase II clinical study in treatment-naïve patients with advanced gastric or GEJ-ADC was carried out with the idea of analyzing the benefit of adding panitumumab to the first-line DOC and CIS treatment in a biweekly regimen. This hypothesis was based on evidence, suggesting that EGFR overexpression is relatively frequent in the esophagogastric ADC as well as demonstrated improvement in survival variables with the use of anti-EGFR monoclonal antibodies combined with chemotherapy in RAS-WT metastatic CRC [14, 15]. The ORR (95% CI) of this study was 27% (15–42%) with 7% and 20% CR and PR, respectively. This ORR proved to be lower than the ORR obtained in the phase II clinical study with metastatic gastric cancer patients treated with DOC and CIS alone (43% [33, 53%]) [16, 17] or in the phase II trial on the first-line biweekly DOC and CIS combination in advanced gastric cancer (42% [29–56]) [7]. Similarly, the phase II ATTAX3 study examined the addition of panitumumab to DOC, CIS, and fluoropyrimidines in patients with recurrent oesophagus, oesophagogastric junction, or stomach cancer [12]. The ORR observed in patients treated with chemotherapy plus panitumumab was 58% (42–72%), which was similar to those treated with only chemotherapy [12]. In the REAL3 phase III study, that began at the same time as this study, combined panitumumab with oxaliplatin, epirubicin, and capecitabine, and demonstrated a similar ORR in both groups (46% in the panitumumab group) [11]. In the EXPAND phase III study, stomach or GEJ-ADC patients were randomized to either capecitabine-CIS chemotherapy or capecitabine-CIS chemotherapy with additional cetuximab; the ORR was also similar in both groups (29% [25, 34] and 30% [26, 34], respectively) and similar to the one observed in this study [10].
The median PFS and OS obtained in our study were 5 (95% CI 4, 7) and 7 (5, 9) months, respectively. These PFS and OS outcomes also appeared to be lower than that observed in the phase II clinical trial where patients were treated with DOC and CIS alone—which had a PFS and OS of 7 (5, 9) and 12 (10, 13) months, respectively [16]. A PFS and OS greater than that observed in this study were also reported in the REAL3 study (chemotherapy/chemotherapy plus panitumumab PFS: 6/7 months and OS: 11/9 months), ATTAX3 (chemotherapy/chemotherapy plus panitumumab PFS: 7/6 months and OS: 12/10 months) and EXPAND (chemotherapy/chemotherapy plus cetuximab PFS: 4/6 months and OS: 9/11 months) [12, 18].
A total of 91%, 96% and 91% of the patients presented at least one AEs associated with panitumumab, DOC, and CIS and 43%, 73%, and 73% reported at least one Grade 3–4 AE. Only two patients (7%) had SAEs related to panitumumab. In this study, there were three deaths (duodenal obstruction, intestinal obstruction, and severe respiratory tract infection) due to an AE, but none were treatment-related. The percentage of patients with Grade 3–4 toxicities showed a trend toward higher values than those ones observed in previous trials. In the phase II study with DOC and CIS as treatment, neutropenia (17%) and nausea/vomiting (13%) were found as the most common Grade 3–4 toxicities [16]. In the REAL3 study, an increase in diarrhea, mucositis, rash, and Grade 3–4 hypomagnesemia was observed in the panitumumab plus EOC group, although lower incidence of Grade 3–5 neutropenia AEs was observed in the panitumumab plus EOC group compared to EOC alone (28% vs. 13%, P < 0.0001) [11]. Noteworthy is the safety alert that the REAL3 trial had during the study which even led to a data review in the ATTAX3 phase II study and a subsequent suspension of new recruitments albeit the lack of evidence for higher number of AEs observed in the panitumumab arm in this study [12].
The main limitation of this phase II study is the lack of a comparator group and the limited sample size. In addition, it is important to note that most patients included had an ECOG performance status of 0 or 1. This may limit the generalizability of these findings to the overall population of patients with advanced gastric or GEJ-ADC population due to a possible overestimation of the treatment effect.
However, the results of the study hereby presented corroborates results obtained from other studies in recent years where EGFR and MET pathway targeting agents have failed to show efficacy or safety benefit [8, 19, 20].
In conclusion, the addition of panitumumab to chemotherapy (DOC + CIS) in patients with advanced gastric or advanced GEJ-ADC showed discrete efficacy outcomes and a poor safety profile in this study, suggesting a minimum to null effect compared to DOC + CIS alone.
References
Stewart BW, Wild CP, editors. World Cancer Report 2014. Lyon, France: International Agency for Research on Cancer. 2014.
Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 2012. https://globocan.iarc.fr. Accessed September 23, 2015.
Al-Batran S-E, Lorenzen S. Management of Locally Advanced Gastro-oesophageal Cancer: Still a Multidisciplinary Global Challenge? Hematol Oncol Clin North Am. 2017;31:441–52. https://doi.org/10.1016/j.hoc.2017.01.004.
Van Cutsem E, Moiseyenko VM, Tjulandin S, Majlis A, Constenla M, Boni C, et al. Phase III study of docetaxel and cisplatin plus fluorouracil compared with cisplatin and fluorouracil as first-line therapy for advanced gastric cancer: a report of the V325 Study Group. J Clin Oncol. 2006;24:4991–7. https://doi.org/10.1200/JCO.2006.06.8429.
Ajani JA, Fodor MB, Tjulandin SA, Moiseyenko VM, Chao Y, Cabral Filho S, et al. Phase II multi-institutional randomized trial of docetaxel plus cisplatin with or without fluorouracil in patients with untreated, advanced gastric, or gastro-oesophageal adenocarcinoma. J Clin Oncol. 2005;23:5660–7. https://doi.org/10.1200/JCO.2005.17.376.
Kim JY, Do YR, Park KU, Kim MK, Lee KH, Bae SH, et al. A multi-center phase II study of docetaxel plus cisplatin as first-line therapy in patients with metastatic squamous cell esophageal cancer. Cancer Chemother Pharmacol. 2010;66:31–6. https://doi.org/10.1007/s00280-009-1130-6.
Quintero-Aldana G, Jorge M, Grande C, Salgado M, Gallardo E, Varela S, et al. Phase II study of first-line biweekly docetaxel and cisplatin combination chemotherapy in advanced gastric cancer. Cancer Chemother Pharmacol. 2015;76:731–7. https://doi.org/10.1007/s00280-015-2839-z.
Woo J, Cohen SA, Grim JE. Targeted therapy in gastro-oesophageal cancers: past, present and future. Gastroenterol Rep. 2015;3:316–29. https://doi.org/10.1093/gastro/gov052.
Cervantes A, Rodríguez Braun E, Pérez Fidalgo A, Chirivella González I. Molecular biology of gastric cancer. Clin Transl Oncol. 2007;9:208–15.
Lordick F, Kang Y-K, Chung H-C, Salman P, Oh SC, Bodoky G, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): A randomised, open-label phase 3 trial. Lancet Oncol. 2013;14:490–9. https://doi.org/10.1016/S1470-2045(13)70102-5.
Waddell T, Chau I, Cunningham D, Gonzalez D, Okines AFC, Frances A, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): a randomised, open-label phase 3 trial. Lancet Oncol. 2013;14:481–9. https://doi.org/10.1016/S1470-2045(13)70096-2.
Tebbutt NC, Price TJ, Ferraro DA, Wong N, Veillard A-S, Hall M, et al. Panitumumab added to docetaxel, cisplatin and fluoropyrimidine in oesophagogastric cancer: ATTAX3 phase II trial. Br J Cancer. 2016;114:505–9. https://doi.org/10.1038/bjc.2015.440.
Pinto C, Di Fabio F, Barone C, Siena S, Falcone A, Cascinu S, et al. Phase II study of cetuximab in combination with cisplatin and docetaxel in patients with untreated advanced gastric or gastro-oesophageal junction adenocarcinoma (DOCETUX study). Br J Cancer. 2009;101:1261–8. https://doi.org/10.1038/sj.bjc.6605319.
Chan JA, Blaszkowsky LS, Enzinger PC, Ryan DP, Abrams TA, Zhu AX, et al. A multicenter phase II trial of single-agent cetuximab in advanced esophageal and gastric adenocarcinoma. Ann Oncol. 2011;22:1367–73. https://doi.org/10.1093/annonc/mdq604.
Gold PJ, Goldman B, Iqbal S, Leichman LP, Zhang W, Lenz H-J, et al. Cetuximab as second-line therapy in patients with metastatic esophageal adenocarcinoma: a phase II Southwest Oncology Group Study (S0415). J Thorac Oncol. 2010;5:1472–6. https://doi.org/10.1097/JTO.0b013e3181e77a92.
Park KW, Ahn JS, Park YS, Lee J, Kang JH, Park JO, et al. Phase II study of docetaxel and cisplatin combination chemotherapy in metastatic gastric cancer. Cancer Chemother Pharmacol. 2007;59:17–211. https://doi.org/10.1007/s00280-006-0253-2.
Aoyagi K, Kouhuji K, Kizaki J, Isobe T, Hashimoto K, Shirouzu K. Molecular targeting to treat gastric cancer. World J Gastroenterol. 2014;20:13741–55. https://doi.org/10.3748/wjg.v20.i38.13741.
Abraham I, Alhossan A, Lee CS, Kutbi H, MacDonald K. “Real-life” effectiveness studies of omalizumab in adult patients with severe allergic asthma: systematic review. Allergy. 2015. https://doi.org/10.1111/all.12815
Pasini F, Fraccon AP, Modena Y, Bencivenga M, Giacopuzzi S, La Russa F, et al. Targeted therapies for advanced and metastatic adenocarcinoma of the gastro-oesophageal junction: is there something new? Gastric Cancer. 2017;20:31–42. https://doi.org/10.1007/s10120-016-0626-0.
Raufi AG, Klempner SJ. Immunotherapy for advanced gastric and esophageal cancer: preclinical rationale and ongoing clinical investigations. J Gastrointest Oncol. 2015;6:561–9. https://doi.org/10.3978/j.issn.2078-6891.2015.037.
Acknowledgements
Writing assistance was supported by Amgen and provided by Cristina Ceballos, MSc, Nerea Gallastegui, PhD, and Neus Valveny, PhD, from TFS Develop. The authors wish to thank to all the investigators of the SPIGA Study.
Funding
This work was supported by Amgen S.A. Amgen did not have any role in study design; collection, analysis, and interpretation of data; writing the report; and the decision to submit the report for publication.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Ethic Committee of Reference (Comité Ético de Investigación Clínica SERGAS, Santiago de Compostela, Spain) and the Spanish Health Authorities (AEMPS), and conducted in accordance with the principles of the Declaration of Helsinki.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Quintero Aldana, G., Salgado, M., Candamio, S. et al. First-line panitumumab plus docetaxel and cisplatin in advanced gastric and gastro-oesophageal junction adenocarcinoma: results of a phase II trial. Clin Transl Oncol 22, 495–502 (2020). https://doi.org/10.1007/s12094-019-02151-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-019-02151-6