
Abstract
Purpose
Colorectal cancer (CRC) is one of the most widely diagnosed cancers in men and women worldwide. With the advancement of next-generation sequencing technologies, many studies have highlighted the involvement of long non-coding RNAs (lncRNAs) in cancer development. Growing evidence demonstrates that lncRNAs play crucial roles in regulating gene and protein expression and are involved in various cancers, including CRC. The field of lncRNAs is still relatively new and a lot of novel lncRNAs have been discovered, but their functional roles are yet to be elucidated. This study aims to characterize the expression and functional roles of a novel lncRNA in CRC.
Method
Several methods were employed to assess the function of LOC285629 such as gene silencing, qPCR, proliferation assay, BrdU assay, transwell migration assay, ELISA and protein profiler.
Results
Via in silico analyses, we identified significant downregulation of LOC285629, a novel lncRNA, across CRC stages. LOC285629 expression was significantly downregulated in advanced stages (Stage III and IV) compared to Stage I (Kruskal–Wallis Test; p = 0.0093). Further in-house validation showed that the expression of LOC285629 was upregulated in colorectal cancer tissues and cell lines compared to the normal counterparts, but was downregulated in advanced stages. By targeting LOC285629, the viability, proliferative abilities, invasiveness and resistance of colorectal cancer cells towards 5-fluorouracil were reduced. It was also discovered that LOC285629 may regulate cancer progression by targeting several different proteins, namely survivin, BCL-xL, progranulin, PDGF-AA, enolase 2 and p70S6 K.
Conclusion
Our findings suggest that LOC285629 may be further developed as a potential therapeutic target for CRC treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) has become the epitome of cancers that are highly associated with death. Being the third most commonly diagnosed cancer and leading cause of cancer-related deaths worldwide, CRC has attracted a lot of attention among researchers and clinicians alike [1]. In Malaysia, CRC accounts for the second most common cancer in males and third most common cancer in females [2]. Most patients are diagnosed with CRC at a late stage, which by then, the treatment available may not be able to eradicate the cancer thoroughly. Though multiple treatments such as surgery, chemotherapy, radiotherapy and targeted therapy are available, the prognosis for CRC patients remains poor.
Over the past few years, the attention on non-coding RNAs has increased gradually [3]. Long non-coding RNA (lncRNA) has piqued the interest of many research groups worldwide. LncRNAs are RNAs that do not code for proteins, are more than 200 bp long and accounts for approximately 80% of the transcripts [4]. The expression of lncRNAs has been associated with various cancers such as cervical cancer [5], prostate cancer [6], lung cancer [7] and CRC [8]. The roles of lncRNAs have been documented to be involved in multiple cellular processes such as viability, proliferation, migration and survival [3]. For instance, a study by Iden et al. discovered that the lncRNA PVT1 was involved in the progression of cervival cancer and was linked to poor prognosis of patients [5]. Similarly, a study conducted by Li et al. found that the lncRNA CASC2 was involved in promoting the proliferation of gastric cancer via the MAPK pathway [9].
In CRC, there are a number of known lncRNAs such as HOTAIR, MALAT1, UCA and TUG [10]. The lncRNA HOTAIR is known to be oncogenic and can be used to predict poor prognosis of colorectal cancer patients [11, 12]. Additionally, patients with high expression of HOTAIR were linked with higher chance of recurrence and metastasis [11]. Likewise, the pattern of expression of MALAT1 was also linked to the poor outcome of CRC patients [13]. It has been shown that MALAT1 contributes to CRC progression by promoting proliferation, migration and invasion [14, 15]. Based on the NONCODE (v25) database, there are around 90,062 lncRNA genes identified in human [16]. However, there are only a handful number of lncRNAs that have been validated and characterized. Hence, we decided to identify promising lncRNAs in CRC via in silico analysis and further characterize it using in vitro assays. Based on our preliminary screening using the cancer genome atlas (TCGA) data, we found that LOC285629 is an interesting lncRNA that shows promising potential in CRC. Therefore, this study is aimed at validating the expression of LOC285629 as well assess the potential of LOC285629 as a therapeutic target in CRC.
Materials and methods
Selection of lncRNA
The Cancer Genome Atlas (TCGA) colorectal cancer expression data obtained from Firebrowse database (http://www.firebrowse.org) was used. Gene expression levels were presented as normalized log2 RSEM (RNA-Seq by Expectation–Maximization) index from Firebrowse database. Kruskal–Wallis test was used to analyze the significance in differential expression among the stages.
Cell lines and culture conditions
The human CRC cell lines (SW480, CCD-112-CoN, COLO-205, HT-29) were purchased from American Type Culture Collection (ATCC). For CCD-112-CoN, the cells were cultured in Eagle Minimum Essential Medium (Invitrogen, USA) and the rest of the cell lines were cultured in RPMI 1640 Medium (Invitrogen, USA). All media were supplemented with 10% fetal bovine serum (Gibco, USA). All of the cells were incubated in humidified air at 37 °C with 5% CO2.
Clinical specimen collection
CRC tissues and normal tissues were obtained from the UKM Medical Molecular Biology Institute (UMBI) Biobank. Written informed consent has been taken from each patient prior to sampling and storage for research. The samples were anonymized before analysis. The pathological stage, grade and nodal status were assessed by an experienced pathologist. Only tumor tissues that contained > 80% of cancerous cells and normal tissues with < 20% of necrotic cells were used for this study. The demographic analysis is presented in Table 1.
Total RNA isolation
For the clinical specimens, total RNA was isolated using the AllPrep DNA/RNA/miRNA Isolation Kit (Qiagen, Germany) according to the manufacturer’s protocol. Meanwhile, for secondary CRC cell lines (HT-29, SW 480, SW 48, CCL 220, HCT 116, and COLO 205) and the normal colonic epithelial cell (CCD-112-CoN), the entire RNAs were isolated using the RNeasy Mini Kit (Qiagen, Germany). The quantity and purity of the RNA were measured using Nanodrop 1000 (Thermo Fisher Scientific, USA). The integrity of the RNA was assessed using the Eukaryote Total RNA Nano Chip on the Bioanalyzer 2100 (Agilent Technologies, USA).
Real-time quantitative PCR (qPCR)
First-strand cDNA synthesis was performed using the RT2 First Strand Kit (Qiagen, Germany) based on the manufacturer’s instructions. Briefly, a total of 500 ng of total RNA was reverse transcribed in a final volume of 100 μl using both oligo-dT and random hexamers. The expression level of LOC285629 in primary CRC tissue and secondary CRC cell lines was determined using the RT2 lncRNA qPCR Assay (Qiagen, Germany). The qPCR reaction was performed using the RT2 SYBR Green Flour qPCR Master Mix (Qiagen, Germany) on a CFX96 Touch™ Real-Time PCR Detection System (Biorad, USA). The qPCR results were analyzed, and the relative messenger RNA expression of CT (threshold cycle) value was then converted to fold changes (2 − ΔΔCt). The expression of LOC285629 was measured in relative to the expression of GAPDH and Beta-Actin.
Transfection of colorectal cancer cell lines
Small interfering RNA (siRNA), known as Lincode Human LOC285629 siRNA, was synthesized (Dharmacon, UK) and transiently transfected into cells using Dharmafect 2 (Dharmacon, UK). For LOC285629 knockdown results, the expression was determined using qPCR after 24-, 48- and 72 h of transfection. The optimization of transfection was determined by the siGLO Green Transfection Indicator (Dharmacon, UK). The siRNA complex was formed in a 24-well plate by adding equal volumes of the stock concentration of siRNA and OptiMEM (Invitrogen, USA) and incubating the mixture at room temperature. After 5 min, the transfection reagent was added along with additional OptiMEM to yield a final total volume of 100 µl/well and incubated at room temperature. After 20 min, the siRNA complex was added, then followed by the resuspended cell lines at the desired concentration. siRNA complex: cell lines mixture was plated and incubated at 37 °C, 5% CO2 for 24 h before the media was changed to fresh RPMI-1640 and EMEM.
Cell viability assay
The cells were transfected in a 96-well culture plate; after 48 h of transfection, the cells were stained by adding 20 µL PrestoBlue reagent (Thermo Fisher Scientific, USA) directly to the sample wells. The plate was then incubated for an additional 30 min in a 37 °C incubator with 5% CO2. Afterwards, the cell viability was determined by measuring the resulting fluorescent signal using the Varioskan™ Flash Multimode Reader (Thermo Fisher Scientific, USA). The percentage of viability (%) was expressed based on the calculation below:
Clonogenic assay
After 48 h of transfection with 25 nM non target control, Lincode LOC285629 siRNA and positive control, 5000 cells were plated in a 6-well plate with complete media and the plate was swirled to ensure an even distribution of the cell. The cells were grown in 37 °C incubator with 5% CO2 for 10 days with media replacement every 3 days. On day 10, the media was removed and cells were washed twice with PBS. The colonies were fixed with 10% acetic acid for 15 min, dried and stained with 0.5% crystal violet solution for 1 h. In order to remove the excess staining, the plate was washed three times with tap water. Images of the stained plates were captured, and the cell colonies containing more than 50 cells were counted. Each treatment was performed in triplicate.
Wound healing assay
Cells were transiently transfected with LOC285629 siRNA and its respective control and harvested at 48 h after transfection. The ibidi Culture-Inserts (ibidi GmbH, Germany) were placed on a cell culture surface and cells were seeded at 3−7 × 105 cells/ml in each of the ibidi Culture-Insert well (70 µl into each well). The cells were incubated at 37 °C and 5% CO2 for the cells to be attached. After 24 h, the inserts were gently removed using sterile tweezers. The cell layer was washed with cell-free medium or PBS to remove cell debris and non-attached cells. Around 2 ml of cell-free medium was filled into the µ-Dish. The dish was viewed under the inverted microscope (Nikon, Japan) and the image was captured. The observation process was started by taking images several times throughout the following hours:0-, 24-, 48- and 72 h. The percentage of wound healing (%) was measured using the T scratch software [17].
In vitro migration assay
Cytoselect 24 well Cell Migration Assay (Cell Biolabs, USA) was used for cell migration assay. The cells were harvested after 48 h of transfection and suspended in serum-free medium. Then 300 μL of cell suspension was added to the upper chamber at a density of 0.5–1.0 × 106 cells/well, and 500 μL of serum-rich medium was added to the lower chamber. The cell inserts were cultured for 2–24 h. The cell inserts were collected for the following treatment. The surface of the cell insert was swabbed with cotton-tipped applicators to remove the cells that did not migrate. The cell inserts were fixed with 100% methanol for 30 min, dried and stained with 0.5% crystal violet solution (Sigma, USA) for 1 h. Distilled water was then applied to wash off excess stain materials. For data analysis, migrated cells were viewed and counted under a light microscope (Nikon, Japan).
Drug sensitivity assay
The cells were seeded in a 96-well plate at a concentration of 5 × 103 cells/well and were incubated at 37 °C and 5% CO2. 5-fluorouracil (Nacalai Tesque, Japan) was added to the wells at a concentration of 3.75 and 1.87 µg/mL. The cells were left to incubate for 24 and 48 h after treatment. After the respective incubation time, 20 µL of PrestoBlue was added to the cells followed by measurement of the resulting absorbance signal using Varioskan™ Flash multimode reader.
BrdU assay
The BrdU proliferation assay was performed according to the manufacturer’s instructions (Milipore, USA). Cells were transiently transfected with si-LOC285629 and were seeded in 96-well plates at a density of 2 × 105 cells/mL. After the respective incubation time, 20 µL of diluted brdU was added to each of the test wells and the plates were further incubated for another 2 h. Then, media from the wells were aspirated and 200 µL of fixing solution was added to the cells for 30 min. After the fixing solution was removed, the plates were washed with the provided washing buffer to a total of three washes. Next, goat anti-mouse IgG was added to the wells for 30 min, followed by the addition of the TMB peroxidase solution. Finally, the reaction was stopped by adding the provided stop solution and the absorbance was measured using Varioskan™ Flash multimode reader (Thermo, USA).
Proteome Profiler™ assay
Cells were transiently transfected with LOC285629 siRNA and its respective control and harvested at 48 h after transfection. Cell lysates were obtained by incubating the cells with lysis buffer. Total protein was measured using Bradford assay at 450 nm (Sigma, USA). Afterwards, 300 µg of protein was incubated with a human apoptosis array membrane that could detect the relative expression of apoptosis-related proteins according to the manufacturer’s protocol (R&D, USA). Next, the array membranes were incubated in the provided antibodies and washed several times. Then, chemiluminescence substrate was added to each array membrane to visualize the dots. The immunoreactive dots were visualized using Molecular Imager ChemiDoc (BioRad, Germany). The intensity of each dot was measured using the ImageLab software (BioRad, Germany) and the fold change was calculated from the LOC285629-knockdown samples against the control samples.
Statistical analysis
Statistical analysis was performed using GraphPad Prism v7 (GraphPad Software, USA). The quantitative data were presented as the mean ± standard deviations (SD). Differences were considered to be statistically significant at values of p < 0.05.
Results
LOC285629 is upregulated in colorectal cancer
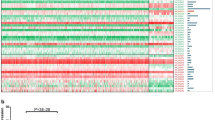
To better understand the LOC285629 expression status in cancer versus their normal counterpart, the FireBrowse gene expression viewer was employed to visualize expression data collected from various RNA sequencing studies. The RSEM expression profiles for each TCGA disease chart is shown in Fig. 1. Using this approach, we observed that LOC285629 expression was detected in almost all cancer types and most normal tissues. Interestingly, majority of the cancer data appeared to have larger error bars in their expression levels compared to their normal counterparts, which is likely a reflection of the heterogeneous nature of the cancer itself. In the colorectal cancer (COAD) study, LOC285629 expression is up-regulated by 1.62-fold. The expression is also upregulated in almost all of the cancer types with exception to kidney (KICH, KIPAN, KIRC and KIRP) as well as stomach cancers (STAD and STES). Further analyses revealed significant downregulation of LOC285629 in advanced stages (Stage III and IV) compared to stage I (Kruskal–Wallis Test; p = 0.0093),

The expression level of LOC285629 as denoted by RSEM value in each TCGA study
We then further examined the level of LOC285629 expression in nine CRC tissues and four normal tissues. As shown in Fig. 2a, the level of expression of LOC285629 was increased in tumor tissues compared to the normal tissues. Moreover, as evidenced in Fig. 2b, when we screened in CRC cell lines, the level of expression of LOC285629 was highest in SW-480 cell line, followed by COLO-205 and HT-29. There was no expression of LOC285629 detected in CCL 220, SW-48 and HCT-116 cells. Based on these results, we chose SW-480 cell line for further studies and validation due to the high expression of LOC285629.

Expression levels of LOC285629 in a clinical specimens and b cancer cell lines detected via real-time PCR
Inhibition of LOC285629 decreased the viability, proliferation and clonogenic potential in SW-480 cells
To investigate the potential of LOC285629 as a therapeutic target, we transiently silenced LOC285629 in SW-480 and CCD-112 CoN cells and monitored the viability and proliferative abilities of the cells. Upon reduction of LOC285629, the viability of SW-480 cells decreased to 91% after 48 h of treatment as shown in Fig. 3a. Similarly, as displayed in Fig. 3b, the percentage of BrdU incorporated in SW-480 cells also reduced when treated with si-LOC285629. Interestingly, for the CCD-112-CoN cells, the reduction of LOC285629 did not affect the cell’s viability and proliferation. Furthermore, as shown in Fig. 3c, the number of colonies formed in SW-480 cells upon treatment with si-LOC285629 also reduced significantly from 360 colonies in the control group to 250 colonies in the si-LOC285629 group.

Inhibition of LOC285629 decreased the a viability, b proliferation and (c and d) clonogenic potential in SW-480 cells after 48 h of treatment
LOC285629 regulates colorectal cancer cell motility
Moreover, we also explored the role of LOC285629 in the motility abilities of SW-480 cells. To investigate this, we performed wound healing assay and transwell migration assay. Based on Fig. 4a, after 48 h of treatment, the percentage of wound closure decreased from 51.38 to 31.27% upon reduction of LOC285629 in SW-480 cells. Additionally, it was also observed in CCD-112-CoN cells that there was no significant difference in terms of wound closure when treated with si-LOC285629 (Fig. 4b). Furthermore, we conducted the transwell migration assay as well. As presented in Fig. 4c and d, in LOC285629-depleted SW480 cells, the number of migrated cells was lower than the untreated cells and in CCD-112-CoN cells, no observable difference was detected.

LOC285629 regulates colorectal cancer cell motility. a After 48 h of treatment, the percentage of wound closure decreased with reduction of LOC285629 in SW-480 cells. b and in CCD-112-CoN cells c The number of migrated cells for CCD-112-CoN cells. d Image example of the migration membrane for SW-480 cells
Reduction of LOC285629 increases colorectal cancer cell sensitivity towards 5-Fluorouracil
To assess whether LOC285629 affects the sensitivity of the cells towards drug treatment, we performed drug sensitivity assay using 5-fluorouracil. As shown in Fig. 5, when the LOC285629-depleted cells were treated with 3.75 µg of 5-fluorouracil, the viability of the cells reduced to 86.7% as opposed to the untreated cells, with 95% viability. Likewise, the same pattern was also observed when the cells were treated with 1.75 µg of 5-fluorouracil.

Reduction of LOC285629 increases colorectal cancer cell sensitivity towards 5-Fluorouracil at both 3.75 and 1.875 µg/mL
LOC285629 affects several cancer-related proteins
To interrogate the role of LOC285629 in regulating other cancer-related players, we conducted the Proteome Profiler Assay for oncogenic proteins. Based on Fig. 6a, upon reduction of LOC285629, the protein expression of several proteins were decreased. The most affected protein was the BCL-X protein with around 2.51-fold of down-regulation. This was followed by PDGF-AA, progranulin, survivin and Enolase 2. Additionally, we also investigated the role of LOC285629 in relation to the p70S6 K tyrosine kinase. As shown in Fig. 5b, the expression of p70S6 K decreased substantially upon loss-of-function of LOC285629.

LOC285629 affects cancer-related proteins. a The most affected protein was the BCL-X protein with 2.51-fold down-regulation. b Expression of p70S6 K tyrosine kinases decreased substantially upon LOC285629 loss-of-expression
Discussion
Colorectal cancer (CRC) is one of the most widely diagnosed cancers in men and women in Malaysia and also worldwide [2]. Long non-coding RNAs are implicated in a lot of cancers including CRC. It has been reported that multiple lncRNAs such as MALAT1, HOTAIR and CCAT1 are involved in the progression of colorectal cancer [10,11,12, 14]. This study was performed to uncover the potential roles of novel lncRNAs in the progression of CRC. We identified LOC285629 as a novel lncRNA from our in silico analysis which showed the expression of LOC285629 to be higher in the early stages of CRC than the late stages of CRC. To verify the expression of LOC285629 in CRC, we screened the level of expression of LOC285629 in both primary CRC tissues and secondary CRC cell lines. Based on our results, the expression of LOC285629 was indeed higher in cancerous tissues than the normal tissues. However, due to the small number size of samples for primary tissue, we were unable to conclude whether LOC285629 was higher in the early stages of CRC. Nevertheless, among all the patients screened, Patient 11 with Dukes A CRC had the highest expression of LOC285629, which is in concordance with our hypothesis. Similarly, in secondary cell lines, the expression of LOC285629 was higher in cancer cells than normal colonic epithelial cells. This was in agreement to what we hypothesized as shown in our in silico analysis. To further understand the role of LOC285629 we proceeded with loss-of-function analysis.
The ability to grow uncontrollably and proliferate indefinitely is one among the major characteristics of cancer cells. To be able to inhibit the proliferation process as well as decrease the cell viability is one among the important considerations for developing treatment for cancer. There are multiple lncRNAs that are involved in the regulation of the viability and proliferation state of cancer cells. For instance, the lncRNA Fer-1-like protein 4 (FER1L4) was found to significantly regulate the proliferation of CRC cells [8]. Similarly, as evidenced by the viability assay, BrdU incorporation assay and clonogenic assay, upon reduction of LOC285629, the viability and proliferative abilities of SW-480 decreased. Nevertheless, there was no difference seen in the normal colonic epithelial cells, CCD-112 CoN cells when LOC285629 was silenced. This could indicate that the effects of LOC285629 were more selective in cancer cells than normal cells. Moreover, as shown in our viability assay, the percentage of dead cells increased marginally in LOC285629-depleted cells. Cell death involves many steps and multiple proteins that can execute the process efficiently. Survivin for instance, is an inhibitor of apoptosis and is highly expressed in cancers [18, 19]. Therefore, to target survivin has been one of the approaches to prevent cancer cells from progressing. Similarly, BCL-xL is also an oncogenic protein that drives tumor progression by inhibiting apoptosis and promoting metastasis [20,21,22]. The decreased expressions of survivin and BCL-xL are indicators that LOC285629 may be involved in cancer progression by inhibiting cell death.
Additionally, it has been shown that a certain percentage of CRC cells have resistance towards standard chemotherapy. Among the most widely used drugs to treat colorectal cancer is 5-fluorouracil [23,24,25]. Resistance towards 5-fluorouracil has been extensively acknowledged and is one of the major hurdles in treating cancer [23]. It has been a major research area to find targets or molecules that could reverse or potentiate the resistance towards standard chemotherapy regimes, especially 5-fluorouracil. Interestingly, here, we show that by targeting LOC285629, the percentage of cells killed by 5-fluorouracil was higher than the control cells. This indicates that one of the ways that LOC285629 is involved in cancer progression is by providing resistance towards anti-cancer drugs.
Moreover, most of the reported cancer-related deaths are associated with the metastatic potential of the cancer. The more aggressive the cancer, the more likely it will metastasize and form secondary tumors. It is estimated that in 1 in 20 males and 1 in 23 females will be diagnosed with invasive colorectal cancer [1]. Among the behavior of metastatic cells that can be analyzed is the aptitude to migrate. By performing cell migration-related assays, we demonstrated that when the expression of LOC285629 was reduced, the ability of the cells to migrate also declined. Furthermore, we assessed the expression of progranulin in the treated and untreated cells. Progranulin is well-known to be associated with breast and ovarian cancers [26]. In CRC, it was recently discovered that progranulin was involved in the proliferation, migration and tubule formation [27]. It was revealed that progranulin increased tumorigenesis by regulating TNFR2/AKT and ERK pathways [27]. As demonstrated by our findings, the expression of progranulin decreased upon reduction of LOC285629 in SW-480 cells. Furthermore, we found that the expression platelet-derived growth factor AA or alternatively known as PDGF-AA was also reduced in the treated cells. PDGF-AA is mostly overexpressed in cancers and is associated with the activation of VEGF [28]. Additionally, PDGF-AA is involved in angiogenesis that when overexpress, increases the probability of pre-cancerous lesions to turn into cancerous tumors [28,29,30]. Moreover, we also found that LOC285629 deregulated the expression of Enolase-2 as well. Enolase-2, also known as neuron-specific enolase (NSE), is an enzyme that is involved in the glycolytic pathway [31]. The expression of enolase-2 has been associated with the progression of cancer [31]. Furthermore, we discovered that expression of p70s6 k was also reduced when the cells were treated with si-LOC285629. P70S6 K is a tyrosine kinase that is involved in various processes that contribute to carcinogenesis [32, 33]. For instance, it has been found that the aberrant expression of P70S6 K in gastric cancer promoted cell growth, invasiveness and metastasis [34]. Therefore, the reduction of progranulin, PDGF-AA, enolase-2 and p70s6 k could further support the potential of LOC285629 as a therapeutic target for CRC. Figure 7 displays the proposed schematic mechanism on how LOC285629 operates.

Proposed schematic mechanism of how LOC285629 regulates cancer progression
Conclusion
LncRNAs serve as promising targets for the diagnosis and treatment of CRC. Collectively, our study has shown that LOC285629 is involved in CRC progression by targeting different pathways and proteins. LOC285629 may be involved in CRC pathogenesis by directly or indirectly associating with several cancer-related proteins such as Enolase and PDGF-AA. Moreover, we were also able to elucidate the function of LOC285629 in impeding the motility of CRC cells in vitro. Though the preliminary results presented seem promising, further in depth analysis should be conducted, especially in terms of molecular regulation of important signaling pathways.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. https://doi.org/10.3322/caac.21332.
Abu Hassan MR, Ismail I, Mohd Suan MA, Ahmad F, Wan Khazim WK, Othman Z, et al. Incidence and mortality rates of colorectal cancer in Malaysia. Epidemiol Health. 2016;38:e2016007. https://doi.org/10.4178/epih.e2016007.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. https://doi.org/10.1038/nrg.2015.10.
Wang Y, Zhang D, Wu K, Zhao Q, Nie Y, Fan D. Long noncoding RNA MRUL promotes ABCB1 expression in multidrug-resistant gastric cancer cell sublines. Mol Cell Biol. 2014;34(17):3182–93. https://doi.org/10.1128/mcb.01580-13.
Iden M, Fye S, Li K, Chowdhury T, Ramchandran R, Rader JS. The lncRNA PVT1 contributes to the cervical cancer Phenotype and associates with poor patient prognosis. PLoS One. 2016;11(5):e0156274. https://doi.org/10.1371/journal.pone.0156274.
Misawa A, Takayama K-i, Urano T, Inoue S. Androgen-induced long noncoding RNA (lncRNA) SOCS2-AS1 promotes cell growth and inhibits apoptosis in prostate cancer cells. J Biol Chem. 2016;291(34):17861–80. https://doi.org/10.1074/jbc.M116.718536.
Wu C-H, Hsu C-L, Lu P-C, Lin W-C, Juan H-F, Huang H-C. Identification of lncRNA functions in lung cancer based on associated protein-protein interaction modules. Scientific Rep. 2016;6:35939. https://doi.org/10.1038/srep35939. http://www.nature.com/articles/srep35939#supplementary-information
Yue B, Sun B, Liu C, Zhao S, Zhang D, Yu F, et al. Long non-coding RNA Fer-1-like protein 4 suppresses oncogenesis and exhibits prognostic value by associating with miR-106a-5p in colon cancer. Cancer Sci. 2015;106(10):1323–32. https://doi.org/10.1111/cas.12759.
Li P, Xue W-J, Feng Y, Mao Q-S. Long non-coding RNA CASC2 suppresses the proliferation of gastric cancer cells by regulating the MAPK signaling pathway. Am J Transl Res. 2016;8(8):3522–9.
Saus E, Brunet-Vega A, Iraola-Guzmán S, Pegueroles C, Gabaldón T, Pericay C. Long non-coding RNAs as potential novel prognostic biomarkers in colorectal cancer. Front Genet. 2016;7:54. https://doi.org/10.3389/fgene.2016.00054.
Wu Z, Wang X, Tang H, Jiang T, Chen J, Lu S, et al. Long non-coding RNA HOTAIR is a powerful predictor of metastasis and poor prognosis and is associated with epithelial-mesenchymal transition in colon cancer. Oncol Rep. 2014;32(1):395–402.
Zhao W, Song M, Zhang J, Kuerban M, Wang H. Combined identification of long non-coding RNA CCAT1 and HOTAIR in serum as an effective screening for colorectal carcinoma. International Journal of Clinical and Experimental Pathology. 2015;8(11):14131–40.
Zheng H-T, Shi D-B, Wang Y-W, Li X-X, Xu Y, Tripathi P, et al. High expression of lncRNA MALAT1 suggests a biomarker of poor prognosis in colorectal cancer. Int J Clin Exp Pathol. 2014;7(6):3174–81.
Xu C, Yang M, Tian J, Wang X, Li Z. MALAT-1: a long non-coding RNA and its important 3′ end functional motif in colorectal cancer metastasis. Int J Oncol. 2011;39(1):169–75.
Yang M-H, Hu Z-Y, Xu C, Xie L-Y, Wang X-Y, Chen S-Y, et al. MALAT1 promotes colorectal cancer cell proliferation/migration/invasion via PRKA kinase anchor protein 9. Biochem Biophys Acta. 2015;1852(1):166–74. https://doi.org/10.1016/j.bbadis.2014.11.013.
Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, et al. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Research. 2016;44(Database issue):203–8. https://doi.org/10.1093/nar/gkv1252.
Gebäck T, Schulz MM, Koumoutsakos P, Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46:265–274. https://doi.org/10.2144/000113083
Jaiswal PK, Goel A, Mittal RD. Survivin: a molecular biomarker in cancer. Indian J Med Res. 2015;141(4):389–97. https://doi.org/10.4103/0971-5916.159250.
Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14(16):5000.
Choi S, Chen Z, Tang LH, Fang Y, Shin SJ, Panarelli NC et al. Bcl-xL promotes metastasis independent of its anti-apoptotic activity. Nature Commun. 2016;7:10384. doi:10.1038/ncomms10384 http://www.nature.com/articles/ncomms10384#supplementary-information
Jeng PS, Cheng EH. Cancer therapeutics: pulling the plug on BCL-xL. Nat Chem Biol. 2013;9(6):351–2. https://doi.org/10.1038/nchembio.1256.
Scherr A-L, Gdynia G, Salou M, Radhakrishnan P, Duglova K, Heller A, et al. Bcl-xL is an oncogenic driver in colorectal cancer. Cell Death Dis. 2016;7:e2342. https://doi.org/10.1038/cddis.2016.233.
Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–8.
Pardini B, Kumar R, Naccarati A, Novotny J, Prasad RB, Forsti A, et al. 5-Fluorouracil-based chemotherapy for colorectal cancer and MTHFR/MTRR genotypes. Br J Clin Pharmacol. 2011;72(1):162–3. https://doi.org/10.1111/j.1365-2125.2010.03892.x.
Wiebke EA, Grieshop NA, Loehrer PJ, Eckert GJ, Sidner RA. Antitumor effects of 5-fluorouracil on human colon cancer cell lines: antagonism by levamisole. J Surg Res. 2003;111(1):63–9. https://doi.org/10.1016/S0022-4804(03)00053-2.
Han JJ, Yu M, Houston N, Steinberg SM, Kohn EC. Progranulin is a potential prognostic biomarker in advanced epithelial ovarian cancers. Gynecol Oncol. 2011;120(1):5–10. https://doi.org/10.1016/j.ygyno.2010.09.006.
Yang D, Wang L-L, Dong T-T, Shen Y-H, Guo X-S, Liu C-Y, et al. Progranulin promotes colorectal cancer proliferation and angiogenesis through TNFR2/Akt and ERK signaling pathways. Am J Cancer Res. 2015;5(10):3085–97.
Shikada Y, Yonemitsu Y, Koga T, Onimaru M, Nakano T, Okano S, et al. Platelet-Derived growth factor-AA Is an essential and autocrine regulator of vascular endothelial growth factor expression in non-small cell lung carcinomas. Can Res. 2005;65(16):7241–8. https://doi.org/10.1158/0008-5472.can-04-4171.
Heldin C-H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal. 2013;11(1):97. https://doi.org/10.1186/1478-811x-11-97.
Heldin C-H, Östman A, Rönnstrand L. Signal transduction via platelet-derived growth factor receptors. Biochim Biophys Acta. 1998;1378:79–113.
Vizin T, Kos J. Gamma-enolase: a well-known tumour marker, with a less-known role in cancer. Radiol Oncol. 2015;49(3):217–26. https://doi.org/10.1515/raon-2015-0035.
Berven LA, Crouch MF. Cellular function of p70S6 K: a role in regulating cell motility. Immunol Cell Biol. 2000;78(4):447–51.
Berven LA, Willard FS, Crouch MF. Role of the p70(S6 K) pathway in regulating the actin cytoskeleton and cell migration. Exp Cell Res. 2004;296:183–95. https://doi.org/10.1016/j.yexcr.2003.12.032.
Xiao L, Wang YC, Li WS, Du Y. The role of mTOR and phospho-p70S6 K in pathogenesis and progression of gastric carcinomas: an immunohistochemical study on tissue microarray. J Exp Clin Cancer Res. 2009;28(1):152. https://doi.org/10.1186/1756-9966-28-152.
Acknowledgement
The authors would like to thank Sri Noraima Othman for the initial involvement in in silico analysis. This study is funded by Universiti Kebangsaan Malaysia under Research University Grant (GUP-2014-068).
Author information
Authors and Affiliations
Contributions
SNN: conducted the experiments, NA, NSAM: data analyses, interpretation, writing of the manuscript, MI: specimen quality control and RNA extraction, IS, LM: colorectal surgeon involved in specimen collection, IMR: pathologist involve in confirming the diagnosis, RJ: principal investigator and critical evaluation of the manuscript
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Research involving patients
All procedures performed in studies involving humans were in accordance with the ethical standards of the institution or practice at which the studies were conducted.
Rights and permissions
About this article

Cite this article
Nasir, S.N., Abu, N., Ab Mutalib, N.S. et al. LOC285629 regulates cell proliferation and motility in colorectal cancer cells. Clin Transl Oncol 20, 775–784 (2018). https://doi.org/10.1007/s12094-017-1788-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-017-1788-x