
Abstract
Publication of this consensus statement is a joint initiative of the Spanish Society of Pathology (SEAP) and the Spanish Society of Medical Oncology (SEOM), intended to revise and update the diagnostic and treatment recommendations published 2 years ago on biomarker use and the management of patients with colorectal carcinoma (CRC), thereby providing an opportunity to improve healthcare efficiency and resource use in these patients. This expert group recommends testing for KRAS and NRAS status in all patients with metastatic CRC being considered for anti-epidermal growth factor receptor (anti-EGFR) therapy, as this type of treatment should only be used in patients not harbouring mutations in these genes. In contrast, testing for BRAF, EGFR, PI3K and PTEN mutation status is not necessary for therapeutic decision making, so does not need to be done routinely.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Patients with colorectal carcinoma (CRC) must be tested for molecular biomarkers that predict the response to specific treatments, as this is essential for developing personalised medicine, thereby avoiding the use of inappropriate therapy associated with undesirable effects and costs. In this context, cooperation between oncologists and pathologists is indispensable, and thus consensus guidelines containing recommendations for biomarker testing in CRC were recently published by the Spanish Society of Pathology (SEAP) and the Spanish Society of Medical Oncology (SEOM) [1]. Those guidelines may be revised from time to time to incorporate new biomarkers, when the evidence level warrants their use for new treatment strategies, especially in the management of patients with metastatic disease. Little has changed in terms of markers of susceptibility to hereditary CRC, or molecular markers of localised carcinoma [microsatellite instability (MSI)], so the parameters published in the previous guidelines remain valid.
However, recent studies have shown that molecular tests in patients with metastatic cancer do not only need to consider KRAS mutations, because other activating mutations such as NRAS can be regarded as negative predictive biomarkers for anti-epidermal growth factor receptor (anti-EGFR) therapy. Testing for all RAS mutations has, therefore, radically changed the treatment strategy in CRC [2]. Also, the addition of new high-throughput methodologies makes it advisable for these to be included in this new version of the consensus guidelines.
It is also worth defining the new biomarkers appearing, and the advisability of testing for them routinely according to the evidence levels [3]. From the pathology perspective, other issues raised address resource optimisation for often scanty samples (because the minimum amount of material the pathologist should receive and retain for further testing and/or new clinical trials needs to be established), and deciding which type of specimen (metastases, primary tumours or both) is most effective for molecular techniques. Lastly, the situations in which it is advisable for the patient to be re-biopsied should be defined. All these issues will be addressed in these guidelines, which, like the previous version, may be revised as required.
Which of the previous guidelines are still valid?
From the point of view of identifying patients with a susceptibility to colon cancer, and deciding on adjuvant therapy in the case of localised colon cancer, there is no change as regards the indication for MSI testing or the recommendations in the previous guidelines.
Gene expression signatures in localised disease
The Oncotype Dx® genetic signature was initially validated with data from the QUASAR study [4], thus establishing the prognostic value of the gene signature by defining three risk categories (high, intermediate and low), with 3-year relapse rates of 22, 18 and 12 % (p = 0.046), respectively. However, the predictive value of the gene signature could not be validated, which compromises the clinical usefulness of this test [5]. The prognostic value of Oncotype Dx® has subsequently been confirmed in another two series of randomised clinical trials (CALGB 9581 and NSABP C07) [6, 7]. The ColoPrint® validation sets have also been extended in a pooled analysis of several series containing 227 stage II T3 patients with MSI [8]. This genetic signature proved able to differentiate tumours with a high-risk signature, with a 79 % 3-year relapse-free survival, from 94 % of tumours with a low-risk signature (HR: 2.74; 95 % CI: 1.54–4.88; p = 0.006). Despite this progress, the recommendation level for using these genetic signatures when deciding on adjuvant chemotherapy in stage II is low. However, they are probably helpful for decision making in cases in which both doctor and patient want more prognostic information, e.g. in cases with MSI and T3 if one wishes to suggest the option of treating the patient, or T4 if greater certainty is desired when deciding not to treat.
Changes in the pathological diagnosis
Changes in TNM
The seventh-edition TNM incorporates changes in all three categories, i.e. T, N and M [9, 10].
-
T category: T4 is subdivided into T4a if the tumour penetrates the visceral peritoneum and T4b if the tumour directly invades other organs or structures.
-
N category: N1 lymph node involvement is subdivided into N1a if only one lymph node is involved and N1b when 2 or 3 are affected. pN1c refers to pN0 patients with discontinuous tumour deposits or satellite nodules in the mesocolic or mesorectal adipose tissue. These deposits represent lymphovascular or perineural invasion rather than true lymph nodes. They are associated with worse disease-free survival and worse overall survival (OS) [11, 12]. pN1c patients will be classified in TNM stage III, by stage group. N2 has been subdivided into N2a if 4–6 lymph nodes are involved and N2b if 7 or more are affected. These changes have prognostic implications in these patient subgroups.
-
M category: subdivided into M1a when metastases exist in a single organ and M1b when they occur in several organs or the peritoneum [10].
Laboratory accreditation
To ensure quality and efficiency, it is advisable for tests for predictive molecular markers to be performed in accredited laboratories involved in regular quality controls [13, 14].
Other recommended changes in the pathology report and diagnostic issues
The latest diagnostic guidelines for CRC from the College of American Pathologists (CAP) and the American Joint Committee on Cancer (AJCC), published in 2013, include the following [15]:
-
A recommendation for the pathology report to state whether the tumour has histological features suggestive of MSI, as this has implications for prognosis and treatment [16]. Histological features of instability are: (i) intratumoral lymphocytic response or tumour-infiltrating lymphocytes, divided into three grades, i.e. none, mild to moderate (0–2 per high-power [×400] field [HPF]), or marked (≥3/HPF); (ii) Crohn-like peritumoral lymphocytic response, divided into none, mild to moderate, or marked; (iii) histological subtype and tumour differentiation, specifying the presence and percentage of mucinous, medullary and high-grade components, location in the right colon, tumour heterogeneity and lack of dirty necrosis [17].
-
Tumour invasion of vascular structures, whether lymphatic vessels or veins, is described under a single category of angiolymphatic invasion.
-
Lymph nodes showing no gross evidence of tumour should be included in their entirety.
-
The number of positive lymph nodes is reported, specifying the number of micro- and macrometastases. Micrometastases measure >0.2 and ≤2.0 mm. They are classified as N1(mic), or M1(mic) in non-regional lymph nodes.
-
Isolated tumour cells (ITCs) occur as isolated cells or in small clusters of <0.2 mm. They tend to be seen with serial sections of lymph nodes or molecular techniques, immunohistochemistry (IHC) or the polymerase chain reaction (PCR). If detected by non-conventional methods, they are classified as pN0, because their clinical significance is unproven.
Which biomarkers should be tested before starting treatment?
Recently, some studies have demonstrated the value of considering NRAS as well as KRAS mutation status for selecting patients eligible for anti-EGFR therapy in metastatic CRC. The PRIME trial, which compared 5-fluorouracil, folinic acid and oxaliplatin (FOLFOX4) against FOLFOX4 and panitumumab, in 512 patients with all wild-type RAS (KRAS and NRAS), demonstrated a progression-free survival (PFS) of 10.1 months with the addition of panitumumab versus 7.9 months in the non-antibody arm (hazard ratio [HR] for progression or death: 0.72; 95 % confidence interval [CI]: 0.58–0.90; p = 0.004) [2]. Likewise, median OS was 26.0 months for the group treated with the biological agent, compared with 20.2 months (HR: 0.78; 95 % CI: 0.62–0.99; p = 0.04).
The PEAK trial compared FOLFOX6 combined with panitumumab versus FOLFOX6 plus bevacizumab as first-line treatment for metastatic colorectal cancer. The outcome of this trial was that patients with all wild-type RAS (KRAS and NRAS, both in exons 2, 3 and 4) in the panitumumab arm had better PFS (13.0 months) than patients treated with bevacizumab, with a median of 9.5 months (HR: 0.65; 95 % CI: 0.44–0.96; p = 0.029) [18]. In terms of OS, the results were again better for patients treated with the anti-EGFR drug. However, this result failed to reach statistical significance, with a median of 41.3 versus 28.9 months in patients given bevacizumab (HR: 0.63; 95 % CI: 0.39–1.02; p = 0.058).
The FIRE-3 trial, recently reported in Lancet Oncology [19], compared first-line treatment with folinic acid, fluorouracil and irinotecan (FOLFIRI) combined with cetuximab versus FOLFIRI with bevacizumab in patients with metastatic colorectal cancer. The results demonstrated that in patients with wild-type KRAS, OS was significantly better for those treated with cetuximab (28.7 months) versus bevacizumab (25.0 months) (HR: 0.77; 95 % CI: 0.62–0.96; p = 0.017). However, there were no differences in PFS, as very similar medians were obtained (10.0 versus 10.3 months, respectively; HR: 1.06; 95 % CI: 0.88–1.26; p = 0.55).
The OPUS study has been reanalysed according to the role of all wild-type RAS as shown in terms of PFS in patients treated with FOLFOX and cetuximab [20]. This treatment group achieved a median PFS of 12.0 months compared with patients treated with chemotherapy alone, with 5.8 months (HR: 0.43; p = 0.018). This represents a 6.2-month increase in this parameter compared with the 1.1-month increase obtained when only KRAS mutations are considered. Likewise, in patients with all wild-type RAS, the CRYSTAL trial showed a PFS value of 11.4 months in patients treated with FOLFIRI and cetuximab versus 8.4 months in those treated with FOLFIRI alone (HR: 0.56; p = 0.0002) [21]. In OS terms, patients with all wild-type RAS given cetuximab showed a median of 28.4 versus 20.2 months in those treated with FOLFIRI alone (HR: 0.69; p = 0.0024).
Based on these studies, this expert panel recommends that:
-
All patients with metastatic CRC being considered for anti-EGFR therapy should be tested for KRAS and NRAS status, as this therapy should only be given when no mutations exist in these genes (Level of Evidence Ia) [3].
Which techniques should be used to test all RAS?
The technologies currently available for addressing these new mutations are the same ones used to date to test for mutations in KRAS exon 2 [22] (Table 1). They consist mainly of sequencing those exons, by the Sanger method, pyrosequencing or other techniques. However, there is continual technological development aimed at producing commercial mutation test kits bearing the European conformity marking for in vitro diagnostics (CE-IVD). There are now many platforms on the market for performing these molecular tests for research use only (RUO). Worthy of note in this group are the technological advances in massively parallel sequencing or next-generation sequencing (NGS), in which various platforms enable such tests to be done simultaneously in the context of clinical trials and research studies.
Which sample should be tested for all RAS, and when?
Cancer is a dynamic process. It is in this context that the concepts of clonal evolution and intratumour heterogeneity should be considered, and their clinical repercussions assessed in a personalised approach for treating this disease [23]. The idea of cancer as an exponential proliferation of monoclonal cancer cells has evolved in the last few decades towards a dual-hierarchy model in which a sub-population of tumour stem cells keeps the tumour proliferating, giving rise to a population of non-tumourigenic cells. Tumour transfection studies in mice have shown the presence of phenotypically distinct populations of stem cells and non-tumourigenic cells in various human tumours [24]. Similarly, it is known that the plasticity of cancer cells allows them to reversibly turn from epithelial cells into mesenchymal cells. Drug resistance is also described as another form of tumour plasticity that causes sensitive cells to become resistant. Lastly, reversible epigenetic changes can also alter the cellular characteristics of any population present in a tumour.
These findings have called into question the hierarchical model based on populations of stem cells and non-tumourigenic cells, to make way for the clonal evolution theory. According to this, a tumour contains distinct cell populations that undergo mutations, resulting in positive or negative selection depending on whether or not these mutations confer an advantage over other cancer cells. The presence of distinct cancer cell populations in a single tumour has been elegantly illustrated by Gerlinger et al. [25], who studied various portions of the same clear-cell renal tumour and its metastases.
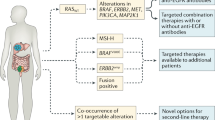
CRC is a heterogeneous disease, in which tumours formed via different carcinogenesis pathways coexist. These distinct pathways are initiated by different mechanisms of molecular alteration such as MSI, hypermethylation and chromosomal instability, and by mutations in driver genes. Personalised treatment of colon cancer is currently targeted by RAS mutation status. The absence of mutations in exons 2, 3 and 4 of the KRAS and NRAS genes identifies patients with colon tumours more susceptible to treatment with antibodies against EGFR. However, still a significant proportion of patients selected this way do not respond to therapy. Potential reasons for this, besides alternative resistance mechanisms, may include intratumour heterogeneity [26], differences between the primary tumour and its metastases [27], and the sensitivity of methods used to identify mutations [28].
According to some study results, as described above, before deciding on the treatment of a patient with metastatic colon cancer, tests should be done for possible mutations in exons 2, 3 and 4 of KRAS, specifically codons 12, 13, 61, 117 and 146, and in the same exons and codons in NRAS. A sample of either the primary tumour or a metastasis can be used in these tests, with a concordance level of over 95 % [29]. Lamy et al. [30] looked for the presence of KRAS and BRAF mutations in a series of 1,130 tumours from 992 patients with CRC. The authors identified discordant KRAS status between the primary tumour and metastases in 11.4 % of cases. They also detected tumours harbouring two concomitant KRAS mutations. Other authors have subsequently confirmed these same findings [26–28, 31]. Based on these studies, it is recommended that tests for RAS status be conducted on metastatic tissue.
What is the optimal amount of sample?
One of the most important factors in the pre-analytical phase is the amount of sample needed to conduct the various tests. Because of issues that can affect the results, such as over-fixation, it is advisable for biomarker tests to be done on material from endoscopic biopsies or, in the case of metastatic tumours, on needle biopsy cores.
Molecular techniques perform less well if these tests are done on tissue from surgical resection specimens, and even worse using surgical specimens from patients given pre-operative chemotherapy/radiotherapy. One of the problems encountered in some Spanish hospitals is that the diagnosis is done first, and the oncologist orders biomarker tests afterwards, so the material is subjected to further processing. Moreover, the profusion of new clinical trials can mean the material is handled for a third time, and often proves insufficient for the external requirements of the laboratories responsible for the strict protocols used in these studies.
Several options, therefore, exist. One is to cut multiple sections at the time of diagnosis, cover them with paraffin, and store them until biomarker tests are ordered [32, 33]. Another solution is to request duplicate endoscopic biopsies, so that at least 2–3 fragments are embedded in one paraffin block for diagnostic histopathological analysis, and another 2–3 additional fragments are embedded in another block, to be used in molecular biomarker tests and perhaps future clinical trials. As far as these are concerned, at least some hospitals are experiencing excessive demands for material (15–20 sections 5 microns thick). Sometimes shipment of a paraffin block is even requested, leading to situations in which the pathology department is left without any type of sample for the patient concerned.
Lastly, some regional biobanks and genetic counselling units also request shipment of a paraffin block for storage and future use in research projects. In these cases, it is better to send the paraffin block obtained from surgical resection specimens. Again, the amount of sample is closely related to the quality of the endoscopic examination. Disappointingly often, multiple fragments of normal tissue are obtained accompanied by scanty tumour material. In these situations, anticipating the need for molecular tests, it may be advisable to ask for the patient to be re-biopsied.
In which situations should the patient be re-biopsied?
Tumour progression is a dynamic, ever-changing phenomenon governed by the laws of the multi-clonality or tumour heterogeneity theory. Different mutation profiles are, therefore, likely to be found at different sites of metastasis. There may also be heterogeneity within a single organ affected by metastasis, and even in different areas of the same metastasis, as shown in a pivotal study by Gerlinger et al. [25] focusing on renal cancer. In CRC, technological innovation in the field of identifying mutations from circulating tumour deoxyribonucleic acid (DNA) has revealed the appearance of “de novo” mutations or molecular alterations in patients with tumours initially labelled KRAS or RAS wild type. To date, mutations in the KRAS gene itself or NRAS, c-met amplifications, and the S492R EGFR mutation, at the site where cetuximab binds to the receptor, have been identified. In all these cases, the appearance of these mutations or molecular anomalies preceded the onset of secondary or acquired resistance to treatment with EGFR-inhibiting monoclonal antibodies [34–38].
The most plausible hypothesis is that the emergence of these “de novo” mutations represents minority resistant clones with undetectable mutations that have acquired a proliferative advantage and managed to grow above the limit of detection of the test methods used [39]. These all seem to support the practice of repeat biopsies or testing for mutations in circulating tumour DNA during the course of disease progression and exposure to different lines of treatment [40, 41]. The aim of performing repeat biopsies would be to anticipate resistance and develop molecular treatments targeted at emerging molecular profiles. The limitation of this approach is the lack of effective treatments for most of these situations, although many clinical trials of these molecular targets are currently in progress, based on rational combinations that have shown synergy and synthetic lethality in pre-clinical models [42–44].
Another important factor is that most pharmacologically actionable mutations known to date in CRC tend to appear at early stages of carcinogenesis and are likely to be found in stored biopsy material or old surgical resection specimens of primary tumour or metastases. The most practical first step is, therefore, to analyse the available tumour material and assume that, if RAS mutations are found, they will still be represented in the existing volume of disease. To date, there has been no suggestion that routine re-biopsy should take place in cases of wild-type tumours, based on analysis of the available archived material. In any case, although KRAS and NRAS mutations are currently all that is required for a standard treatment strategy, with EGFR-inhibitor drugs administered only in cases of unmutated tumours, in the near future there will be treatment options targeted against these and other mutations and alterations such as BRAF, c-met, ERBB2, ERBB3, and EGFR. For this reason, it is becoming increasingly common to suggest that patients should be re-biopsied, and testing for these mutations in circulating tumour DNA might even be considered in future.
Which other biomarkers are currently of interest?
BRAF
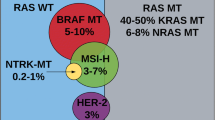
The BRAF gene, which encodes one of the main intracellular effectors of KRAS, is mutated in 5–10 % of patients with CRC [45]. The most common mutation (~90 %) is the V600E substitution, located in the kinase domain of the protein, in exon 15. In general, BRAF mutations and KRAS mutations are mutually exclusive, and BRAF mutations are associated with the presence of high microsatellite instability (MSI-H) [46, 47]. In other contexts, particularly in advanced disease, the presence of BRAF mutations is associated with a worse prognosis [45–51].
The role of BRAF mutations as a predictive factor for response to anti-EGFR therapies is more controversial. In uncontrolled studies, it has been observed that the efficacy of anti-EGFR therapies in patients with KRAS wild-type, but BRAF-mutated tumours is substantially lower than in KRAS wild-type, BRAF wild-type tumours (overall response rate [ORR]: 8 vs. 38 %; HR: 0.15; p = 0.0012. PFS: 8 vs. 26 weeks; HR: 3.74; p < 0.0001. OS: 26 vs. 54 weeks; HR: 3.03; p < 0.0001) [45]. However, the small amount of data available from randomised trials suggests that these worse outcomes are independent of the treatment received [48, 49, 51].
In the CRYSTAL study, which evaluated the administration of FOLFIRI with and without cetuximab in CRC patients, ORR, PFS and OS rates increased significantly when cetuximab was added to conventional chemotherapy in KRAS and BRAF wild-type patients (n = 566) [51]. A similar but smaller trend, which did not attain statistical significance, was seen in KRAS wild-type, BRAF-mutated patients (n = 59) amongst a group of patients treated with FOLFIRI with and without cetuximab, respectively (ORR: 19 vs. 15 %; PFS: 8.0 vs. 5.6 months; and OS: 14.1 vs. 10.3 months). Similarly, in the CAIRO2 study, the presence of BRAF mutations was significantly associated with worse PFS and OS, irrespective of the treatment received, with or without cetuximab [52]. Likewise, some more recent subgroup analyses in the context of randomised clinical trials, such as the PRIME study, evaluating the administration of FOLFOX with or without panitumumab [2], or trial 20020408/NCT00113763 testing the use of panitumumab versus supportive care [53], again suggest that the presence of BRAF mutations has more prognostic than predictive value.
EGFR
Initially, anti-EGFR therapies were developed in tumours expressing EGFR on the cell surface, as detected by IHC techniques in 85 % of CRCs. It was subsequently observed, however, that there was no good correlation between EGFR protein expression and response to these drugs. Nevertheless, mutations affecting the spatial configuration of the extracellular domain of the receptor have recently been described, associated with acquired resistance to cetuximab, but not panitumumab [37].
Other factors that have been suggested as potential predictive biomarkers of response to anti-EGFR therapies include over-expression of the receptor’s natural ligands, such as epiregulin and amphiregulin [54], certain ligand polymorphisms (EGF61A/G vs. EGF61A/A or EGF61G/G) [55], amplification of the EGFR gene or chromosome 7 polysomy (both of which are associated with increased copy number of the gene) [56]. Likewise, polymorphisms in the Fc gamma receptors (FcγR) of immune effector cells (FcγRIIa-131H/H and/or FcγRIIIa-158V/V) have also been found to be associated with a better response to cetuximab, even in tumours harbouring KRAS mutations [57]. This suggests that antibody-dependent cellular cytotoxicity (ADCC) might play an important role in this context, a hypothesis currently being tested in the MUTEX trial (ClinicalTrials.gov identifier: NCT01450319). Much of these data, however, come from small retrospective series, without proper controls, so no definite conclusions can be drawn concerning their clinical usefulness.
Other mutations
The PI3K–AKT–mTOR pathway is another major intracellular signalling effector pathway activated by EGFR stimulation. Fifteen percent of CRCs harbour PIK3CA-activating mutations, 69 % of them in exon 9 and 20 % in exon 20 [45]. Mutations in exon 20 seem to be associated with a worse response to cetuximab in uncontrolled series [45]. In agreement with this, subgroup analysis in the FIRE randomised trial, comparing FOLFIRI with cetuximab versus bevacizumab found that in patients with PI3K mutations, PFS was greater in patients treated with bevacizumab than those given cetuximab, although no differences in ORR or OS were seen between the two arms [58]. Another way in which this pathway can be activated is loss of PTEN function, which some authors have also associated with less response to anti-EGFR treatments [59, 60]. However, the literature data on this are inconsistent, so these results should be confirmed by means of prospective studies properly designed for that purpose.
Based on the above, this expert panel recommends that:
-
These patients should not be tested routinely for BRAF, EGFR, PI3K and PTEN status, as this is not necessary for therapeutic decision making (Level of Evidence IIb for BRAF; Level of Evidence IIIc for EGFR, PI3K and PTEN) [3].
Are there any new technological developments in biomarker testing?
The new technologies seek to detect specific sequences with greater sensitivity. Beads, Emulsions, Amplification and Magnetics-PCR (BEAMing-PCR) is based on single-molecule PCR on microparticles in drops of water-in-oil emulsion [61]. The sensitivity of this approach is calculated at 0.01 %, so it has been proposed for detecting free mutated sequences in peripheral blood, with an overall concordance level of 93 % in a small series.
Digital PCR is another new technology now becoming more widespread in genetic diagnostics, although it was first described in 1999 [62]. It involves amplifying a DNA template in extremely dilute samples, thereby generating amplicons from just a single DNA molecule. Any change in sequence can thus be detected by fluorescence analysis in that mini-reaction. This method also permits a detection level of 0.01 %. A third approach is the COLD-PCR method [63]. This is a new form of PCR that amplifies alleles present in tiny numbers in a sample containing wild-type sequences, using a co-amplification that favours the specific sequences sought, by means of lower denaturation temperatures. In this way, the sensitivity of this technique too allows levels of 0.01 % to be attained.
Lastly, a number of technologies enable a multitude of genes to be analysed simultaneously (multiplexing), and may provide better characterisation of the molecular profile of many of the tumours being tested today (Table 2). With these techniques, a vast number of genes can be addressed, which may not need testing at the time, but will allow greater understanding of that cancer in prospective terms. A notable example of these technologies is Sequenom® iPLEX® Gold. This is a multiplex PCR technique in which the amplicons obtained are subsequently examined by mass spectrometry (MALDI-TOF MS), allowing 238 mutations in 17 known oncogenes to be analysed simultaneously, with a sensitivity level of 5–10 % [64]. Similarly, other methodologies that are becoming established in the research environment, but will enter the healthcare setting in the future, are the massive sequencing platforms, already available today in the form of various kits or gene panels designed to offer more detailed understanding of the molecular profile of patients with CRC. These technologies have a sensitivity of about 1–5 %.
Conclusions
Great progress has been made in understanding the biological alterations essential to carcinogenesis and tumour development in CRC. This is a heterogeneous disease, in which tumours formed via different carcinogenesis pathways coexist. These distinct pathways are initiated by different mechanisms of molecular alteration, including MSI, hypermethylation and chromosomal instability, and by mutations in driver genes.
Genome studies reveal the complexity of molecular heterogenicity in CRC, and although various types of molecular classifications have been proposed, no standardised, validated classification is available in clinical practice. Knowledge of some genomic events has allowed prognostic and predictive biomarkers to be developed. These guidelines review biomarkers in CRC, making clinical- and pathology-related recommendations based on the level of scientific evidence for each of them, and defining the quality controls and methodology to be used in each case. Because of rapidly developing understanding of the molecular events involved in this disease, these guidelines must be reviewed from time to time and the recommendations updated. In terms of markers of susceptibility to hereditary CRC and molecular markers of localised carcinoma, the recommendations contained in the previous guidelines remain unchanged, as no substantial progress has been made.
However, there has been an important change in how to identify patients with metastatic CRC eligible for anti-EGFR treatment, warranting the updating of these guidelines. Currently, personalised treatment of colon cancer is targeted according to RAS mutation status. The presence of mutations in exons 2, 3 and 4 of the KRAS and NRAS genes distinguishes between patients with colon tumours susceptible to treatment with antibodies against EGFR and those in whom such treatment has a detrimental effect. Therefore, the new recommendation for administering anti-EGFR therapy in patients with metastatic CRC is to test for all RAS (KRAS and NRAS) mutation status, as this therapy should only be given when no mutations exist in these genes.
Testing for RAS mutation status should be done with the relevant quality controls using the right sample. To ensure quality and efficiency, it is advisable for tests for predictive molecular markers to be done in accredited laboratories involved in regular quality controls. As regards the choice of sample, the use of metastatic tissue is recommended, to avoid discordance with the primary tumour, because it must be understood that tumour progression in CRC is a dynamic, ever-changing phenomenon governed by the laws of the multi-clonality or tumour heterogeneity theory. In this respect, there have been reports of patients with wild-type KRAS in whom appearance of these mutations or molecular anomalies preceded the onset of secondary or acquired resistance to treatment with EGFR-inhibiting monoclonal antibodies. The most plausible hypothesis is that the emergence of these “de novo” mutations represents minority resistant clones with undetectable mutations that have acquired a proliferative advantage and managed to grow above the limit of detection of the test methods used. These all seem to support the practice of repeat biopsies or testing for mutations in circulating tumour DNA during the course of disease progression.
In clinical practice, testing for a wider mutation profile, such as BRAF, EGFR, PI3K and PTEN status, should not be done routinely in these patients, as it is not necessary for therapeutic decision making. However, it does provide important information for identifying subgroups and stratifying patients in clinical trials.
This update of the consensus statement on biomarkers in CRC is the result of oncologists and pathologists working together in a multidisciplinary fashion to optimise their clinical use with all the pathology-related quality criteria, laying the foundation for the future development of personalised treatment.
References
Garcia-Alfonso P, Salazar R, Garcia-Foncillas J, Musulen E, Garcia-Carbonero R, Paya A, et al. Guidelines for biomarker testing in colorectal carcinoma (CRC): a national consensus of the Spanish Society of Pathology (SEAP) and the Spanish Society of Medical Oncology (SEOM). Clin Transl Oncol. 2012;14:726–39.
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34.
Sackett DL. Rules of evidence and clinical recommendations on the use of antithrombotic agents. Chest. 1986;89:2S–3S.
Gray RG, Barnwell J, Hills R, McConkey C, Williams N, Kerr D. QUASAR: a randomized study of adjuvant chemotherapy (CT) vs observation including 3238 colorectal cancer patients. ASCO Meeting Abstracts. 2004;22:3501.
Gray RG, Quirke P, Handley K, Lopatin M, Magill L, Baehner FL, et al. Validation study of a quantitative multigene reverse transcriptase polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J Clin Oncol. 2011;29:4611–9.
Venook AP, Niedzwiecki D, Lopatin M, Ye X, Lee M, Friedman PN, et al. Biologic determinants of tumor recurrence in stage II colon cancer: validation study of the 12-gene recurrence score in cancer and leukemia group B (CALGB) 9581. J Clin Oncol. 2013;31:1775–81.
Yothers G, O’Connell MJ, Lee M, Lopatin M, Clark-Langone KM, Millward C, et al. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J Clin Oncol. 2013;31:4512–9.
Tabernero J, Moreno V, Rosenberg R, Nitsche U, Bachleitner-Hofmann T, Lanza G, et al. Clinical and technical validation of a genomic classifier (ColoPrint) for predicting outcome of patients with stage II colon cancer. ASCO Meeting Abstracts. 2012;30:384.
Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A (eds). AJCC cancer staging manual. 7th ed. New York: Springer; 2010.
Washington MK, Berlin J, Branton P, Burgart LJ, Carter DK, Fitzgibbons PL, et al. Protocol for the examination of specimens from patients with primary carcinoma of the colon and rectum. Based on AJCC/UICC TNM. 7th edn; 2013.
Lo DS, Pollett A, Siu LL, Gallinger S, Burkes RL. Prognostic significance of mesenteric tumor nodules in patients with stage III colorectal cancer. Cancer. 2008;112:50–4.
Puppa G, Maisonneuve P, Sonzogni A, Masullo M, Capelli P, Chilosi M, et al. Pathological assessment of pericolonic tumor deposits in advanced colonic carcinoma: relevance to prognosis and tumor staging. Mod Pathol. 2007;20:843–55.
Fitzgibbons PL, Bradley LA, Fatheree LA, Alsabeh R, Fulton RS, Goldsmith JD, et al. Principles of analytic validation of immunohistochemical assays: guideline from the college of american pathologists pathology and laboratory quality center. Arch Pathol Lab Med. 2014;138:1432–43.
Schrijver I, Aziz N, Jennings LJ, Richards CS, Voelkerding KV, Weck KE. Methods-based proficiency testing in molecular genetic pathology. J Mol Diagn. 2014;16:283–7.
Bartley AN, Hamilton SR, Alsabeh R, Ambinder EP, Berman M, Collins E, et al. Template for reporting results of biomarker testing of specimens from patients with carcinoma of the colon and rectum. Arch Pathol Lab Med. 2014;138:166–70.
Ribic CM, Sargent DJ, Moore MJ, Thibodeau SN, French AJ, Goldberg RM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–57.
Greenson JK, Bonner JD, Ben-Yzhak O, Cohen HI, Miselevich I, Resnick MB, et al. Phenotype of microsatellite unstable colorectal carcinomas: well-differentiated and focally mucinous tumors and the absence of dirty necrosis correlate with microsatellite instability. Am J Surg Pathol. 2003;27:563–70.
Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Hecht JR, et al. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014;32:2240–7.
Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–75.
Tejpar S, Lenz H-J, Kohne C-H, Heinemann V, Ciardiello F, Esser R, et al. Effect of KRAS and NRAS mutations on treatment outcomes in patients with metastatic colorectal cancer (mCRC) treated first-line with cetuximab plus FOLFOX4: New results from the OPUS study. ASCO Meeting Abstracts. 2014;32:LBA444.
Ciardiello F, Lenz H-J, Kohne C-H, Heinemann V, Tejpar S, Melezinek I, et al. Treatment outcome according to tumor RAS mutation status in CRYSTAL study patients with metastatic colorectal cancer (mCRC) randomized to FOLFIRI with/without cetuximab. ASCO Meeting Abstracts. 2014;32:3506.
Hernández-Losa J, Sanz J, Landolfi S, López-Ríos F, Palacios J, Bautista MD, et al. Recomendaciones para la determinación de mutaciones de K-RAS en cáncer de colon. Rev Esp Patol. 2012;45:76–85.
Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–82.
Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328–37.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92.
Kosmidou V, Oikonomou E, Vlassi M, Avlonitis S, Katseli A, Tsipras I, et al. Tumor heterogeneity revealed by KRAS, BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor mutation profile differences and their therapeutic implications. Hum Mutat. 2014;35:329–40.
Bossard C, Kury S, Jamet P, Senellart H, Airaud F, Ramee JF, et al. Delineation of the infrequent mosaicism of KRAS mutational status in metastatic colorectal adenocarcinomas. J Clin Pathol. 2012;65:466–9.
Perez K, Walsh R, Brilliant K, Noble L, Yakirevich E, Breese V, et al. Heterogeneity of colorectal cancer (CRC) in reference to KRAS proto-oncogene utilizing WAVE technology. Exp Mol Pathol. 2013;95:74–82.
Baas JM, Krens LL, Guchelaar HJ, Morreau H, Gelderblom H. Concordance of predictive markers for EGFR inhibitors in primary tumors and metastases in colorectal cancer: a review. Oncologist. 2011;16:1239–49.
Lamy A, Blanchard F, Le Pessot F, Sesboue R, Di Fiore F, Bossut J, et al. Metastatic colorectal cancer KRAS genotyping in routine practice: results and pitfalls. Mod Pathol. 2011;24:1090–100.
Improta G, Zupa A, Possidente L, Tartarone A, Pedicini P, Nappi A, et al. Coexistence of two different mutations in codon 12 of the Kras gene in colorectal cancer: Report of a case supporting the concept of tumoral heterogeneity. Oncol Lett. 2013;5:1741–3.
Engel KB, Moore HM. Effects of preanalytical variables on the detection of proteins by immunohistochemistry in formalin-fixed, paraffin-embedded tissue. Arch Pathol Lab Med. 2011;135:537–43.
Wester K, Wahlund E, Sundstrom C, Ranefall P, Bengtsson E, Russell PJ, et al. Paraffin section storage and immunohistochemistry. Effects of time, temperature, fixation, and retrieval protocol with emphasis on p53 protein and MIB1 antigen. Appl Immunohistochem Mol Morphol. 2000;8:61–70.
Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–73.
Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40.
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6.
Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–3.
Vilar E, Tabernero J. Cancer: pinprick diagnostics. Nature. 2012;486:482–3.
Santini D, Vincenzi B, Addeo R, Garufi C, Masi G, Scartozzi M, et al. Cetuximab rechallenge in metastatic colorectal cancer patients: how to come away from acquired resistance? Ann Oncol. 2012;23:2313–8.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra224.
Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20:430–5.
Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–35.
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3.
Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, et al. MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res. 2012;72:3228–37.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62.
Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29:1261–70.
Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466–74.
Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22:1535–46.
Knijn N, Mekenkamp LJ, Klomp M, Vink-Borger ME, Tol J, Teerenstra S, et al. KRAS mutation analysis: a comparison between primary tumours and matched liver metastases in 305 colorectal cancer patients. Br J Cancer. 2011;104:1020–6.
Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, et al. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol. 2009;27:5931–7.
Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–9.
Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–72.
Peeters M, Oliner KS, Parker A, Siena S, Van Cutsem E, Huang J, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013;19:1902–12.
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25:3230–7.
Spindler KL, Andersen RF, Jensen LH, Ploen J, Jakobsen A. EGF61A>G polymorphism as predictive marker of clinical outcome to first-line capecitabine and oxaliplatin in metastatic colorectal cancer. Ann Oncol. 2010;21:535–9.
Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6:279–86.
Rodriguez J, Zarate R, Bandres E, Boni V, Hernandez A, Sola JJ, et al. Fc gamma receptor polymorphisms as predictive markers of Cetuximab efficacy in epidermal growth factor receptor downstream-mutated metastatic colorectal cancer. Eur J Cancer. 2012;48:1774–80.
Stintzing S, Jung A, Rossius L, Modest DP, Fischer von Weikersthal L, Decker T, et al. Mutations within the EGFR signaling pathway: influence on efficacy in FIRE-3—a randomized phase III study of FOLFIRI plus cetuximab or bevacizumab as first-line treatment for wild-type (WT) KRAS (exon 2) metastatic colorectal cancer (mCRC) patients. J Clin Oncol. 2014;32:abstract 445.
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol. 2009;27:5924–30.
Negri FV, Bozzetti C, Lagrasta CA, Crafa P, Bonasoni MP, Camisa R, et al. PTEN status in advanced colorectal cancer treated with cetuximab. Br J Cancer. 2010;102:162–4.
Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006;3:551–9.
Vogelstein B, Kinzler KW. Digital PCR. Proc Natl Acad Sci USA. 1999;96:9236–9241.
Li J, Wang L, Mamon H, Kulke MH, Berbeco R, Makrigiorgos GM. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat Med. 2008;14:579–84.
Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–51.
Acknowledgments
The authors are grateful for the editorial assistance of Ana Martín of HealthCo (Madrid, Spain) in the production of this manuscript. SEOM and SEAP are grateful for financial support for this project in the form of unrestricted grants from Merck, Sanofi and Roche.
Conflict of interest
The authors declare that, when writing and revising the text, they did not know the names of the pharmaceutical companies that provided financial support for this project, so this support has not influenced the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
García-Alfonso, P., García-Foncillas, J., Salazar, R. et al. Updated guidelines for biomarker testing in colorectal carcinoma: a national consensus of the Spanish Society of Pathology and the Spanish Society of Medical Oncology. Clin Transl Oncol 17, 264–273 (2015). https://doi.org/10.1007/s12094-014-1252-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-014-1252-0