
Abstract
The Rsu1 protein contributes to cell adhesion and migration via its association with the adaptor complex of Integrin linked kinase (ILK), PINCH, and Parvin (IPP), which binds to the cytoplasmic domain of β1 integrins joining integrins to the actin cytoskeleton. Rsu1 binding to PINCH in the IPP complex is required for EGF-induced adhesion, spreading and migration in MCF10A mammary epithelial cells. In addition, Rsu1 expression inhibits Jun kinase but is necessary for the activation of MKK4 and p38 Map kinase signaling essential for migration in MCF10A cells. The data reported here examines the links between MKK4-p38-ATF2 signaling and AKT regulation in MCF10A cells. Ectopic Rsu1 inhibited AKT1 phosphorylation while Rsu1 depletion induced AKT activation and AKT1 phosphorylation of MKK4 on serine 80, blocking MKK4 activity. Rsu1 depletion also reduced the RNA for lipid phosphatase PTEN thus implicating PTEN in modulating levels of activated AKT in these conditions. ChIP analysis of the PTEN promoter revealed that Rsu1 depletion prevented binding of ATF2 to a positive regulatory site in the PTEN promoter and the enhanced binding of cJun to a negatively regulatory PTEN promoter site. These results demonstrate a mechanism by which Rsu1 adhesion signaling alters the balance between MKK4-p38-ATF2 and cJun activation thus altering PTEN expression in MCF10A cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The IPP complex of proteins that links integrins to the actin cytoskeleton consists of Integrin linked kinase (ILK), Parvin, and PINCH (Legate et al. 2006). Rsu1, a 33 kDa small leucine rich repeat protein, binds to the LIM domain protein PINCH1 (Kadrmas et al. 2004; Dougherty et al. 2005), thus associating with the integrin adhesome (Horton et al. 2015) (Byron and Frame 2016). Rsu1 is required for cell spreading, adhesion and migration as well as the formation of mature focal adhesions (Simpson et al. 2008; Winograd-Katz et al. 2009; Gonzalez-Nieves et al. 2013; Horton et al. 2015). The latter occurs by stabilization of PINCH1 and the PINCH1-ILK complex (Gonzalez-Nieves et al. 2013) and the loss of Rsu1, or PINCH or ILK, causes cell detachment (Gonzalez-Nieves et al. 2013). Rsu1 links cell adhesion to intracellular signaling. siRNA-mediated depletion of Rsu1 in mammary epithelial cells elevates JNK and phosphorylation of c-Jun, but it inhibits MKK4 and p38 activation resulting in decreased ATF2 phosphorylation (Gonzalez-Nieves et al. 2013; Kim et al. 2015). Conversely, ectopic Rsu1 expression inhibits the activation of JNK in multiple cells and tissues (Masuelli and Cutler 1996; Montanez et al. 2012; Porcheri et al. 2014). The intermediary role of Rsu1 in directing signaling from the adhesome to the stress kinases also requires Rac signaling (Dougherty et al. 2008; Ito et al. 2010; Donthamsetty et al. 2013; Gonzalez-Nieves et al. 2013).
The levels of other proteins in the IPP complex, including ILK, affect JNK and p38 signaling in multiple cell types (Durbin et al. 2009) (Yu et al. 2014) (Smeeton et al. 2010). In addition, ILK is a critical regulator of AKT1 activation in breast and other cancer cells (Troussard et al. 2006), and in cells with mutant PTEN, depletion of ILK causes apoptosis (Edwards et al. 2005) (Persad et al. 2000). The loss of PINCH has also been linked to decreased AKT1 activation (Fukuda et al. 2003) (Meder et al. 2011) and PINCH expression can block AKT1 dephosphorylation (Eke et al. 2010). In contrast, our studies demonstrated that ectopic Rsu1 expression inhibited activation of AKT while decreasing the growth of MCF7 breast cancer cells (Vasaturo et al. 2000). Hence, Rsu1 and the IPP proteins have opposing roles in adhesion-dependent survival signaling.
Cell adhesion and migration are core activities modulated during tumor growth, survival and metastatic dissemination. The p38-ATF2 and JNK signaling pathways altered by Rsu1 expression are well documented contributors to oncogenic initiation and progression. Rsu1 was identified based on its inhibition of oncogenic Ras-induced anchorage independent growth (Cutler et al. 1992). Rsu1 is deleted in subsets of human hepatocellular carcinomas and gliomas (Nalesnik et al. 2012) (Wu et al. 2014). An altered Rsu1 spliced product that fails to bind PINCH1 was detected in some human gliomas and cell lines particularly in the context of Ras activation (Chunduru et al. 2002) (Dougherty et al. 2008) and ectopic Rsu1 blocked tumor formation by a glioma cell line in a xenograph model (Tsuda et al. 1995). Reduction of Rsu1 expression due to targeting by stromal derived miRs was observed in human prostate tumors and then modeled in cell lines and xenographs (Josson et al. 2014a, b). GWAS studies identified the Rsu1 locus as a predictor of response to hormone therapy in breast cancer (Onishi et al. 2018) and Rsu1 is a component of a novel four gene signature predicting therapeutic response (Quist et al. 2019). In addition, reports of elevated Rsu1 RNA in some breast tumors and the finding that the Rsu1 binding partner PINCH1 is elevated at the leading invasive edge of common solid tumors indicates that the contribution of these proteins to tumor initiation and progression is not completely understood (Gkretsi et al. 2017) (Wang-Rodriguez et al. 2002; Donthamsetty et al. 2013).
Based on the previously observed effects of Rsu1 expression on biochemical pathways, we examined interaction between Rsu1, MKK4-p38, JNK and AKT signaling pathways in non-transformed mammary epithelial cells. This approach identified Rsu1-dependent effects on PTEN expression that are regulated by MKK4-p38-ATF2 and JNK signaling. These findings link Rsu1 expression levels and cell adhesion to changes in AKT activation.
Materials and methods
Cell lines and cell culture
MCF10A mammary epithelial cells maintained in DMEM-F12 medium and MCF10A cells expressing Rsu1or dominant negative mutant Rsu1 (Rsu1-N92D) tagged with myc were described previously (Gonzalez-Nieves et al. 2013). EGF and SB203580 were purchased from Sigma-Aldrich (St. Louis, MO).
siRNA-induced gene silencing
Rsu1 depletion in MCF10A cells was described previously (Gonzalez-Nieves et al. 2013). p38α (MapK14), p38β1 (MapK11), p38γ (MapK12), MKK3 (Map2K3), MKK6 (Map2K6), MKK4 (Map2K4), MEKK1 (Map3K1) and ASK1 (Map3K5) siRNA were purchased from Qiagen (Valencia, CA). The control siRNA, Allstars negative control siRNA, was purchased from Qiagen (Valencia, CA).
Western blotting and antibodies
Western blotting assays were performed as described(Gonzalez-Nieves et al. 2013). Antibodies purchased from Cell Signaling Technology (Beverly, MA) include: anti-p-p38 (T180/Y182, #4511), p-MKK4/SEK1 (S257, #4514; S80, #9155), p-MKK3/6 (S189/S207, #9236), p-JNK (T183/Y185, #4668), p-ATF2 (T71, #5112), p-c-Jun (#2361), c-Jun (#9165 for immunoblot and ChIP), PTEN (#9188), p38α (#9218), p38β (#2339) and p38γ (#2307). The antibodies obtained from Millipore (Billerica, MA) include: anti-AKT (#05–591), p-AKT(T308, #05–802), ILK (#05–575), p38 (#05–454, reacted with p38α/β2). In addition, anti-p-AKT (S473, sc-7985), α-tubulin (sc-8035), MKK3/6 (sc-136982), p-p38 (Y182, sc-80035), p38α (sc-7985), MKK4 (sc-166168), MEKK1 (sc-252), JNK (sc-571), P-ERK (Y204, sc-7383) and ATF2 (sc-187, sc-187X) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-p-MKK4/SEK1 (T261, #GWB-ASB802) and anti-PINCH (#GWB-MX035H) were purchased from Genway. The anti-vinculin (#v4505) was from Sigma-Aldrich (St. Louis, MO). The Rsu1 antibody was described previously (Dougherty et al. 2008; Gonzalez-Nieves et al. 2013).
Western blots were repeated multiple times. Images were collected using LAS-4000 Imager. To ensure linearity of the signal, protein quantification was determined using the Multi Gauge program (Fujifilm). For quantitation, band intensity analysis was performed by determination of ratio for Rsu1/ tubulin or actin, phosphospecific/ non-phosphorylated antibodies. For fold changes, each ratio was compared by the other representative images. The control band was set as 1.
RNA isolation and quantitative real-time RT-PCR
Total RNA was isolated with TriPure (Roche, Indianapolis, IN) and cDNA synthesis was performed using the GeneAmp RNA PCR Core Kit (Applied Biosystems, Foster City, CA). Primer sequences for Real-time RT-PCR for human PTEN are:
5’-GATGTGGCGGGACTCTTTAT-3′ (forward) and 5’-AGCGGCTCAACTCTCAAACT-3′ (reverse); human Rsu1: 5’-GGATGTCAACGGCCTCTTT-3′ (forward) and 5’-TGGCACCATTGTTAGCTTGT-3′ (reverse). Amplification for real-time PCR was performed in triplicate with FastStart Universal SYBR Green Master (Roche, Indianapolis, IN) and was analyzed using the ABI 7500 (Applied Biosystems, Foster City, CA). 18S ribosomal RNA was used as an internal control (forward primer: 5’-GGATCCATTGGAGGGCAAGT-3′ and reverse primer 5’-AATATACGCTATTGGAGCTGGAATTAC-3′) to normalize the results. A complete list of primers sequences are included in Supplementary file 1.
ChIP (chromatin immunoprecipitation) assay
ChIP assays were performed using reagents from Active Motif (Carlsbad, CA) as recommended by the manufacturer. In brief, cells were cross-linked with 10% formaldehyde in cell culture medium for 10 min at room temperature then washed with ice-cold PBS and glycine stop solution to end the fixation. The cells were scraped and lysed with cold-lysis buffer. The nuclear pellet was collected, digested, and chromatin was sheared enzymatically for 15 min at 37 °C. The sheared chromatin DNA samples were centrifuged at 18,000 RCF at 4 °C for 10 min and phenol/chloroform extracted. The pre-cleared chromatin was incubated overnight at 4 °C with specific antibodies or normal rabbit IgG and protein G beads. After incubation at 4 °C overnight, the protein G beads were collected, washed and the DNA was eluted. Protein-DNA cross-links were reversed 15 min at 95 °C and the samples were treated with proteinase K for 1 h at 37 °C. The DNA samples were analyzed by PCR using AmpliTaq DNA polymerase kit (Life Technologies) with the following human PTEN promoter-specific primers. Site 1: 5’-TCGACTACTTGCTTTGTAGA-3′ (forward) and 5’-TTTACAGCCCCGATTGGGCT-3′ (reverse). Site 2: 5’-CAGACTTGACAGGTTTGTTC-3′ (forward) and 5’-TCCAGTCACTACCCCTGAGC-3′ (reverse). PCR conditions were as follows: 94 °C for 3 min; 40 cycles at 94 °C for 20 s for denaturation; 59 °C for 30 s for annealing; 72 °C for 30 s for elongation; and a final extension at 72 °C for 10 min. The PCR products were analysed on a 3% agarose gel electrophoresis in TAE buffer.
Results
The depletion of Rsu1 inhibits activation of MKK4 in response to EGF stimulation of MCF10A cells
Rsu1 contributes to the control of cell signaling and migration in MCF10A mammary epithelial cells (Gonzalez-Nieves et al. 2013) and, as shown previously, the siRNA-mediated depletion of Rsu1 in MCF10A cells inhibited EGF stimulation of both MKK4 and p38 phosphorylation (Gonzalez-Nieves et al. 2013) (Kim et al. 2015). This pathway, which also controls phosphorylation of ATF2, is critical for migration of MCF10A cells. The results reported here confirm and extend those findings. Western blot analysis of lysates from control- or Rsu1 siRNA transfected cells confirmed that Rsu1 depletion inhibited MKK4 phosphorylation in response to EGF, but not phosphorylation of MKK3 and MKK6, indicating that MKK4 is the likely immediate upstream activator of p38 in this experimental condition (Fig. 1a). The phosphorylation of the MKK4 targets, p38 and Jun kinase, in response to EGF were examined. p38α is the prominently phosphorylated isoform in response to EGF stimulation in MCF10A cells and, as reported previously, p38 phosphorylation is inhibited in the absence of Rsu1(Gonzalez-Nieves et al. 2013). In contrast, Rsu1 depletion resulted in the enhanced phosphorylation of JNK in response to EGF (Fig. 1b).

The Rsu1 is necessary for activation of p38 Mapk and MKK4 but not MKK3/6 in response to EGF. (a, b) Cell lysates were isolated from control siRNA and Rsu1 siRNA transfected MCF10A cells following EGF treatment (100 ng/ml) for the indicated time. Immunoblot analysis was performed with equal amounts of protein samples as indicated in Materials and Methods and the levels of phospho-p38 (T180/Y182), phospho-MKK4 (S257 and T261), phospho-MKK3/6 (S189/S207), phospho-JNK (T183/Y185), were detected. Total p38, JNK, MKK4 and MKK3/6 were measured and α-tubulin and Rsu1 antibodies were used for loading controls and depletion levels, respectively. c MCF10A cells stably expressing control vector (pBabe), Rsu1-myc (Rsu1) or Rsu1-N92D-myc (Rsu1-N92D) were transfected with negative control siRNA or endogenous Rsu1 specific siRNA and treated with EGF (100 ng/ml) for 10 min as described in Materials and Methods. The lysates were subjected by western blot and analyzed with phosphorylation specific antibodies indicated. d Lysates from MCF10A cells depleted of MKK4, MKK3 or MKK6 were examined for levels of phospho-p38, phospho-JNK and phospho-Erk. Quantitation is expressed as fold stimulation compared to T = 0 for control siRNA transfected cells set as 1
PINCH1 is the adaptor protein linking Rsu1 to ILK and the focal adhesions (Dougherty et al. 2008). Our previous studies demonstrated that depletion of PINCH1 in MCF10A cells did not block EGF-induced phosphorylation of p38 and ATF2 (Gonzalez-Nieves et al. 2013). Studies using a mutant of Rsu1, Rsu1-N92D, which does not bind PINCH1, indicated that the pathway exhibits additional complexity. First, in cells depleted of endogenous Rsu1, wild type Rsu1 rescued focal adhesion formation but the cells expressing Rsu1-N92D were less adherent and without fully developed and functional FAs (Gonzalez-Nieves et al. 2013). In the experiment shown here, MCF10A cell lines constructed to express wtRsu1 or the Rsu1-N92D mutant were depleted of endogenous but not the vector-encoded Rsu1 RNA. The expression of wtRsu1 restored MKK4 (S257) and p38 phosphorylation in response to EGF. However, expression of Rsu1-N92D did not support MKK4 activation but led to constitutive p38 phosphorylation (Fig. 1c).
The physical detachment of MCF10A cells from substrate activates MKK6 and phosphorylation of p38 (Wen et al. 2011). Data in Fig. 1d show that MKK6 contributes to EGF-induced p38 phosphorylation in MCF10A cells as its depletion decreases phospho-p38. Hence, cells expressing Rsu1-N92D have a profile similar to detached cells i.e. constitutive phosphorylation of p38 that is likely due to activation of MKK3 and/or MKK6 (Fig. 1c, boxed). Because, the non-binding mutant (Rsu1-N92D) did not fully rescue EGF-induced MKK4 (S257) (Fig. 1c, boxed), it appears that Rsu1-PINCH1 interaction, or stable cell adhesion, is necessary for growth factor-induced MKK4 activation and phosphorylation of p38.
JNK activation is inhibited by ectopic expression of Rsu1 and Rsu1 depletion enhances JNK activation by EGF (Fig. 1b). MKK4 can phosphorylate JNK in MCF10A and depletion of MKK4 or MKK3, but not MKK6, reduced JNK activation. Hence, MKK4 may contribute to Rsu1 effect on JNK activation by EGF (Fig. 1d).
An analysis of the potential activators of MKK4 was performed in MCF10A cells. The results of siRNA-mediated depletion of MEKK1, ASK1 and HGK indicate that reduction of MEKK1 reduces EGF induced phosphorylation of MKK4 and p38 (in Supplementary Fig. 1). Currently the effect of Rsu1 level on MEKK1 activation or stability is not known.
AKT activation correlates with Rsu1 expression and contributes to MKK4 function
Because Rsu1 functions through MKK4 and p38 we examined regulatory events involving MKK4 activity. PI3K/AKT signaling pathways regulate cellular functions including cell migration, proliferation and survival through multiple downstream targets, including the inhibition of MKK4. This includes AKT phosphorylation of MKK4 on serine 80, a negative regulatory site for MKK4 activity (Park et al. 2002; Song and Lee 2005). A previously identified link between Rsu1 expression and AKT was observed when ectopic expression of Rsu1 in the MCF7 breast tumor cell line inhibited AKT phosphorylation (Vasaturo et al. 2000). In the current study AKT was examined in cells in which levels of Rsu1 were elevated or reduced. MCF10A and human breast cancer cell lines T47D and MDA-MB-231 were infected with adenovirus encoding Rsu1; these cells exhibited decreased phospho-AKT (ser473) similar to that observed in the MCF7 cells (Fig. 2a). Conversely, the depletion of Rsu1 from MCF10A cells resulted in elevated phospho-AKT primarily at serine 473 whereas phosphorylation at threonine 308 was less affected by Rsu1 level (Fig. 2b). These findings suggested a role for Rsu1 in the regulation of the AKT phosphorylation. In MCF10A cells the depletion of Rsu1 and increase in AKT (ser473) phosphorylation correlated with phosphorylation of MKK4 on serine 80, the negative regulatory site for MKK4 activity (Fig. 2b). Hence, these data suggest that Rsu1 depletion both blocks the activation of MKK4 and inhibits MKK4 kinase activity by serine 80 phosphorylation.

Rsu1-dependent changes in AKT signaling pathway. a MCF10A, T47D and MDA-MB-231 cells were infected with adenoviral vector encoding HA-tagged Rsu1 or empty vector. At 72 h post infection cell lysates were prepared and analyzed by western blot with phosphorylation specific antibodies, phospho-AKT (T308) and phospho-AKT (S473). Antibodies directed against total AKT, ILK, Rac1 and Rsu1were used to determine expression levels of those proteins. Quantitation shows the level of phosphorylated AKT normalized to total AKT and the level set in the control lane = 1. b MCF10A lysates were isolated from control siRNA and Rsu1 siRNA transfected MCF10A cells after EGF treatment for the indicated times. Immunoblot analysis was performed with equal amounts of protein samples and separated by SDS-PAGE for the levels of phospho-MKK4 (S80), phospho-AKT (p S473), total MKK4, AKT and PTEN. α-tubulin and Rsu1 antibodies were used for internal and depletion controls. c MCF10A RNA from control siRNA and Rsu1 siRNA transfected cells was used to determination level of PTEN RNA by quantitative real-time PCR. Quantitation is expressed as fold stimulation or relative amount compared to T = 0 for control siRNA transfected cells set as 1
Next, experiments were performed to determine the cause of elevated phospho-AKT in MCF10A cells depleted of Rsu1. The tumor suppressor and lipid phosphatase, PTEN, reduces levels of PIP3 thus attenuating PI3K/AKT signaling and the phosphorylation of AKT. Because PI3K/AKT signaling is tightly regulated by PTEN and PTEN controls migration (Tamura et al. 1998), we investigated the effect of Rsu1 on PTEN expression. The siRNA-mediated depletion of Rsu1 decreased both PTEN RNA and protein expression (Fig. 2b and c). Because this occurs in conjunction with increased phosphorylation of AKT on serine 473 in MCF10A cells, it appears that Rsu1 depletion results in a decrease in PTEN, elevation of phospho-AKT, and increased phosphorylation of MKK4 at serine 80, a negative regulatory site targeted by AKT. Hence, Rsu1 depletion interferes with MKK4 activation at multiple steps.
PTEN is a frequently altered gene in human cancers and it can be regulated in multiple ways including by phosphorylation (Ser380/Thr382/Thr383). We examined levels of phosphorylated and non-phosphorylated PTEN in cells depleted of Rsu1 following EGF stimulation. The results in Supplementary Fig. 2 indicate that there is no significant difference in PTEN phosphorylation between control and Rsu1 depleted cells. While PTEN can modulate cell migration in some cell backgrounds, only the complete deletion of PTEN altered the growth phenotype of MCF10A cells (Vitolo et al. 2009). Hence, we did not expect changes in adhesion due to Rsu1-induced changes in PTEN level and siRNA mediated reduction in PTEN in MCF10A cells did not alter adhesion (data not shown).
The contribution of MKKs to PTEN expression was tested by siRNA-mediated depletion of these kinases in MCF10A cells; the results indicated that MKK3 and MKK4, but not MKK6, contributed to control of PTEN expression (Fig. 3a). The specific inhibitor of p38 (SB203580) decreased expression of PTEN in MCF10A cells in a dose-dependent way confirming that reduced p38 phosphorylation correlated with the reduced level of PTEN RNA in MCF10A cells (Fig. 3b and c). These data link a decrease in a MKK4-p38 signaling with reduced PTEN transcription in MCF10A cells.

PTEN gene regulation by MKK4-p38 Mapk signaling and Rsu1-dependent expression of PTEN in MCF10A cells. a Determination of PTEN RNA levels was measured by quantitative real-time PCR following transfection with siRNAs for the following: Rsu1, p38α, MKK3, MKK4 and MKK6. The 18S RNA was used for normalization. Bar graphs show the fold change of PTEN expression quantified as the ratios to the control siRNA level. The data show means ± SD, n = 3. b RNA levels of PTEN were measured by quantitative real-time PCR following SB 203580 treatment of MCF10A cells (0, 5 or 20 μM). The 18S RNA was used for normalization. Bar graphs show the fold change of PTEN expression quantified as the ratios to the control, the means ± SD, n = 3. c Immunoblot analysis was performed with equal amounts of protein samples for the levels of phospho-AKT (S473) and phospho-p38 (T180/Y182), total AKT, p38, PTEN expression and α-tubulin was for internal controls
Signal transduction modifications in Rsu1-depleted cells alter cJun and ATF2 binding to the PTEN promoter
The PTEN gene is transcriptionally regulated by the MKK4, p38 Mapk and Jun kinase signaling pathway via the binding of the transcription factor(s) ATF2 and cJun to two putative AP-1 sites in its promoter region (Shen et al. 2006) (Qian et al. 2012). Rsu1 depletion reduces MKK4, p38 and ATF2 activation while elevating Jun kinase activation. MCF10A cells depleted of Rsu1 exhibit a decrease in ATF2 phosphorylation at threonine 71 (Fig. 4d) and a slight increase in c-Jun phosphorylation at serine 63 following EGF stimulation when compared to control cells.

ATF2 and cJun bind on the ATF2 site of the PTEN promoter. a Schematic representation of putative two ATF2 binding sites on the PTEN promoter. b Chromatin immunoprecipitation (ChIP) assay was performed for occupancy of the binding factors in the two ATF2 sites in the PTEN promoter. Purified chromatin DNA from either control siRNA (C) or Rsu1-specific siRNA (R) MCF10A cells was immunoprecipitated with anti-ATF2 (Top) and anti-cJun (Bottom) antibodies for binding to the promoter of PTEN. The immunoprecipitated complexes were amplified by PCR using the primer sets (either site 1 or site 2) as described in Materials and Methods. The PCR products were analyzed on the agarose gel with duplicated samples (C; control siRNA and R; Rsu1 siRNA transfected MCF10A cells). Input is shown as positive control and immunoprecipitation with anti-normal rabbit IgG antibodies was used as negative control (Negative control). c ChIP experiment was performed with EGF stimulated MCF10A cells. Cells were maintained in media without EGF for 16 h then stimulated for 30 min. Real-time PCR analysis of chromatin bounded ATF2 (left) or cJun (right) were measured by quantitative PCR methods with either control siRNA (control) or Rsu1-specific siRNA (Rsu1) MCF10A cells with or without EGF stimulation. d Immunoblot analysis was performed using cell lysates from control siRNA (control) and Rsu1 specific siRNA (Rsu1) MCF10A cells for the levels of phospho-ATF2 (p-ATF2, T71), phospho-cJun (p-cJun, S63), total ATF2 and cJun. α-tubulin and Rsu1 antibodies were used for internal controls and depletion levels, respectively
Hence, we examined the effect of Rsu1 level on ATF2-dependent and cJun-dependent PTEN gene regulation in MCF10A cells maintained in EGF-containing media. Using ChIP assay, we tested the binding of ATF2 and cJun to potential PTEN promoter binding sites essential for PTEN gene regulation (Fig. 4a). As shown in Fig. 4b, the binding of ATF2 to site 2 in PTEN gene promoter was regulated by Rsu1 level, but no alteration in ATF2 binding was detected at PTEN promoter site 1. Moreover, while ATF2 binding to site 2 was decreased in Rsu1-siRNA transfected MCF10A cells, c-Jun binding was reciprocally increased at site 2. These data indicate that ATF2 positive regulation of PTEN gene expression in MCF10A cells depends on Rsu1 level. In addition, in the absence of Rsu1, a condition that leads to elevated Jun kinase and phospho-cJun, negative regulation of PTEN promoter activation also contributes to decreased PTEN transcription (Fig. 4b). This is predicted by previous work demonstrating negative regulation of PTEN expression by cJun (Hettinger et al. 2007).
We next investigated whether the binding of ATF2 at site 2 is regulated by EGF stimulation. As demonstrated in Fig. 4c, the binding of ATF2 was enhanced by EGF treatment in the control, but not in the absence of Rsu1, indicating that ATF2 plays an important role for the PTEN gene regulation by EGF. EGF stimulation had less effect on cJun binding to site 2. This is consistent with the reduction in expression of PTEN RNA as determined by quantitative real time PCR experiment (Fig. 2c). Based on the data it appears that the inhibition of the p38-ATF2 pathway by Rsu1 depletion is responsible for the decrease in ATF2 binding to site 2 in the PTEN promoter and reduction in PTEN expression.
Discussion
Rsu1 and the proteins in the IPP complex control adhesion and migration in part through alterations in signal transduction pathways. The pathways that are modulated are those that contribute to membrane alterations during integrin clustering as well as stress and survival signaling. Our previous work identified a role for Rsu1 in the activation of MKK4-p38 signaling during EGF-induced stimulation and migration of MCF10A cells. The results here confirm and expand the findings to links with the AKT1 pathway, changes in which were previously observed following ectopic Rsu1 expression (Vasaturo et al. 2000). The lipid phosphatase PTEN controls levels of activated AKT1 and its expression can be modulated by ATF2 and cJun. ATF2 activation and binding to a site in the PTEN promotor is necessary for PTEN transcription (Shen et al. 2006; Qian et al. 2012). Our data demonstrate that MKK4-p38-ATF2 signaling is disrupted following Rsu1-depletion. In addition, Rsu1-depletion causes elevation of JNK and phospho-cJun, which binds at negative regulatory site in the PTEN promoter blocking transcription (Hettinger et al. 2007) (Fig. 5). Hence, the reduction of Rsu1 caused decreased levels of PTEN and elevated phospho-AKT, while the converse is seen in cells with elevated Rsu1 (Vasaturo et al. 2000).

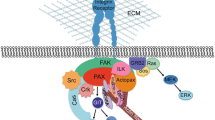
Schematic model of the regulation of PTEN promoter in MCF10A cells by Rsu1 and IPP complex. Rsu1 plays a role in the regulation of PTEN gene transcription via MKK4-p38-ATF2 and MKK4-JNK-cJun. p38 MAPK can be constitutively activated by detachment or by blocking Rsu1-PINCH1 interaction and this is mediated by MKK3/6 signaling. However, EGF-induced p38 MAPK phosphorylation is dependent on Rsu1 and occurs through MKK4 and possibly MEKK1. In MCF10A cells, PTEN gene transcription is regulated through the ATF2/AP1 by the binding of AFT2 and cJun
Reciprocal changes in ATF2 and cJun function have been reported in other contexts. Expression of an N-terminal peptide of ATF2 reduced ATF2-dependent transcriptional activity, while increasing expression and the activity of cJun in melanoma (Bhoumik et al. 2002; Bhoumik et al. 2004). Likewise, inhibiting ATF2 via RNAi increased cJun expression and stimulated melanoma cells to apoptosis, suggesting the change in the balance of ATF2/cJun allowing replacement of other cJun heterodimer partners (Huang et al. 2008).
JNK activity has been identified as a contributor to tumor progression but depending on the cell context it can block tumor formation as well. In the study by Hettinger et al. cJun negatively regulated PTEN expression by binding to the AP1 site 2 and promoted tumor cell survival (Hettinger et al. 2007). In prostate, a more complex role for PTEN and JNK is illustrated by the finding that PTEN inactivation is associated with elevated JNK which may block progression by contributing to senescence (Vivanco et al. 2007; Hubner et al. 2012). While AKT1 can inhibit MEKK1-MKK4-JNK via phosphorylation in the N terminal region of MKK4 (Park et al. 2002), MEKK1 loss enhances PI3K signaling via stabilization of IRS1 and IRS1 binding to p85 subunit of PI3K (Avivar-Valderas et al. 2018). The loss of Rsu1 increases JNK activity and elevated Rsu1 levels block Jun kinase activation in multiple cells and tissues (Masuelli and Cutler 1996) (Montanez et al. 2012; Porcheri et al. 2014). Hence, while Rsu1 loss causes changes in adhesion and migration there is also potential for loss or mutation of Rsu1 to contribute to cancer development or progression via decreased PTEN signaling or altered JNK activity.
Sequencing breast tumor DNA revealed that luminal A breast tumors exhibit sets of mutually exclusive mutations; these include activating mutations of PIK3CA, PIK3R1, loss or inactivation of PTEN, or inactivating mutations of Map3K1/MEKK1 and Map2K4/MKK4 that block activation of p38 and JNK (Ellis et al. 2012; TCGA 2012). ATF2, a target of p38 kinase, can suppress tumorigenesis (Bhoumik et al. 2008; Gozdecka et al. 2014). In breast cancer, analysis of the level of phosphorylation and activation of ATF2 revealed that high phospho-ATF2 was linked to both tamoxifen sensitivity of luminal A tumors and survival while low phospho-ATF2 was associated with recurrence and failure of tamoxifen therapy (Rudraraju et al. 2014). Hence, the MKK4-p38-ATF2 signaling pathway may be critical for response to hormone therapy in luminal A breast tumors. A recent GWAS studies identified the Rsu1 locus as a predictor of response to hormone therapy in breast cancer (Onishi et al. 2018). Also, tumor cells with low levels of p38 and phospho-ATF2 are associated with metastasis in HER2+ disease (Harper et al. 2016)
RSU1 deletion has been identified in glioma and hepatocellular carcinoma (Nalesnik et al. 2012; Wu et al. 2014) and a subset of genes whose expression is altered by Rsu1 depletion can be seen in Table 1. Multiple RSU1 transcripts resulting from alternative splicing are detected in human tumors, including some forms that are specific to tumors with Ras mutations, and some of these RNAs encode proteins that fail to bind to PINCH1 (Chunduru et al. 2002; Dougherty et al. 2008). So while there are reports of RSU1 elevation in breast and other tumors, further analysis, including links to JNK, p38 and ATF2, or to the expression, mutation or deletion of PTEN, MEKK1, MKK4, PIK3 and PIK3R1, will be necessary to understand the contribution of RSU1 to tumor development or progression.
References
Avivar-Valderas A, McEwen R, Taheri-Ghahfarokhi A, Carnevalli LS, Hardaker EL, Maresca M, Hudson K, Harrington EA, Cruzalegui F (2018) Functional significance of co-occurring mutations in PIK3CA and MAP3K1 in breast cancer. Oncotarget 9:21444–21458
Bhoumik A, Huang TG, Ivanov V, Gangi L, Qiao RF, Woo SL, Chen SH, Ronai Z (2002) An ATF2-derived peptide sensitizes melanomas to apoptosis and inhibits their growth and metastasis. J Clin Invest 110:643–650
Bhoumik A, Jones N, Ronai Z (2004) Transcriptional switch by activating transcription factor 2-derived peptide sensitizes melanoma cells to apoptosis and inhibits their tumorigenicity. Proc Natl Acad Sci U S A 101:4222–4227
Bhoumik A, Fichtman B, Derossi C, Breitwieser W, Kluger HM, Davis S, Subtil A, Meltzer P, Krajewski S, Jones N, Ronai Z (2008) Suppressor role of activating transcription factor 2 (ATF2) in skin cancer. Proc Natl Acad Sci U S A 105:1674–1679
Byron A, Frame MC (2016) Adhesion protein networks reveal functions proximal and distal to cell-matrix contacts. Curr Opin Cell Biol 39:93–100
Chunduru S, Kawami H, Gullick R, Monacci WJ, Dougherty G, Cutler ML (2002) Identification of an alternatively spliced RNA for the Ras suppressor RSU-1 in human gliomas. J Neuro-Oncol 60:201–211
Cutler ML, Bassin RH, Zanoni L, Talbot N (1992) Isolation of rsp-1, a novel cDNA capable of suppressing v-Ras transformation. Mol Cell Biol 12:3750–3756
Donthamsetty S, Bhave VS, Mars WM, Bowen WC, Orr A, Haynes MM, Wu C, Michalopoulos GK (2013) Role of PINCH and its partner tumor suppressor Rsu-1 in regulating liver size and tumorigenesis. PLoS One 8:e74625
Dougherty GW, Chopp T, Qi SM, Cutler ML (2005) The Ras suppressor Rsu-1 binds to the LIM 5 domain of the adaptor protein PINCH1 and participates in adhesion-related functions. Exp Cell Res 306:168–179
Dougherty GW, Jose C, Gimona M, Cutler ML (2008) The Rsu-1-PINCH1-ILK complex is regulated by Ras activation in tumor cells. Eur J Cell Biol 87:721–734
Durbin AD, Somers GR, Forrester M, Pienkowska M, Hannigan GE, Malkin D (2009) JNK1 determines the oncogenic or tumor-suppressive activity of the integrin-linked kinase in human rhabdomyosarcoma. J Clin Invest 119:1558–1570
Edwards LA, Thiessen B, Dragowska WH, Daynard T, Bally MB, Dedhar S (2005) Inhibition of ILK in PTEN-mutant human glioblastomas inhibits PKB/Akt activation, induces apoptosis, and delays tumor growth. Oncogene 24:3596–3605
Eke I, Koch U, Hehlgans S, Sandfort V, Stanchi F, Zips D, Baumann M, Shevchenko A, Pilarsky C, Haase M, Baretton GB, Calleja V, Larijani B, Fassler R, Cordes N (2010) PINCH1 regulates Akt1 activation and enhances radioresistance by inhibiting PP1alpha. J Clin Invest 120:2516–2527
Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, Crowder R, Snider J, Ballman K, Weber J, Chen K, Koboldt DC, Kandoth C, Schierding WS, McMichael JF, Miller CA, Lu C, Harris CC, McLellan MD, Wendl MC, DeSchryver K, Allred DC, Esserman L, Unzeitig G, Margenthaler J, Babiera GV, Marcom PK, Guenther JM, Leitch M, Hunt K, Olson J, Tao Y, Maher CA, Fulton LL, Fulton RS, Harrison M, Oberkfell B, Du F, Demeter R, Vickery TL, Elhammali A, Piwnica-Worms H, McDonald S, Watson M, Dooling DJ, Ota D, Chang LW, Bose R, Ley TJ, Piwnica-Worms D, Stuart JM, Wilson RK, Mardis ER (2012) Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486:353–360
Fukuda T, Chen K, Shi X, Wu C (2003) PINCH-1 is an obligate partner of integrin-linked kinase (ILK) functioning in cell shape modulation, motility, and survival. J Biol Chem 278:51324–51333
Gkretsi V, Stylianou A, Louca M, Stylianopoulos T (2017) Identification of Ras suppressor-1 (RSU-1) as a potential breast cancer metastasis biomarker using a three-dimensional in vitro approach. Oncotarget 8:27364–27379
Gonzalez-Nieves R, Desantis AI, Cutler ML (2013) Rsu1 contributes to regulation of cell adhesion and spreading by PINCH1-dependent and - independent mechanisms. J Cell Commun Signal 7:279–293
Gozdecka M, Lyons S, Kondo S, Taylor J, Li Y, Walczynski J, Thiel G, Breitwieser W, Jones N (2014) JNK suppresses tumor formation via a gene-expression program mediated by ATF2. Cell Rep 9:1361–1374
Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, Avivar-Valderas A, Nagi C, Girnius N, Davis RJ, Farias EF, Condeelis J, Klein CA, Aguirre-Ghiso JA (2016) Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature 540:588–592
Hettinger K, Vikhanskaya F, Poh MK, Lee MK, de Belle I, Zhang JT, Reddy SA, Sabapathy K (2007) C-Jun promotes cellular survival by suppression of PTEN. Cell Death Differ 14:218–229
Horton ER, Byron A, Askari JA, Ng DH, Millon-Fremillon A, Robertson J, Koper EJ, Paul NR, Warwood S, Knight D, Humphries JD, Humphries MJ (2015) Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol 17:1577–1587
Huang Y, Minigh J, Miles S, Niles RM (2008) Retinoic acid decreases ATF-2 phosphorylation and sensitizes melanoma cells to taxol-mediated growth inhibition. J Mol Signal 3:3
Hubner A, Mulholland DJ, Standen CL, Karasarides M, Cavanagh-Kyros J, Barrett T, Chi H, Greiner DL, Tournier C, Sawyers CL, Flavell RA, Wu H, Davis RJ (2012) JNK and PTEN cooperatively control the development of invasive adenocarcinoma of the prostate. Proc Natl Acad Sci U S A 109:12046–12051
Ito S, Takahara Y, Hyodo T, Hasegawa H, Asano E, Hamaguchi M, Senga T (2010) The roles of two distinct regions of PINCH-1 in the regulation of cell attachment and spreading. Mol Biol Cell 21:4120–4129
Josson S, Gururajan M, Hu P, Shao C, Chu GC, Zhau HE, Liu C, Lao K, Lu CL, Lu YT, Lichterman J, Nandana S, Li Q, Rogatko A, Berel D, Posadas EM, Fazli L, Sareen D, Chung LW (2014a) miR-409-3p/−5p promotes tumorigenesis, epithelial-to-mesenchymal transition, and bone metastasis of human prostate Cancer. Clin Cancer Res 20:4636–4646
Josson S, Gururajan M, Sung SY, Hu P, Shao C, Zhau HE, Liu C, Lichterman J, Duan P, Li Q, Rogatko A, Posadas EM, Haga CL and Chung LW (2014b) Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 34:2690–2699
Kadrmas JL, Smith MA, Clark KA, Pronovost SM, Muster N, Yates JR 3rd, Beckerle MC (2004) The integrin effector PINCH regulates JNK activity and epithelial migration in concert with Ras suppressor 1. J Cell Biol 167:1019–1024
Kim YC, Gonzalez-Nieves R, Cutler ML (2015) Rsu1 contributes to cell adhesion and spreading in MCF10A cells via effects on P38 map kinase signaling. Cell Adhes Migr 9:227–232
Legate KR, Montanez E, Kudlacek O, Fassler R (2006) ILK, PINCH and parvin: the tIPP of integrin signalling. Nat Rev Mol Cell Biol 7:20–31
Masuelli L, Cutler ML (1996) Increased expression of the Ras suppressor Rsu-1 enhances Erk-2 activation and inhibits Jun kinase activation. Mol Cell Biol 16:5466–5476
Meder B, Huttner IG, Sedaghat-Hamedani F, Just S, Dahme T, Frese KS, Vogel B, Kohler D, Kloos W, Rudloff J, Marquart S, Katus HA, Rottbauer W (2011) PINCH proteins regulate cardiac contractility by modulating integrin-linked kinase-protein kinase B signaling. Mol Cell Biol 31:3424–3435
Montanez E, Karakose E, Tischner D, Villunger A, Fassler R (2012) PINCH-1 promotes Bcl-2-dependent survival signalling and inhibits JNK-mediated apoptosis in the primitive endoderm. J Cell Sci 125:5233–5240
Nalesnik MA, Tseng G, Ding Y, Xiang GS, Zheng ZL, Yu Y, Marsh JW, Michalopoulos GK, Luo JH (2012) Gene deletions and amplifications in human hepatocellular carcinomas: correlation with hepatocyte growth regulation. Am J Pathol 180:1495–1508
Onishi H, Udagawa C, Kubo M, Nakamura S, Akashi-Tanaka S, Kuwayama T, Watanabe C, Takamaru T, Takei H, Ishikawa T, Miyahara K, Matsumoto H, Hasegawa Y, Momozawa Y, Low SK, Kutomi G, Shima H, Satomi F, Okazaki M, Zaha H, Onomura M, Matsukata A, Sagara Y, Baba S, Yamada A, Shimada K, Shimizu D, Tsugawa K, Shimo A, Hartman M, Chan CW, Lee SC, Endo I, Zembutsu H (2018) A genome-wide association study identifies three novel genetic markers for response to tamoxifen: a prospective multicenter study. PLoS One 13:e0201606
Park HS, Kim MS, Huh SH, Park J, Chung J, Kang SS, Choi EJ (2002) Akt (protein kinase B) negatively regulates SEK1 by means of protein phosphorylation. J Biol Chem 277:2573–2578
Persad S, Attwell S, Gray V, Delcommenne M, Troussard A, Sanghera J, Dedhar S (2000) Inhibition of integrin-linked kinase (ILK) suppresses activation of protein kinase B/Akt and induces cell cycle arrest and apoptosis of PTEN-mutant prostate cancer cells. Proc Natl Acad Sci U S A 97:3207–3212
Porcheri C, Suter U, Jessberger S (2014) Dissecting integrin-dependent regulation of neural stem cell proliferation in the adult brain. J Neurosci 34:5222–5232
Qian J, Ling S, Castillo AC, Long B, Birnbaum Y, Ye Y (2012) Regulation of phosphatase and tensin homolog on chromosome 10 in response to hypoxia. Am J Physiol Heart Circ Physiol 302:H1806–H1817
Quist J, Mirza H, Cheang MCU, Telli ML, O'Shaughnessy J, Lord CJ, Tutt ANJ and Grigoriadis A (2019) A four-gene decision tree signature classification of triple-negative breast cancer: implications for targeted therapeutics. Mol Cancer Ther 18:204–212
Rudraraju B, Droog M, Abdel-Fatah TM, Zwart W, Giannoudis A, Malki MI, Moore D, Patel H, Shaw J, Ellis IO, Chan S, Brooke GN, Nevedomskaya E, Lo Nigro C, Carroll J, Coombes RC, Bevan C, Ali S, Palmieri C (2014) Phosphorylation of activating transcription factor-2 (ATF-2) within the activation domain is a key determinant of sensitivity to tamoxifen in breast cancer. Breast Cancer Res Treat 147:295–309
Shen YH, Zhang L, Gan Y, Wang X, Wang J, LeMaire SA, Coselli JS, Wang XL (2006) Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling. A cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J Biol Chem 281:7727–7736
Simpson KJ, Selfors LM, Bui J, Reynolds A, Leake D, Khvorova A, Brugge JS (2008) Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat Cell Biol 10:1027–1038
Smeeton J, Zhang X, Bulus N, Mernaugh G, Lange A, Karner CM, Carroll TJ, Fassler R, Pozzi A, Rosenblum ND, Zent R (2010) Integrin-linked kinase regulates p38 MAPK-dependent cell cycle arrest in ureteric bud development. Development 137:3233–3243
Song JJ, Lee YJ (2005) Dissociation of Akt1 from its negative regulator JIP1 is mediated through the ASK1-MEK-JNK signal transduction pathway during metabolic oxidative stress: a negative feedback loop. J Cell Biol 170:61–72
Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM (1998) Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280:1614–1617
TCGA (2012) Comprehensive molecular portraits of human breast tumours. Nature 490:61–70
Troussard AA, McDonald PC, Wederell ED, Mawji NM, Filipenko NR, Gelmon KA, Kucab JE, Dunn SE, Emerman JT, Bally MB, Dedhar S (2006) Preferential dependence of breast cancer cells versus normal cells on integrin-linked kinase for protein kinase B/Akt activation and cell survival. Cancer Res 66:393–403
Tsuda T, Marinetti MR, Masuelli L, Cutler ML (1995) The Ras suppressor RSU-1 localizes to 10p13 and its expression in the U251 glioblastoma cell line correlates with a decrease in growth rate and tumorigenic potential. Oncogene 11:397–403
Vasaturo F, Dougherty GW, Cutler ML (2000) Ectopic expression of Rsu-1 results in elevation of p21CIP and inhibits anchorage-independent growth of MCF7 breast cancer cells. Breast Cancer Res Treat 61:69–78
Vitolo MI, Weiss MB, Szmacinski M, Tahir K, Waldman T, Park BH, Martin SS, Weber DJ, Bachman KE (2009) Deletion of PTEN promotes tumorigenic signaling, resistance to anoikis, and altered response to chemotherapeutic agents in human mammary epithelial cells. Cancer Res 69:8275–8283
Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ, Jiao J, Rose J, Xie W, Loda M, Golub T, Mellinghoff IK, Davis RJ, Wu H, Sawyers CL (2007) Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell 11:555–569
Wang-Rodriguez J, Dreilinger AD, Alsharabi GM, Rearden A (2002) The signaling adapter protein PINCH is up-regulated in the stroma of common cancers, notably at invasive edges. Cancer 95:1387–1395
Wen HC, Avivar-Valderas A, Sosa MS, Girnius N, Farias EF, Davis RJ, Aguirre-Ghiso JA (2011) p38alpha signaling induces anoikis and lumen formation during mammary morphogenesis. Sci Signal 4:ra34
Winograd-Katz SE, Itzkovitz S, Kam Z, Geiger B (2009) Multiparametric analysis of focal adhesion formation by RNAi-mediated gene knockdown. J Cell Biol 186:423–436
Wu HT, Hajirasouliha I, Raphael BJ (2014) Detecting independent and recurrent copy number aberrations using interval graphs. Bioinformatics 30:i195–i203
Yu L, Yuan X, Wang D, Barakat B, Williams ED, Hannigan GE (2014) Selective regulation of p38beta protein and signaling by integrin-linked kinase mediates bladder cancer cell migration. Oncogene 33:690–701
Funding
The following funding agencies provided support: the Murtha Cancer Center at Walter Reed National Military Medical Center through Uniformed Services University under the auspices of the Henry M. Jackson Foundation for the Advancement of Military Medicine (to MLC) and W81XWH-10-1-0024 from the Congressionally Directed Medical Research Breast Cancer Program (pre-doctoral fellowship to RG-N).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclaimer
The opinions expressed here are those of the authors and should not be construed as official policy or reflecting the views of the Uniformed Services University of the Health Sciences or the Department of the Navy, Army, or the Department of Defense. In addition, any opinion, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation or the US Government.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Kim, YC., Gonzalez-Nieves, R. & Cutler, M.L. Rsu1-dependent control of PTEN expression is regulated via ATF2 and cJun. J. Cell Commun. Signal. 13, 331–341 (2019). https://doi.org/10.1007/s12079-018-00504-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-018-00504-4