
Abstract
Sugar beet (Beta vulgaris L.) meets the 21% of world sugar production. Soil pollution, biotic and abiotic factors in production areas greatly reduce product quantity and quality. Sugar beet responds to biotic and abiotic stresses such as drought, salt, heat, light, and infections of nematode, bacteria and fungi at the molecular level. Understanding molecular mechanisms require comprehensive genomics studies in order to control these mechanisms to increase the yield and quality. Transcriptome studies performed under stress conditions can shed light on the responses of plants at the molecular level. In addition, meta-analysis can help to find common responses under different stress conditions. In this study four different stress-related transcriptome data were used: two of them are related with biotic stress (nematode and fungi infection) and two of them are related with abiotic stress (ABA treatment and salt stress). In this study, we performed meta-analysis of studies conducted under biotic and abiotic stress conditions. Our results revealed 460 commonly regulated genes from biotic stress related data and 1031 commonly regulated genes from abiotic stress related data. Our data also showed that expression of ten genes is controlled regardless of the type of stress condition. The data can be useful for understanding the molecular aspect of adaptive stress response in sugar beet.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Environmental and anthropogenic stressors are one of the main causes of stress for organisms. Biotic and abiotic stress in plants can negatively affect the organisms (Ku et al. 2018). While biotic stress is related with the infection of plants by pathogens (viruses, nematodes, bacteria, and fungi) or infestation by herbivorous parasites, abiotic stress is usually related with the exposure of drought, heat, cold, nutrient misbalance, and excess salt or toxic metals. These stress conditions adversely affect plants and reduce crop yields by altering their physiology, phenotype, and biochemical and molecular structures. Through evolution, plants developed complex defense mechanisms to respond to environmental stresses.
Beta vulgaris L. is the most important sugar crop in the temperate zone and accounts for almost 21% of world sugar production (World Sugar Balance - November 2021 | International Sugar Organization, n.d.). It belongs to the order Caryophyllales. Beta vulgaris subspecies include leafy beets (chard), garden beet (red, white, yellow, or golden), fodder beet (fodder), and sugar beet. In addition to being a food source, sugar beet has been used as animal feed, and it has been a promising source for production of biofuels, alcohol, pharmaceuticals, and baker’s yeast.
Majority of the studies regarding with impact of stress on Beta vulgaris has been focused on responses to salt and drought. Sugar beet considered as a moderately tolerant species among several other crop species. Beta vulgaris has the capacity to tolerate NaCl ions that has minor effects on the yield. Some of the key traits that contribute to salt tolerance include leaf relative water content, leaf area index, biomass accumulation, and photosynthesis (Shams and Khadivi 2023). Under salt stress, the genes involved in signal transduction, phosphorylation, and redox balance play an important role (Lv et al. 2019; Rasouli et al. 2020). In addition, expression of the genes associated with the ROS scavenging system are significantly differentiated along with photosynthesis-related genes(Lv et al. 2019). Response to biotic stress has been poorly studied in Beta vulgaris. In summary, there are studies covering the biotic and abiotic stress related genetic mechanisms on sugar beet. The data provided in previous studies can be a source for the search of insight on adaptive stress response in sugar beet through meta-analysis (Stracke et al. 2014; Zou et al. 2020a, b; Liu et al. 2020b; Xing et al. 2020; Ghaemi et al. 2020; Holmquist et al. 2021; Ibrahim et al. 2021). Meta-analysis is a set of quantitative methodological tools that combine the results of similar but independent studies to arrive at an overall conclusion and to evaluate the similarities between the studies. It is a promising approach to avoid controversial aspects of the study and for creating a qualified data summary (Nakagawa and Santos 2012).
A better understanding of an organism’s response to stress conditions requires using molecular techniques and bioinformatics approaches. Development of new molecular strategies to reduce the negative effects of biotic and abiotic stress would benefit from these approaches (Zhang et al. 2016; Yu et al. 2020). Transcriptome profiling is a powerful tool for discovering the stress response genes. In previous studies of Beta vulgaris, individual transcriptome profiles were analyzed to discover stress-related genes. There are novel transcriptome studies to enhance our understanding on improving stress tolerance levels in sugar beet (Zou et al. 2020a; Liu et al. 2020b). In this study, we aimed to find the common stress response genes that are differentially expressed under different biotic and abiotic stress conditions using meta-analysis approach.
Materials and Methods
Study Data
Eleven stress-related data of Beta vulgaris were selected from the NCBI database on March 30th, 2021, with the following filters: Source: “RNA” and Platform: “Illumina”. Related data downloaded by SRA archive and compressed data was converted to “fastqc” format by SRA Toolkit (v. 2.11.0) fasterq-dump tool (Leinonen et al. 2011) (Table 1). The.
The quality-control of data was by FastQC (v. 0.11.9) (Andrews 2010). Cleaned raw data was mapped to RefBeet-1.2.2 reference genome from Ensembl plants database using Hisat2 (v. 2.1.0). (Kim et al. 2019). The data with less than 60% of alignment rate was eliminated from the study. FeatureCounts (v. 2.0.0) (Liao et al. 2014) was used to obtain gene count numbers. The required GTF file was downloaded from Ensembl plants (Ref-Beet-1.2.2) (Dohm et al. 2012).
Differential Gene Expression Analysis
For differentially expressed gene analysis DESeq2 (v. 1.30.1) package for R version 4.0.5 (Love et al. 2014) was used. Studies were compared to their control groups and the p-adjusted value was set to 0.05. Raw counts were normalized by the negative binomial model. To work with better efficiency, the Log2 transformation was applied using R. The same process was repeated for all data. Ensembl IDs for differentially expressed genes of Beta vulgaris were converted to GO and KEGG ENZYME IDs through biomaRt (v. 2.46.3) R package (Durinck et al. 2005) based on the datasets parsed from the Ensemble plants database.
Normalization Process
Eleven stress-related data of Beta vulgaris were used for this study (Table 1). The reference genome for mapping the raw RNA-Seq data was downloaded from the Ensemble plant database (RefBeet-1.2.2.). Sixty-percent alignment rate cut-off threshold was assigned to use the raw data for meta-analysis. According to the mapping results, four of the datasets were selected with an alignment rate above the threshold. These are studies based on effects of fungal pathogens (SRP095578) (Holmquist et al. 2021), nematodes (SRP217806) (Ghaemi et al. 2020), salinity stress (SRP149098) (Liu et al. 2020b), and ABA treatment (SRP235645) (Xing et al. 2020). Expression quantifications were carried out for each dataset for the analysis of differentially expressed genes (DEG) using R package DESeq2. Individual DEG analysis results obtained for each dataset were used for meta-analysis.
Meta-Analysis of Transcriptome data
Differentially expressed genes (DEGs) data was collected for meta-analysis. For the meta-analysis, R package metaRNASeq version 1.0.7 (Marot et al. 2015) was used. The results of the meta-analysis converted to GO and KEGG ENZYME IDs. For Ensembl ID to ENTREZ ID conversion, the NCBI E-utilities (v. 2.0) was used (Sayers 2009).
Results and Discussion
Meta-analysis of Transcriptomes
In this study meta-analysis was carried out under three different groups: i-biotic stress, ii-abiotic stress, iii-cumulative (biotic and abiotic stresses). The meta-analysis was carried out for transcriptome data for each biotic stress and abiotic stress to evaluate the DEGs separately in addition to the meta-analysis of the data from cumulative (biotic and abiotic) stress conditions.
Meta-analysis of Transcriptomes for Biotic and Abiotic Stress Response
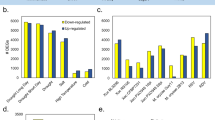
Meta-analysis of biotic-stress related data resulted with 460 common DEGs (Fig. 1a). Analysis of GO term distributions for biotic stress related DEGs showed 268 “biological process”, 146 “cell component”, and 350 “molecular function” terms (Fig. 2). 1031 common DEGs were retrieved during the meta-analysis of abiotic-stress related data (Fig. 1b). Analysis of abiotic stress related DEGs for GO term distributions showed 736 biological process terms, 415 cellular component terms, and 1020 molecular function terms. A summarized representation of GO term distributions is provided in Fig. 3.

Venn diagrams of meta-analysis results for transcriptome profiles related with biotic stress (a), abiotic stress (b), and cumulative (biotic and abiotic) stress (c)

GO term distribution of meta-analysis of data related with biotic stress. 10 most abundant terms are represented in each major GO Terms

GO term distribution of meta-analysis of data related with abiotic stress. 10 most abundant terms are represented in each major GO Terms
The cellular component GO terms of abiotic stress have been mainly related with terms under cell membrane and chloroplast. Membrane-related conditions are likely to be associated with balancing the cellular osmotic pressure. Since salt treatment affects the osmotic pressure of cells, transmembrane transport also appears to be abundant in abiotic stress data (Liang et al. 2018). Chloroplast-related terms are also abundant in abiotic stress data compared to biotic stress data. Chlorophyll levels are known to decrease under salt pressure (Hubbard and Cohen 1993). Among the DEGs obtained from meta-analysis of either biotic or abiotic stress related data, some of the DEGs associated with biotic stress was exclusive for the “Extracellular domain” GO term. It is well known that the concept of the “extracellular domain” is related to the space between cells, and the genes covered under this term are shown to be upregulated during the infection of a parasite (Bult et al. 2018).
The number of GO terms related with the defense response is the second most abundant term under ”Biological process” term for biotic stress meta-analysis data, on the other hand there is no GO term about defense response in meta-analysis results for the abiotic stress conditions suggesting that these genes are specific for biotic stress. Also, “RNA binding” term that is under “molecular functions” is abundant. Molecular function terms of biotic data can be thought of as being primarily related to transcription and protein synthesis. As expected, data suggest a molecular activation of transcription during the biotic stress response (Cohen and Leach 2019).
Under the “biological process” abiotic data have 54 terms related to transmembrane transport, whereas biotic data have 6 related terms. The genes covered under “transmembrane transport processes” term are usually associated with plant physiology such as nutrition, solute storage, cell metabolism, signaling, osmoregulation, cell growth, and stress responses, in general. Data suggest that impact of abiotic stress is more severe in terms of changes in gene expression to change plant physiology. It is likely that plants have developed more stress-related responses against the abiotic stress due to the high density and diversity of abiotic stressors compared to biotic stressors. Under “molecular function”, the GO terms are covered as “iron ion binding”, “monooxygenase activity”, and” redox activity”. Iron is an essential element for plants and plays important roles such as chlorophyll synthesis, and photosynthesis (Kobayashi et al. 2019). Data suggest activation of photosynthetic reactions under salt stress. GO terms associated with monooxygenase are also higher in abiotic stress data. The flavin and choline monooxygenase genes are known to increase drought resistance in Arabidopsis and increase abiotic stress tolerance in rice and spinach (Shirasawa et al. 2006; Lee et al. 2012). Our results are consistent with these studies since they also showed higher abundance under abiotic stress.
Meta-analysis of Cumulative Transcriptome Data
DEG results from 4 different studies were used for meta-analysis. Cumulative analysis of the data showed that 10 genes were differentially expressed for every stress condition analyzed (Fig. 1c). Among the DEGs, cytochrome P450 81E8-like, thioredoxin domain-containing protein 2, probable mannitol dehydrogenase, glutathione transferase GST 23-like, an uncharacterized protein, and a hypothetical protein encoding genes were upregulated. On the other hand, one probable leucine-rich repeat receptor-like protein kinase encoding gene and three hypothetical protein encoding genes were down-regulated according to meta-analysis of biotic and abiotic stress related data, cumulatively (Table 2).
We were able to retrieve the gene identifiers for 6 DEGs by searching their Ensembl IDs through the NCBI database. These identifiers then converted to KEGG gene identifiers (Table 3). The rest of the genes remained unknown in the KEGG database.
Meta-analysis of biotic and abiotic data shows that expression of genes especially related with photosynthesis and oxidative stress are affected in both stress types.
Our results show the up regulation of mannitol dehydrogenase gene under biotic and abiotic stress. Mannitol is also well-known ROS scavenger. Mannitol is a by-product of photosynthesis, and they move from leaf to root. Mannitol dehydrogenase is a catalyst for the catabolism of mannitol (Upadhyay et al. 2015). It is mainly used to defense the plants against pathogens by mannitol to catabolize the pathogen’s secreted mannitol under biotic stress and work as osmoprotectant and/or osmoregulator under abiotic stress conditions (Upadhyay et al. 2015). Thus, our data highlight the role of mannitol dehydrogenase in general stress response in sugar beet. Previous studies showed that induction of ROS scavengers would enhance the capacity of Beta vulgaris to tolerate stress like excessive salt. It is shown that enhancement of the photosynthesis, water status, antioxidant system and ion homeostasis by exogenous metabolites such as and melatonin and allantoin have positive impact on improving the tolerance levels. It is possible that these mechanisms may have overlapping components on mannitol dehydrogenase gene regulation and/or general stress response (Liu et al. 2020a; Zhang et al. 2021).
Under stress conditions, ROS increases dramatically. The increase in ROS is associated with the plant’s defense mechanisms (Houghton 2005). While ROS are damaging the cells molecular structures, they are also used as signaling molecules. In this study we found the gene products that are mainly related to ROS as we expected. Firstly, our data show that mitochondrial thioredoxin, which is a redox protein essential for the proper function of metabolic pathways, expressing gene is upregulated under all stress conditions. And it also takes roles in signaling pathways include stomatal opening, antioxidant metabolism, drought, salt exposure, and detoxifying hydrogen peroxide to reduce the damage of ROS to cell physiology. In addition, our data shows an increase in Glutathione S-transferases (GST), Cytochrome P450, and thioredoxin domain-containing protein 2 expression. These genes are known detoxifying enzymes along with their role in regulation of oxidative stress (Xu et al. 2015). A novel study also showed differentiated expression of genes related with ROS metabolism, carbohydrate metabolism and hormone signaling under salt stress which increases the low temperature tolerance for Beta vulgaris (Liu et al. 2023). Those genes can be a source to increase tolerance against different stress conditions when combined with salt stress in Beta vulgaris.
Increase of free radicals in plant tissues is a natural phenomenon and plants have active defense mechanisms to fight against damaging effects of these radicals. Limiting the role of these mechanisms to reduction of ROS would be underestimating their values. Studies showed that there is a direct correlation between the expression of the genes and/or activating antioxidant enzyme mechanisms related with mitigating oxidative damage and improving stress tolerance in sugar beet (Zou et al. 2020a; Xing et al. 2020; Liu et al. 2023). Scavenging of ROS has an impact on plant physiology by controlling diverse mechanisms related with photosynthesis, lipid and energy metabolism, cell wall structure, and signaling to improve plants’ capacity to tolerate diverse stress conditions (Foyer and Shigeoka 2011; Rasouli et al. 2020; Liu et al. 2023).
A Leucine-rich repeat receptor kinase and three hypothetical proteins were down-regulated in the meta-analysis of biotic and abiotic stress related data. Leucine-rich repeat receptor kinases are the largest subfamily of transmembrane receptor-like kinases in plants. They regulate a wide range of defense-related processes (Torii 2004). Previous studies show the importance of transmembrane receptor-like kinases under biotic and abiotic stresses (Wu et al. 2015). While expression of most of known Leucine-rich repeat receptor kinases are increased under stress, leucine-rich repeat (LRR)-RLK gene (Leaf Panicle 2 (LP2) was a negative-regulator drought stress and higher expression of LP2 inhibits stoma closure. It is possible that Leucine-rich repeat receptor kinase encoding gene found in this study can have similar function as LP2 protein.
The overall results may show increased photosynthesis under biotic and abiotic stress conditions. Plants tend to decrease in the rate of their photosynthesis under stress, especially salt stress (Ashraf and Harris 2013). They close their stoma under stressful conditions to stop evaporation, preventing them from absorbing CO2. Beta vulgaris, on the other hand, is known to be a salt-resistant plant, which maintains osmotic pressure and absorbs more water than other plants (Ferreira da Silva et al. 2018). Therefore, our data suggest that evaporation does not significantly affect sugar beets under stress. The reduction of the Leucine-rich repeat receptor kinase and increase of mannitol dehydrogenase genes supports the phenomenon. Chloroplast is known as an environmental sensor and creates the first response for stress conditions. The relationship between photosynthesis and ROS is a complicated process. ROS can be thought as double-edged sword. They can damage cells at high concentrations or they can also act as signaling molecules to enhance photosynthesis. (Foyer and Shigeoka 2011). Therefore, plant cells maintain ROS levels in an adequately required level and adjust the photosynthesis rate depending on the impact of all surrounding conditions by controlling the expression of several genes (Xu et al. 2015; Yu et al. 2020; Zou et al. 2020a). Since we covered the genes that are regulated in all stress conditions tested, our data may suggest photosynthetic rate can fluctuate depending on the type and the duration of the stress factors.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Andrews S (2010) FASTQC A Quality Control tool for High Throughput Sequence Data. In: Babraham Institute. http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Ashraf M, Harris PJC (2013) Photosynthesis under stressful environments: an overview. Photosynthetica 51:163–190. https://doi.org/10.1007/s11099-013-0021-6
Bult C, Blake J, Smith C et al (2018) Mouse genome database (MGD) 2019. https://doi.org/10.1093/nar/gky1056. Nucleic acids research 47:
Cohen SP, Leach JE (2019) Abiotic and biotic stresses induce a core transcriptome response in rice. Sci Rep 9:6273. https://doi.org/10.1038/s41598-019-42731-8
Dohm JC, Lange C, Holtgräwe D et al (2012) Palaeohexaploid ancestry for Caryophyllales inferred from extensive gene-based physical and genetic mapping of the sugar beet genome (Beta vulgaris): Sugar beet maps and palaeohexaploid ancestry. Plant J 70:528–540. https://doi.org/10.1111/j.1365-313X.2011.04898.x
Durinck S, Moreau Y, Kasprzyk A et al (2005) BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21:3439–3440. https://doi.org/10.1093/bioinformatics/bti525
Ferreira da Silva P, Matos R, Borges V et al (2018) Water consumption of Beta vulgaris L. cultivated in greenhouse under fertigation and types of foundation fertilization. Aust J Crop Sci 12:1335–1341. https://doi.org/10.21475/ajcs.18.12.08.PNE1177
Foyer CH, Shigeoka S (2011) Understanding oxidative stress and antioxidant functions to enhance photosynthesis. Plant Physiol 155:93–100. https://doi.org/10.1104/pp.110.166181
Ghaemi R, Pourjam E, Safaie N et al (2020) Molecular insights into the compatible and incompatible interactions between sugar beet and the beet cyst nematode. BMC Plant Biol 20:483. https://doi.org/10.1186/s12870-020-02706-8
Holmquist L, Dölfors F, Fogelqvist J et al (2021) Major latex protein-like encoding genes contribute to Rhizoctonia solani defense responses in sugar beet. Mol Genet Genomics 296:155–164. https://doi.org/10.1007/s00438-020-01735-0
Houghton J (2005) Global warming. Rep Prog Phys 68:1343–1403. https://doi.org/10.1088/0034-4885/68/6/R02
Hubbard MJ, Cohen P (1993) On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem Sci 18:172–177. https://doi.org/10.1016/0968-0004(93)90109-z
Ibrahim HMM, Kusch S, Didelon M, Raffaele S (2021) Genome-wide alternative splicing profiling in the fungal plant pathogen Sclerotinia sclerotiorum during the colonization of diverse host families. Mol Plant Pathol 22:31–47. https://doi.org/10.1111/mpp.13006
Kim D, Paggi JM, Park C et al (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37:907–915. https://doi.org/10.1038/s41587-019-0201-4
Kobayashi T, Nozoye T, Nishizawa NK (2019) Iron transport and its regulation in plants. Free Radic Biol Med 133:11–20. https://doi.org/10.1016/j.freeradbiomed.2018.10.439
Larkin MA, Blackshields G, Brown NP et al (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. https://doi.org/10.1093/bioinformatics/btm404
Lee M, Jung J-H, Han D-Y et al (2012) Activation of a flavin monooxygenase gene YUCCA7 enhances drought resistance in Arabidopsis. Planta 235:923–938. https://doi.org/10.1007/s00425-011-1552-3
Leinonen R, Sugawara H, Shumway M (2011) The sequence read Archive. Nucleic Acids Res 39:D19–D21. https://doi.org/10.1093/nar/gkq1019
Liang W, Ma X, Wan P, Liu L (2018) Plant salt-tolerance mechanism: a review. Biochem Biophys Res Commun 495:286–291. https://doi.org/10.1016/j.bbrc.2017.11.043
Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. https://doi.org/10.1093/bioinformatics/btt656
Liu L, Liu D, Wang Z et al (2020a) Exogenous allantoin improves the salt tolerance of sugar beet by increasing putrescine metabolism and antioxidant activities. Plant Physiol Biochem 154:699–713. https://doi.org/10.1016/j.plaphy.2020.06.034
Liu L, Wang B, Liu D et al (2020b) Transcriptomic and metabolomic analyses reveal mechanisms of adaptation to salinity in which carbon and nitrogen metabolism is altered in sugar beet roots. BMC Plant Biol 20:138. https://doi.org/10.1186/s12870-020-02349-9
Liu L, Gai Z, Qiu X et al (2023) Salt stress improves the low-temperature tolerance in sugar beet in which carbohydrate metabolism and signal transduction are involved. Environ Exp Bot 208:105239. https://doi.org/10.1016/j.envexpbot.2023.105239
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Lv X, Chen S, Wang Y (2019) Advances in understanding the physiological and molecular responses of Sugar Beet to Salt stress. Front Plant Sci 10:1431. https://doi.org/10.3389/fpls.2019.01431
Marot G, Jaffrezic F, Rau A (2015) metaRNASeq: Differential meta-analysis of RNA-seq data. https://cran.r-project.org/web/packages/metaRNASeq/vignettes/metaRNASeq.pdf
Mistry J, Chuguransky S, Williams L et al (2021) Pfam: the protein families database in 2021. Nucleic Acids Res 49:D412–D419. https://doi.org/10.1093/nar/gkaa913
Nakagawa S, Santos ESA (2012) Methodological issues and advances in biological meta-analysis. Evol Ecol 26:1253–1274. https://doi.org/10.1007/s10682-012-9555-5
Rasouli F, Kiani-Pouya A, Li L et al (2020) Sugar Beet (Beta vulgaris) Guard cells responses to salinity stress: a proteomic analysis. Int J Mol Sci 21:2331. https://doi.org/10.3390/ijms21072331
Sayers E (2009) A General Introduction to the E-utilities. https://www.ncbi.nlm.nih.gov/books/NBK25497/. Accessed 10 Jul 2023
Shams M, Khadivi A (2023) Mechanisms of salinity tolerance and their possible application in the breeding of vegetables. BMC Plant Biol 23:139. https://doi.org/10.1186/s12870-023-04152-8
Shirasawa K, Takabe T, Takabe T, Kishitani S (2006) Accumulation of glycinebetaine in Rice plants that overexpress choline monooxygenase from spinach and evaluation of their tolerance to abiotic stress. Ann Bot 98:565–571. https://doi.org/10.1093/aob/mcl126
Stracke R, Holtgräwe D, Schneider J et al (2014) Genome-wide identification and characterisation of R2R3-MYB genes in sugar beet (Beta vulgaris). BMC Plant Biol 14:249. https://doi.org/10.1186/s12870-014-0249-8
Torii KU (2004) Leucine-rich repeat receptor kinases in plants: structure, function, and signal transduction pathways. Int Rev Cytol 234:1–46. https://doi.org/10.1016/S0074-7696(04)34001-5
Upadhyay R, Meena M, Prasad V et al (2015) Mannitol metabolism during pathogenic fungal–host interactions under stressed conditions. Front Microbiol 6
Wu F, Sheng P, Tan J, Chen X, Lu G, Ma W, Heng Y, Lin Q, Zhu S, Wang J, Wang J, Guo X, Zhang X, Lei C, Wan J (2015) Plasma membrane receptor-like kinase leaf panicle 2 acts downstream of the drought and salt tolerance transcription factor to regulate drought sensitivity in rice. J Exp Bot 66:271–281. https://doi.org/10.1093/jxb/eru417
Xing W, Pi Z, Liu J et al (2020) Comparative transcriptome analysis reveals an ABA-responsive regulation network associated with cell wall organization and oxidation reduction in sugar beet. Plant Growth Regul 91:127–141. https://doi.org/10.1007/s10725-020-00592-6
Xu J, Wang X, Guo W (2015) The cytochrome P450 superfamily: Key players in plant development and defense. J Integr Agric 14:1673–1686. https://doi.org/10.1016/S2095-3119(14)60980-1
Yu B, Chen M, Grin I, Ma C (2020) Mechanisms of Sugar Beet response to biotic and abiotic stresses. In: Zharkov DO (ed) Mechanisms of Genome Protection and Repair. Springer International Publishing, Cham, pp 167–194
Zhang Y, Nan J, Yu B (2016) OMICS Technologies and Applications in Sugar Beet. Front Plant Sci 7
Zhang P, Liu L, Wang X et al (2021) Beneficial Effects of Exogenous Melatonin on Overcoming Salt stress in Sugar beets (Beta vulgaris L). Plants (Basel) 10:886. https://doi.org/10.3390/plants10050886
Zou C, Liu D, Wu P et al (2020a) Transcriptome analysis of sugar beet (Beta vulgaris L.) in response to alkaline stress. Plant Mol Biol 102:645–657. https://doi.org/10.1007/s11103-020-00971-7
Zou C, Wang Y, Wang B et al (2020b) Long non-coding RNAs in the alkaline stress response in sugar beet (Beta vulgaris L). BMC Plant Biol 20:227. https://doi.org/10.1186/s12870-020-02437-w
Funding
Not available.
Author information
Authors and Affiliations
Contributions
Burak Bulut: Conception and design, sampling, data collection and analysis, evaluation of results, and writing/editing the manuscript. Songül Gürel: Data evaluation and editing the manuscript. Ömer Can Ünüvar: Evaluation of results, and writing/editing the manuscript. Ekrem Gürel: Data evaluation and editing the manuscript. Yunus Şahin: R Coding and evaluation of results. Uğur Çabuk: Datanormalization. Ercan Selçuk Ünlü: Conception and design, sampling, Perl coding, data collection and analysis, evaluation of results, and writing/editing/revising the manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Conflicts of interest/Competing Interests
Authors declare that they have no conflict of interest/competing interest.
Additional information
Communicated by Ray Ming.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bulut, B., Gürel, S., Ünüvar, Ö.C. et al. Meta Analysis of Sugar Beet (Beta vulgaris L.) Transcriptome Profiles Under Different Biotic and Abiotic Stress Conditions. Tropical Plant Biol. 16, 199–207 (2023). https://doi.org/10.1007/s12042-023-09344-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12042-023-09344-y