
Abstract
Interstitial 6p25.1p24.3 microdeletions are rare events and a clear karyotype/phenotype correlation has not yet been determined. In this study, we present the clinical and molecular description of a child with a de novo 6p25.1p24.3 microdeletion, characterized by array-CGH, associated with mild intellectual disability, facial dysmorphisms, hypopigmentation of the skin of the abdomen, heart defects, mild pontine hypoplasia and hypotonia. This deleted region contains 14 OMIM genes (NRN1, F13A1, RREB1, SSR1, RIOK1, DSP, BMP6, TXNDC5, BLOC1S5, EEF1E1, SLC35B3 and HULC). To the best of our knowledge until now only six cases have been reported presenting an interstitial microdeletion, but a unique case carries a deleted region containing the same genes of our patient. We compared clinical features and genetic data with that of the previously reported patient. We also analysed the gene content of the deleted region to investigate the possible role of specific genes in the clinical phenotype of our patient.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Deletions of the distal part of 6p cause a clinically recognized syndrome (chromosome 6pter-p24 deletion syndrome, OMIM: 612582) characterized by developmental delay/cognitive impairment, brain malformations, conductive hearing loss, ocular anomalies, cardiac abnormalities and craniofacial dysmorphisms (DeScipio 2007). More than 30 cases with larger deletion have been reported, mostly detected by cytogenetic or subtelomeric screening, involving the terminal portions (0.3–14 Mb in size) with breakpoints within 6p25.3p23 region (Linhares et al. 2015). Conversely, isolated interstitial deletions within 6p25p24 are very rare and, to the best of our knowledge, only six cases have been reported so far (Davies et al. 1999; Mirza et al. 2004; Kuipers et al. 2013; Qi et al. 2015). Of note, small interstitial deletions/duplications are usually not detected by standard cytogenetic techniques, while microarray technology may uncover intragenic deletions as small as dozen bases (Boone et al. 2010).
Here, we present the phenotypic and genomic findings in a young female patient who carries an interstitial deletion within the 6p25.1p24.3 region.
Clinical report
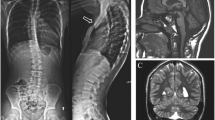
The patient is a 15-year-old female, born as the first child of nonconsanguineous parents (figure 1). The father is affected by melanoma, while the 7-year-old sister and a first cousin were diagnosed with speech disorder. Our patient was born at term by normal delivery after an uneventful pregnancy. Birth weight was 3000 g (21° centile; –0.80 standard deviation score (SDS)), length 49 cm (45° centile; –0.12 SDS), head circumference 34 cm (33° centile; –0.45 SDS), and APGAR scores were 10 and 10 at 1 and 5 min, respectively. At neonatal examination, the axial hypotonia, poor sucking, facial dysmorphisms with submucous cleft palate (not deserving surgically treatment) and a sectorial hypopigmentation of the skin of the abdomen were noted. Due to breastfeeding problems leading to failure to thrive, an artificial feeding was started. Frequent regurgitation was present until 18 months of age. During the first one and a half years, the proposita suffered from a gastro oesophageal reflux disease, controlled by pharmacotherapy. From a neurodevelopmental point of view, proposita showed a moderate delay, achieving autonomous walking at 22 months, first words of significance and anal sphincter control at 3 years of age. Bladder control was never achieved, and she remained enuretic even after treatment with oxybutynin and suffered frequent urinary infections. The voiding cystourethrogram revealed incomplete bladder emptying and irregular bladder wall profiles with pseudodiverticula, without evidence of vesicoureteric reflux, consistent with a neurogenic bladder (figure 2a). At two years of age, proposita was operated for obstruction of the lacrimal ducts. A dysplastic tricuspid valve and mild patent foramen ovale were detected, which did not require surgery. Brain MRI performed at 3.5 years and repeated at 13 years demonstrated mild pontine hypoplasia (figure 2b), while brain MR spectroscopy performed at the level of the basal ganglia was normal; a suspicion of pituitary adenoma had been raised up but not confirmed and for this reason the patient is still in clinical and radiological follow-up. Spinal MRI was normal except for the presence of tall and slight foreshortened vertebral bodies at the lumbar level (figure 2c). Hand and feet radiograph performed at 2.7 years of age demonstrated slightly delayed bone age and abnormal morphology of both distal phalanges of the feet (figure 2, d&e). Metabolic screening and analysis of FMR1 gene (MIM: 309550) resulted normal. At 5 years of age, orthopaedic evaluation revealed mild dorsolumbar scoliosis that was treated with an orthopaedic corset.

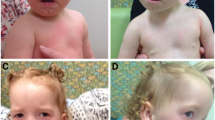
(a) Patient's frontal profile where the almond shaped eyes with long eye-lashes and everted lower lids, short philtrum and a mild micrognathia are evident. (b) A right later face profile view in which the presence of a moderately flat facies with low set ears and the presence of (bilateral) square helices are notable. (c) Long fingers and presence of a minimal syndactyly. (d) Clinodactyly of finger feet and also presence of a mild to moderate syndactyly, more evident between the second and third fingers.

(a) Voiding cystourethrogram, frontal view, reveals irregular bladder wall profiles with pseudo-diverticula (empty arrows), without evidence of vesico-ureteric reflux. (b) Brain MRI, sagittal T1-weighted image, demonstrates mild pontine hypoplasia with reduction of the cranio-caudal diameter of the pons (arrow). (c) Spinal MRI, sagittal T2-weighted image, shows tall and slightly foreshortened vertebral bodies at the lumbar level (empty arrows). There is also minimal, nonsignificant dilatation of the central canal of the dorsal spinal cord (arrow). (d, e) Hand and feet radiograph performed at 2.7 years of age demonstrated slightly delayed bone age. Bone age in this case is 2 years as per Tanner. Note the short squared distal phalanges of the feet (arrowheads).
At the last follow-up, age 14 and half years, she was 161 cm tall (57° centile; 0.2 SDS) and had a weight of 42.3 kg (49° centile; –1.47 SDS). Neurological examination showed hyperlaxity and hypermobility, with no focal neurological signs but presence of mild left palpebral ptosis, clumsiness, hyperlaxity and hypermobility. Facial dysmorphisms included flat facies, low-set ears, square helices, almond shape eyes with everted low lids, long eye lashes, short philtrum micrognathia and fingers with mild syndactyly and clinodactyly (figure 1).
Menarche appeared at 12 years and 8 months. Last neurological examination performed at 14 years of age showed bilateral palpebral ptosis (more evident on the left side), muscular hypotonia, muscular weakness (more evident in the left upper limb), hyperlaxity, and clumsiness, poor coordination with easy fatigability. She had a mild intellectual disability with total intelligence quotient of 67 scored with Wechsler Intelligence scale fourth edition and she showed autistic features and behavioural problems consistent with an oppositional defiant disorder that has been treated with valproic acid (10 mg/kg per day) and periciazine (1.5 mg/day).
Materials and methods
Molecular karyotyping
Molecular karyotyping (array-CGH) was performed on DNA samples extracted from the peripheral blood of the proposita and her parents, according to standard methods using a whole-genome 180 K Agilent array with ~13 kb overall median probe spacing (Human Genome CGH Microarray, Agilent Technologies, Santa Clara, USA), according to the manufacturer’s protocol. Data were analysed using Agilent Cytogenomics. All genomic positions were reported according to the human genome assembly (GRCh37/hg19).
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal. The paper is exempt from ethical committee approval.
Results
Array-CGH analysis revealed a de novo interstitial deletion of 6p25.1p24.3 spanning about 4 Mb of genomic DNA from positions 5,895,544–9,961,470 according to UCSC Genome Browser (hg19; GRChBuild 37.1, February 2009) (arr 6p25.1p24.3(5,895,544–9,961,470)x1dn) (figure 3, a&b). This deleted region contains 14 OMIM genes: NRN1 (MIM: 607409; neuritin 1, transcript variant 1), F13A1 (MIM: 134570; coagulation factor XIII A chain) AR, AD, LY86 (MIM: 605241; lymphocyte antigen 86), RREB1 (MIM: 602209; RAS-responsive element binding protein 1), SSR1 (MIM: 600868; signal sequence receptor, alpha), CAGE1 (MIM: 608304; cancer/testis antigen 3), RIOK1 (MIM: 617753; RIO kinase 1), DSP (MIM: 125647; desmoplakin) AD, BMP6 (MIM: 112266; bone morphogenetic protein 6), TXNDC5 (MIM: 616412; thioredoxin domain-containing protein 5), BLOC1S5 (MIM: 607289; biogenesis of lysosome-related organelles complex 1, subunit 5), EEF1E1 (MIM: 609206; eukaryotic translation elongation factor 1, epsilon-1), SLC35B3 (MIM: 610845; solute carrier family 35 (3-prime-phosphoadenosine 5-prime-phosphosulfate transporter), member B3), and HULC (MIM: 612210; highly upregulated in liver cancer).

Results of array-CGH analyses. (a) Array-CGH profile of patient’s chromosome 6 showing the 6p deletion. (b) Zoom view of short arm of chromosome 6 shows a de novo 6p25.1p24.3 deletion spanning about 4 Mb of genomic DNA from positions 5,895,544 to 9,961,470 according to UCSC Genome Browser (hg19; GRChBuild 37.1, February 2009) [arr 6p25.1p24.3(5,895,544-9,961,470)x1dn]. (c) Overview of the 6p25.1p24.3 region and its gene content according to the UCSC Genome Browser [GRCh37/hg19 assembly]. The red bar indicates the deleted region of our patient, the two reported by Kuipers et al. (2013), the patient reported by Qui et al. (2015) and the one reported by Mirza et al. (2004).
A second CNV on chromosome X was inherited from proposita’s mother arr[GRCh37] Xp21.1(37,223,427-37,519,772)x3 mat. Array-CGH analysis of proposita’s father was normal, while her mother showed three CNVs: arr[GRCh37] 2q21.2(133,440,191-133,618,068)x1 NCKAP5 (MIM: 608789; NCK-associated protein 5), arr[GRCh37] 10p15.1(5,174,667-5,256,896)x1, AKR1C4 (MIM: 600451; aldo-keto reductase family 1, member C4), and arr[GRCh37] Xp21.1(37,223,427-37,519,772)x3 PRRG1 (MIM: 300935; proline-rich gamma-carboxy-glutamic acid protein 1).
Discussion
Array-CGH is a powerful diagnostic tool that facilitates the detection of new syndromes starting from the diagnosis of a deletion or duplication of a DNA region. In this study, we describe a 15-year young female with intellectual disability, dysmorphisms, and other phenotypic features. The patient carried a de novo interstitial deletion 6p25.1p24.3 spanning about 4 Mb of genomic DNA involving 14 OMIM genes. While terminal or subtelomeric deletions of 6p25.3p23 are relatively frequent, the isolated interstitial deletions within 6p25p24 are very rare and, to the best of our knowledge, only six cases have been reported so far (Davies et al. 1999; Mirza et al. 2004; Kuipers et al. 2013; Qi et al. 2015).
Interestingly, in the literature we found a unique case carrying a deleted region containing the same genes as of our patient (Kuipers et al. 2013). They have described two cases with overlapping deletions in 6p25.1p24.3. Particularly interesting is their ‘Case 2’ that contains the same genes present in young female from our study (figure 3c). Other reported cases present only some deleted genes in common with our patient (Kuipers et al. 2013, case 1; Mirza et al. 2004, case3) or more genes deleted (Qi et al. 2015).
Here, we focus on ‘Case 2’ of Kuipers et al. (2013) and compare the phenotypic features with our case. The two patients present in common: mild intellectual disability, facial dysmorphisms as flat facies, low-set ears, square helices, almond shape eyes with everted low lids, long eye lashes, short philtrum, micrognathia, long fingers with mild clinodactyly, and hair abnormalities (table 1).
In our opinion the deleted region contains some genes that can play an important role in our patient’s phenotype. An interesting gene deleted in our proposita is neuritin (NRN1, also identified as the candidate plasticity gene 15; cpg15), that is highly expressed in the nervous system (Fujino et al. 2008; Putz et al. 2005) especially in sensory neurons (Karamoysoyli et al. 2008), hippocampus, visual cortex and external granular layer of the cerebellum (An et al. 2014). It promotes synaptic growth, axonal regeneration, and nerve cell maturation (Bravo et al. 2013; Karamoysoyli et al. 2008). Also, the RRBE1 gene seems to have a role in the axonal degeneration as reported in a study by Farley et al. (2018).
An excess of hematic iron was referred in our proposita (146 g/dL; N.V. 45–120). Interestingly, the bone morphogenetic protein 6 (BMP6) has been showed to have an essential role in the maintenance of iron homeostasis, at least in mice Bmp6 -/- that had a phenotype resembling hereditary hemochromatosis, with reduced hepcidin expression and tissue iron overload (Kautz et al. 2008; Meynard et al. 2009; Andriopoulos et al. 2009).
The pigmentation pathway of the skin represents a complex and strictly regulated network of various hormones, corresponding receptors and other factors. Among others, the BLOC1S5 is involved in the genesis of the melanosome, which is important for the pigmentation process (Rossberg et al. 2016). Further, this gene seems to have a role in synapses and may participate in neurodevelopmental processes, at least in mouse (Larimore et al. 2014).
Desmoplakin gene (DSP) was shown to be responsible for the autosomal dominant form of the keratosis palmoplantaris striata II (MIM 612908). Our patient showed a sectorial hypopigmentation of the skin of the abdomen and diffused hyperkeratosis.
In conclusion 6p25.1p24.3 microdeletion is extremely rare. We have identified and reported a new individual with very similar interstitial deletion to one reported in the literature. Our study could help in better understanding of karyotype/phenotype correlation in 6p25.1p24.3 microdeletion, and to determine the clinical implication of the genes involved in chromosomal region.
References
Andriopoulos B., Corradini E., Xia Y., Faasse S. A., Chen S., Grgurevic L. et al. 2009 BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 41, 482–487.
An K., Jung J. H., Jeong A. Y., Kim H. G., Jung S. Y., Lee K. et al. 2014 Neuritin can normalize neural deficits of Alzheimer’s disease. Cell Death Dis. 5, e1523.
Boone P. M., Bacino C. A., Shaw C. A., Eng P. A., Hixson P. M., Pursley A. N. et al. 2010 Detection of clinically relevant exonic copy-number changes by array CGH. Hum. Mutat. 31, 1326–1342.
Bravo R., Parra V., Gatica D., Rodriguez A. E., Torrealba N., Paredes F. et al. 2013 Chapter five - Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int. Rev. Cell Mol. Biol. 301, 215–290.
Davies A. F., Mirza G., Flinter F. and Ragoussis J. 1999 An interstitial deletion of 6p24-p25 proximal to the FKHL7 locus and including AP-2alpha that affects anterior eye chamber development. J. Med. Genet. 36, 708–710.
DeScipio C. 2007 The 6p subtelomere deletion syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 145C, 377–382.
Farley J. E., Burdett T. C., Barria R., Neukomm L. J., Kenna K. P., Landers J. E. et al. 2018 Transcription factor Pebbled/RREB1 regulates injury-induced axon degeneration. Proc. Natl. Acad. Sci. USA 115, 1358–1363.
Fujino T., Wu Z., Lin W. C., Phillips M. A. and Nedivi E. 2008 cpg15 and cpg15-2 constitute a family of activity-regulated ligands expressed differentially in the nervous system to promote neurite growth and neuronal survival. J. Comp. Neurol. 507, 1831–1845.
Karamoysoyli E., Burnand R. C., Tomlinson D. R. and Gardiner N. J. 2008 Neuritin mediates Nerve Growth Factor–induced axonal regeneration and is deficient in experimental diabetic neuropathy. Diabetes 57, 181–189.
Kautz L., Meynard D., Monnier A., Darnaud V., Bouvet R. and Wang R. H. 2008 Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad 7, Id1, and Atoh8 in the mouse liver. Blood 112, 1503–1509.
Kuipers B. C., Vulto-van Silfhout A. and T., Marcelis C., Pfundt R., de Leeuw N. and de Vries B. B. 2013 Two patients with intellectual disability, overlapping facial features, and overlapping deletions in 6p25.1p24.3. Clin. Dysmorphol. 22, 18–21.
Larimore J., Zlatic S. A., Gokhale A., Tornieri K., Singleton K. S., Mullin A. P. et al. 2014 Mutations in the BLOC-1 subunits dysbindin and muted generate divergent and dosage dependent phenotypes. J. Biol. Chem. 289, 14291–14300.
Linhares N. D., Svartman M., Rodrigues T. C., Rosenberg C. and Valadares E. R. 2015 Subtelomeric 6p25 deletion/duplication: Report of a patient with new clinical findings and genotype-phenotype correlations. Eur. J. Med. Genet. 58, 310–318.
Meynard D., Kautz L., Darnaud V., Canonne-Hergaux F., Coppin H. and Roth M. P. 2009 Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 41, 478–481.
Mirza G., Williams R. R., Mohammed S., Clark R., Newbury-Ecob R., Baldinger S. et al. 2004 Refined genotype-phenotype correlations in cases of chromosome 6p deletion syndromes. Eur. J. Hum. Genet. 12, 718–728.
Putz U., Harwell C. and Nedivi E. 2005 Soluble CPG15 expressed during early development rescues cortical progenitors from apoptosis. Nat. Neurosci. 8, 322–333.
Qi Z., Jeng L. J., Slavotinek A. and Yu J. 2015 Haploinsufficiency and triploinsensitivity of the same 6p25.1p24.3 region in a family. BMC Med. Genomics 8, 38.
Rossberg W., Saternus R., Wagenpfeil S., Kleber M., März W., Reichrath S. et al. 2016 Human pigmentation, cutaneous vitamin D synthesis and evolution: variants of genes (SNPs) involved in skin pigmentation are associated with 25(OH)D serum concentration. Anticancer Res. 36, 1429–1437.
Acknowledgements
This work was supported by ‘Cinque per mille dell’IRPEF-Finanziamento della ricerca sanitaria’ and ‘Finanziamento Ricerca Corrente’, Ministero Salute (contributo per la ricerca intramurale).
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: H. A. Ranganath
Rights and permissions
About this article

Cite this article
Tassano, E., Uccella, S., Severino, M. et al. Expanding the phenotype associated with interstitial 6p25.1p24.3 microdeletion: a new case and review of the literature. J Genet 100, 9 (2021). https://doi.org/10.1007/s12041-021-01261-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12041-021-01261-x