
Abstract
Sorghum downy mildew caused by Peronosclerospora sorghi is a major disease of maize and resistance is under the control of polygenes which necessitated identification of quantitative-trait loci (QTLs) for initiating marker-assisted introgression of resistant QTLs in elite susceptible inbred lines. In the present study, QTLs for sorghum downy mildew (SDM) resistance in maize were identified based on cosegregation with linked simple sequence repeats in 185 F2 progeny from a cross between susceptible (CM500-19) and resistant (MAI105) parents. F3 families were screened in the National Sorghum Downy Mildew Screening Nursery during 2010 and 2011. High heritability was observed for the disease reaction. The final map generated using 87 SSR markers had 10 linkage groups, spanning a length of 1210.3 cM. Although, we used only 87 SSR markers for mapping, the per cent of genome within 20 cM to the nearest marker was 88.5. Three putative QTLs for SDM resistance were located on chromosomes 3 (bin 3.01), 6 (bin 6.01) and 2 (bin 2.02) using composite interval mapping. The locus on chromosome 3 had a major effect and explained up to 12.6% of the phenotypic variation. The other two QTLs on chromosomes 6 and 2 had minor effects with phenotypic variation of 7.1 and 2%. The three QTLs appeared to have additive effects on resistance. The QTLs on chromosomes 3 and 6 were successfully used in the marker-assisted selection programme for introgression of resistance to SDM in eight susceptible maize lines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sorghum downy mildew (SDM) is a destructive systemic disease of maize worldwide and is caused by an obligate pathogen Peronosclerospora sorghi (Weston and Uppal) C. G. Shaw. The other maize downy mildew pathogens like P. philippinensis (Philippine DM), P. maydis (Java DM), P. sacchari (sugarcane DM) and Sclerophthora rayssiae zeae (brown stripe DM) (Sharma et al. 1993), cause severe disease symptoms in southeast Asia (Frederiksen and Renfro 1977). SDM occurs on maize and sorghum in warm, humid areas of the world causing significant yield reduc tion. This disease became a prominent disease of maize globally during the years when rapid spread of sorghum as grain and forage crop. SDM is prevalent in the peninsular India, in the states of Karnataka, Tamil Nadu and Andhra Pradesh causing yield losses of 30% and higher (Payak 1975; Krishnappa et al. 1995). Though conventional disease management practices (Frederiksen and Renfro 1977; Williams 1984; Craig and Odvody 1992) have been successful in controlling the downy mildews in most of the maize growing regions of the world, SDM of maize remains a serious problem. The disease appears in severe form on maize crop treated with metalaxyl under severe infestation (Raymundo 2000). Added to this, a recent report of metalaxyl resistant P. sorghi (Isakeit et al. 2003) suggests the need for alternative control methods like host resistance.
Genetic analysis of host resistance to SDM in maize has indicated that resistance is under polygenic control and additive effects contribute predominantly to resistance (Borges and Orange 1987; Geetha and Jayaraman 2002; Nallathambi et al. 2010). Three quantitative-trait loci (QTLs) that contribute resistance to P. sorghi were identified in a population of recombinant inbred lines derived from a cross between inbred lines G62 (resistant) and G58 (susceptible) (Agrama et al. 1999). Two of the loci map close together on chromosome 1, while the third one was on chromosome 9. George et al. (2003) reported six QTLs on five chromosomes (1, 2, 6, 7 and 10) in a RIL population from the cross, Ki3 (resistant) × CML139 (susceptible) based on tests in India, Indonesia, Thailand and Philippines. Nair et al. (2005) detected SDM resistance loci on maize chromosomes 2, 3 and 6 in the Indian maize line NAI116 and verified that the locus on chromosome 6 also contributed resistance to diverse downy mildews. In another study, three putative QTLs were detected in different environments with one locus on chromosome 2 had a major effect and explained up to 70% of the phenotypic variation in Thailand where disease pressure was highest and the other two QTLs on chromosomes 3 and 9 had minor effects; each explained not more than 4% of the phenotypic variation (Sabry et al. 2006).
Given the complexity of quantitative traits, mapping populations must be carefully analysed over different years and environments to unravel important components of gene interaction. Identification of simple and accurately scored molecular markers for genes that contribute to SDM resistance of maize could greatly benefit future efforts to prevent disease losses, especially if there are differences in the pathogen populations or environment by genotype interactions in different locations. The major objective of this study was to validate the QTL information generated previously by using different mapping populations over years, to identify other major QTLs, if any, conferring resistance to SDM and introgression of major QTLs in breeding lines.
Materials and methods
Mapping population
Based on response to sorghum downy mildew in screening trials comprising of 57 lines originating from India and Mexico conducted over years, two inbred maize lines were selected for this study. MAI105 (yellow), which was consistently resistant, was selected as the resistant parent (RP) and CM500-19 (yellow) served as the susceptible parent (SP). The F1 from the cross CM500-19 × MAI105 was self-pollinated and 185 F2 individuals were produced. Leaf samples were collected from individual F2 plants before they were selfpollinated to produce F3 families. Healthy young leaves were collected from the parents and F2 plants. Samples were frozen in liquid nitrogen and grounded using pestle and mortar. The fine powder obtained from each sample was used for DNA extraction.
Phenotyping of the mapping population
Seeds from 185 F3 families and two parental lines were grown in the two field trials during 2010 and 2011 postmonsoon period to evaluate the responses to SDW in the National Sorghum Downy Mildew Screening Nursery, Mandya, Karnataka, India (12°N; 76°E; 695 m above mean sea level; average rainfall of 705 mm per year). The field experimental design was randomized block design with two replications, with one row per replication. The test entries were planted in 3 m long rows, with 15–20 plants per row and 0.75 m between rows. Seeds of SDM susceptible maize line CM500 were planted as spreader rows on all sides of the experimental block, 30 days prior to the planting of the test entries. One bed of spreader row was planted for every two beds of test entries. The conidial suspension of SDM was sprayed on the whorl of each seedling of the spreader rows (5–7 days after germination) between 3 and 4 am (Craig et al. 1977). The suspension was prepared by collecting conidia from SDM-infected plants at 2–3 am and suspended in water with a concentration of approximately 40,000–50,000 conidia per mL. The inoculation procedure was repeated for three consecutive days to ensure that no plant escaped artificial infection. Same procedure was repeated on test entries also for uniform disease expression. As a susceptible check, CM500 seeds were planted after every 10th row of test materials. Severe infection (97–100% DM incidence) in the check rows across the experimental block indicated uniform and strong pathogen pressure, leaving no possibility for ‘disease escapes’. The disease reaction was assessed at 25 and 35 days after plant emergence by scoring for systemic infection in the individual plants. Percentage disease incidence in each test entry was determined. Inoculated plants that did not show systemic symptoms of DM (emergence of characteristic chlorotic leaves) were considered to be resistant (Rao et al. 1984).
SSR assay
A set of 384 simple sequence repeat (SSR) primers covering various ‘bin’ locations in the maize genome was selected from http://www.maizegdb.org/ssr.php. The primers were synthesized from M/s Sigma-Aldrich, Bengaluru, India. DNA used in the SSR analysis was extracted from the leaves of 1-week-old F2 plants according to the procedure of Saghai-Maroof et al. (1984) and as modified by Hoisington et al. (1994). Approximately 40 ng of DNA was used as the template for PCR in a 20 μL reaction volume. PCR was carried out in the Eppendorf Thermal Cycler (Eppendorf, Hamburg, Germany) with the following cycling profile: an initial denaturation at 94°C for 2 min, followed by 40 cycles of amplification at 94°C for 1 min, 55–65°C (based on the annealing temperatures standardized for different SSR primers) for 2 min and 72°C for 2 min, with a final extension step at 72°C for 7 min followed by termination of the cycle at 4°C. The amplification products were separated on 3.5% superfine resolution (SFR) agarose gel. Electrophoresis was done at 100 V for 2–3 h.
Marker analysis
The program MAPMAKER ver. 2.0 (Lander et al. 1987) was used to establish linked marker groups and to create genetic map. A LOD score of 3.0 and a maximum recombination frequency of 0.40 were used to declare linkage between two markers. After linkage groups were determined, the recombination frequencies between marker loci were estimated by multipoint analysis.
Data analysis
Phenotypic data: The observations were recorded as the percentage of plants infected with SDM during both years. The percentage values ranged between 0 and 100. Analysis of variance was conducted on arcsine transformed phenotypic data (Little and Hills 1978) for individual environments using PROC GLM of SAS. Components of variance for the F3 families in both experiments were computed. Transformed entry means were used to compute the combined analyses of variance (Bohn et al. 1996). Estimates of variance components of F3 families were calculated as explained by Searle (1971). Broad sense heritability (H) on a F3 family was estimated as described by Hallauer and Miranda (1981).QTL mapping: The QTL analysis was carried out on the set of 185 F2:3 individuals with phenotypic data for SDM using PLABQTL (http://www.uni-hohenheim.de/~ipspwww/soft.html), a computer program to map QTL (ver. 1.2). The genotypic data consisted of 87 SSR marker loci, and the phenotypic data comprised SDM percentage incidence. The program employs the interval mapping approach (Lander and Botstein 1989). Commands were designed following MAPMAKER/QTL (Lincoln et al. 1993). In contrast to this and other programs, we used a multiple regression approach with flanking markers according to the procedure described by Haley and Knott (1992). The method of composite interval mapping (CIM; Zeng 1994) as implemented in QTL CARTOGRAPHER ver. 2.0 (Wang et al. 2004) was used to map QTLs and estimate their genetic effects. The best estimate of QTL location was assumed to correspond to the position having the peak significance level. Additive effects of the detected QTLs were also estimated by the PLABQTL procedure. The R 2 values obtained through this analysis indicate the percentage phenotypic variance explained by each QTL. The QTL positions identified in the present study were compared with previously reported ones (Agrama et al. 1999; George et al. 2003; Nair et al. 2005; Sabry et al. 2006). QTLs within a marker interval size of less than 20 cM (falling in the same bin) are considered to be common across the experiments.
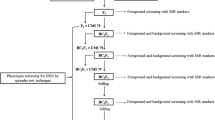
Marker-assisted introgression of QTLs implicated in resistance to SDM
To initiate marker-assisted selection (MAS), eight susceptible inbred lines were selected. These inbreds are parents of many experimental hybrids with high grain-yield potential. The inbreds CML212 and CML153 have high overall general combining ability. The flanking markers of two QTLs on chromosomes 3 and 6 were screened on this set of eight susceptible inbred lines and the resistant inbred MAI105 for initiating marker-assisted backcross breeding. During rainy season of 2012, the SDM-resistant parent MAI105 was crossed to eight susceptible inbred lines and the F1s were raised during postrainy season, 2012–2013. These F1s were backcrossed to susceptible parents. The first backcross generation (BC1F1) was planted during summer 2013 (table 1). We have screened 70–100 plants from all eight BC1F1 populations using flanking markers linked to QTLs on chromosomes 3 and 6. In total 13, 18, 15, 11, 17, 14, 9 and 24 plants were selected. The selected plants were selfed and BC1F2 plants were screened for SDM incidence during rainy season of 2013 and resistant plants were selfed to raise BC1F3 progenies during winter 2013 in the sorghum downy mildew screening nursery. Three-hundred BC1F3 progenies from the cross (CM500-19 × MAI105) × CM500-19 were evaluated for their response to SDM when both or either of QTLs from chromosomes 3 and 6 were present.
Results
Parental polymorphism
Of the 378 SSR primer pairs used, 108 were polymorphic between the parental lines. These SSRs were tried on entire F2 population and 87 fit 1 : 2 : 1 Mendelian segregation ratio which were then used for linkage analysis. Thirteen markers showed deviation from 1 : 2 : 1 ratio and eight failed to show polymorphism. Hence, they were not used for linkage mapping and QTL analysis.
Linkage mapping
The final map generated using 87 SSR markers had 10 linkage groups, spanning a length of 1210.3 cM at an average marker interval of 31.0 cM. Although, we used only 87 SSR markers for mapping, the per cent of genome within 20 cM to the nearest marker was 88.5. The linkage map obtained in the present study was in agreement with published SSR maps (http://www.maizegdb.org/maizedb.php).
Phenotypic data analysis and heritability estimates
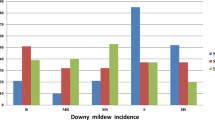
The parental lines differed significantly in their reaction to SDM disease incidence. The F1 was also found to be susceptible. The genotypic component of variance was highly significant for infection in both environments and over environments. This is indicative of the presence of relatively high genetic variance for resistance to SDM in the population (table 2). The genotype × environment interaction was also significant. Broad-sense heritability (H) values were estimated to be 67.13% and 66.31% during 2010 and 2011, respectively. The F3 families were not normally distributed with respect to their responses to SDM. The distribution for SDM was skewed towards susceptibility. The distribution was made near normal to make the means and variances independent and normally distributed using the arcsine transformation.
QTL mapping
Three QTLs for SDM resistance were discovered in the mapping population by marker regression analysis (table 3). These QTLs were located on chromosomes 2 (bin 2.02), 3 (bin 3.01) and 6 (bin 6.01) (figures 1 and 2). Additive effects were significant for all the QTLs detected. The QTL for SDM resistance detected on chromosome 3 had the largest effect, with the MAI105 allele at this locus decreasing the percentage of disease incidence by 12.6%. The QTL on chromosome 6 contributed 7.1% to phenotypic variance in SDM, while the one on chromosome 2 contributed 2%.

Linkage map showing the chromosomes 2, 3 and 6. The solid mark indicates the marker intervals with QTLs. The numbers to the left of the chromosomes indicate the distance in cM relative to the first marker. The SSR marker names are given to the right of each chromosome.

QTL likelihood maps indicating LOD scores for SDM incidence on chromosomes 3 and 6.
Marker-assisted introgression of QTLs in eight susceptible inbreds
The BC1F3 progenies recorded resistance to moderate resistance reaction confirming the presence of resistant QTLs in the selected plants. The progenies with QTL on chromosome 3 alone recorded moderate resistant reaction (0–25% disease incidence) and with both QTLs recorded resistant reaction. The progenies with QTL on chromosome 6 were found to be moderately susceptible (table 4). These QTLs segregated as nine (no QTLs); three (QTL from chromosome 3); three (QTL from chromosome 6) and one (QTLs from chromosome 3 and 6) in the BC1F3 population generated from the cross (CM500-19 × MAI105) × CM500-19. The resistant progenies from eight BC1F3 populations will be advanced to develop SDM-resistant inbreds with better combining ability.
Discussion
SDM disease cause severe yield losses in maize despite the use of chemical control measures. Host resistance is more effective in the SDM management and resistance has been reported to be under polygenic control necessitating identification of resistant QTLs. In this study, resistant inbred MAI105 was crossed with susceptible line CM500-19 and the F3 progenies were screened for response to SDM. There was absence of a normal distribution in the phenotypic values of the mapping population and this kind of absence of normal distribution is frequent, particularly for diseases such as SDM. Agrama et al. (1999), George et al. (2003), Nair et al. (2005) and Sabry et al. (2006) also reported the absence of a normal distribution in phenotypic data, with the distribution also skewed towards the susceptible parent. Heritability values were high and differences in heritability values measured in the different seasons can be attributed to differences in disease pressure. High heritability for SDM was reported earlier (Singburaudom and Renfro 1982; Sabry et al. 2006). The genetic map was constructed using 87 SSRs spanning a length of 1210.3 cM. Although, 108 SSRs were polymorphic between parents, eight produced monomorphism in the mapping population and the possible reason for this is that these alleles might have been fixed due to sampling error. Thirteen markers showed deviation from expected Mendelian segregation ratios of molecular markers and this kind of distortions have been reported in maize (Bentolila et al. 1992; Gardiner et al. 1993; Murigneux et al. 1993; Pereira and Lee 1995), as well as in many other plant species. These distortions may result from a selection process during gametogenesis, fertilization or germination (Lyttle 1991). The region on chromosome 3 (bin 3.01) was associated with downy mildew resistance in both test environments suggests a presence of major resistance gene complex. This region from the resistant parent contributes resistance to SDM that affect maize. It is interesting to note that Nair et al. (2005) detected a QTL on the same chromosome (bins 3.04–3.05) that contributed resistance to SDM in a recombinant-inbred population derived from NAI116 (resistant) × CML139 (susceptible). But the percentage phenotypic variance explained by this QTL was lower in the present study than in that of Nair et al. (2005) in which a different resistant parent was investigated. Second major QTL was detected in the bin 6.01, whereas George et al. (2003) and Nair et al. (2005) identified the QTL in the bin 6.05. The use of CIM also revealed one more QTL on chromosome 2 (bin 2.02) with less effect. The fact that George et al. (2003), Nair et al. (2005) and Sabry et al. (2006) also found a QTL for downy mildew resistance on the same chromosome but in different bins. Although our population and populations developed by George et al. (2003) and Nair et al. (2005) have parents from the Indian germplasm, the presence of QTLs in different bins might be due to the fact that our population had fewer recombination events (Balint-Kurti et al. 2008; Zwonitzer et al. 2010; Chung et al. 2011). It is also interesting to note that bin 3.01 has QTL influencing resistance to southern leaf blight (Jiang et al. 1999). An array of resistance genes are present on bin 6.01, including the gene mdm1 which confers resistance to the maize dwarf mosaic virus (MDMV) (Simcox et al. 1995); wsm1, which confers resistance to a potyvirus, wheat streak mosaic virus (WSMV) (McMullen and Louie 1991); rhm1, which confers resistance to the fungal pathogen Cochlilobus heterostrophus (Zaitlin et al. 1993); a QTL for resistance to sugarcane mosaic virus (SCMV) (de Souza et al. 2008). The bin 2.02 also colocalized QTLs for resistance to gray leaf spot and northern leaf blight (Zwonitzer et al. 2010). Clustering of genes and QTLs for resistance against diseases and pests appears to be a widespread phenomenon in maize, being reported in all 10 chromosomes (McMullen and Simcox 1995). The QTLs, one located on chromosome 3 (bin 3.01) near the marker umc2255 and the other on chromosome 6 (bin 6.01) linked to the marker phi077, were constant in both environments. The estimated effects of QTL in the present study are inconsistent with earlier reports. Various reasons for the inconsistency of estimated QTL effects are (i) different QTL segregating in different mapping populations, (ii) QTL × genetic background interaction, (iii) QTL × environment interaction and (iv) the Beavis effect (Beavis 1994; Xu 2003). In a simulation study, Beavis (1994) showed that the average estimates of phenotypic variances associated with QTLs were greatly overestimated if the size of the population is less and fairly close to the actual magnitude when large progenies were evaluated.
MAS along with phenotypic selection for polygenically-controlled SDM disease resistance would not only be cost-effective and time-effective but could also aid in recovery of a large proportion of the recurrent parent genome along with favourable alleles from the donor parent with significantly reduced linkage drag (Ribaut and Bertran 1999). The major QTL on chromosome 3 is important as the BC1F3 progenies with this QTL exhibited moderate resistance to resistance reaction either alone or in combination with the QTL on chromosome 6. Validation of major QTLs on chromosome 3 (3.01) and 6 (bin 6.01) has considerable significance in the implementation of MAS for the transfer of resistance to SDM in elite, but DM-susceptible, maize germplasm. Hence, we have attempted MAS programme that lead to incorporation of SDM resistance in the susceptible inbreds CML132, CML212, CML153, CML169, CML439, CML335-B-B-#, CML326 and CML304. In the same line, molecular markers were successfully utilized in line conversions through a backcross approach in maize at CIMMYT for the introgression of the opaque2 (o2) gene on chromosome 7 for the development of QPM lines, favourable QTL for earliness and grain yield (Bouchez et al. 2002) and transfer of a major QTL identified on the short arm of chromosome 1 that is associated with maize streak virus resistance (Prasanna and Hoisington 2003). The CIMMYT also conducted several experiments on QTL analysis and MAS for transfer of drought tolerance to tropical maize, and obtained encouraging results (Ribaut et al. 2002; Ribaut and Ragot 2007). Despite a wealth of published literature on QTL mapping, particularly in recent years, successful examples of effective utilization of QTL information in maize breeding through MAS are limited.
References
Agrama H. A., Moussa M. E., Naser M. E., Tarek M. A. and Ibrahim A. H. 1999 Mapping of QTL for downy mildew resistance in maize. Theor. Appl. Genet. 99, 519–523.
Balint-Kurti P. J., Wisser R. and Zwonitzer J. C. 2008 Use of an advanced intercross line population for precise mapping of quantitative trait loci for gray leaf spot resistance in maize. Crop Sci. 48, 1696–1704.
Beavis W. D. 1994 The power and deceit of QTL experiments: lessons from comparative QTL studies. In Proceedings of the 49th Annual Corn Sorghum Research Conference, American Seed Trade Association, pp. 250–266. Chicago, Washington, USA.
Bentolila S., Hardy T. and Guitton C. 1992 Comparative genetic analysis of F2 plants and anther culture derived plants of maize. Genome 35, 575–582.
Bohn M., Khairallah M. M., Jiang C., Gonzalez-De-Leon D., Hoisington D. A., Utz H. F. et al. 1996 QTL mapping in tropical maize: I. Genomic regions affecting leaf feeding resistance to sugarcane borer, other traits. Crop Sci. 36, 1352–1361.
Borges F. and Orange L. 1987 Diallel analysis of maize resistance to sorghum downy mildew. Crop Sci. 27, 178–180.
Bouchez A., Hospital F., Causse M., Gallais A. and Charcosset A. 2002 Marker assisted introgression of favorable alleles at quantitative trait loci between maize elite lines. Genetics 162, 1945–1959.
Chung C. L., Poland J., Kump K., Benson J., Longfellow J., Walsh E. et al. 2011 Targeted discovery of quantitative trait loci for resistance to northern leaf blight and other diseases of maize. Theor. Appl. Genet. 123, 307–326.
Craig J. and Odvody G. N. 1992 Current status of sorghum downy mildew control. In Sorghum and millets diseases: a second world review (ed. W. A. J. Milliano, R. A. Frederiksen and G. D. Bengston), pp. 213–217. ICRISAT, Patancheru, India.
Craig J., Bockholt A. J., Frederiksen R. A. and Zuber M. S. 1977 Reaction of important corn inbred lines to Sclerospora sorghi. Plant Dis. Rep. 61, 563–564.
de Souza I. R. P., Schuelter A. R., Guimaraes C. T., Schuster I, de Oliveira E. and Redinbaugh M. 2008 Clustering of QTL conferring SCMV resistance in tropical maize. Hereditas 145, 167–173.
Frederiksen R. A. and Renfro B. L. 1977 Global status of maize downy mildew. Ann. Rev. Phytopathol. 15, 249–275.
Gardiner J., Coe E. H., Melia-Hancock S., Hoisington D. A. and Chao S. 1993 Development of a core RFLP map in maize using an immortalized-F2 population. Genetics 134, 917–930.
Geetha K. and Jayaraman N. 2002 Inheritance of sorghum downy resistance in maize. Ind. J. Agric. Res. 36, 234–240.
George M. L., Prasanna B. M., Rathore R. S., Setty T. A., Kasim F., Azrai M et al. 2003 Identification of QTLs conferring resistance to downy mildews of maize in Asia. Theor. Appl. Genet. 107, 544–551.
Haley C. S. and Knott S. A. 1992 A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69, 315–324.
Hallauer A. R. and Miranda J. B. 1981 Quantitative genetics in maize breeding. Iowa State University Press, Ames, USA.
Hoisington D., Khairallah M. and Gonzalez-de-Leon D. 1994 Laboratory protocols: CIMMYT applied molecular genetics laboratory, 2nd edition. CIMMYT, Mexico.
Isakeit T., Odvody G., Jahn R. and Decanini L. 2003 Peronosclerospora sorghi resistant to metalaxyl treatment of sorghum seed in Texas. Phytopathology 93, S39.
Jiang C., Edmeades G. O., Armstead I., Lafitte H. R., Hayward M. D. and Hoisington D. 1999 Genetic analysis of adaptation differences between highland and lowland tropical maize using molecular markers. Theor. Appl. Genet. 99, 1106– 1119.
Krishnappa M., Naidu B. S. and Seetharam A. 1995 Inheritance of host resistance to downy mildew in maize. Crop Improv. 22, 33–37.
Lander E. S. and Botstein D. 1989 Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121, 185–199.
Lander E. S., Green P., Abrahamson J., Barlow A., Daly M. J., Lincoln S. A. and Newburg L. 1987 Mapmaker: an interactive computer package for constructing primary linkage maps of experimental and natural populations. Genomics 1, 174–181.
Lincoln S. E., Daly M. J. and Lander E. S. 1993 Mapping genes controlling quantitative traits using MAPMAKER/QTL version 1.1: a tutorial and reference manual. Whitehead Inst., Cambridge, USA.
Little T. M. and Hills F. J. 1978 Agricultural experimentation design and analysis. Wiley, New York, USA.
Lyttle T. W. 1991 Segregation distorters. Annu. Rev. Genet. 25, 511–557.
McMullen M. D. and Louie R. 1991 Identification of a gene for resistance to wheat streak mosaic virus in maize. Phytopathology 81, 624–627.
McMullen M. D. and Simcox K. D. 1995 Genomic organization of disease and insect resistance genes in maize. Mol. Plant Microbe Interact. 8, 811–815.
Murigneux A., Baud S. and Beckert M. 1993 Molecular and morphological evaluation of doubled-haploid lines in maize. 2. Comparison with single-seed-descent lines. Theor. Appl. Genet. 87, 278–287.
Nair S. K., Prasanna B. M., Garg A., Rathore R. S., Setty T. A. S. and Sing N. N. 2005 Identification and validation of QTLs conferring resistance to sorghum downy mildew (Peronosclerospora sorghi) and Rajasthan downy mildew (P. heteropogoni) in maize. Theor. Appl. Genet. 110, 1384–1392.
Nallathambi P., Sundaram K. M. and Arumugachamy S. 2010 Inheritance of resistance to sorghum dowmy mildew (Peronosclerospora sorghi) in maize (Zea mays L.). Int. J. Agric. Environ. Biotech. 3, 285–293.
Payak M. M. 1975 Downy mildews of maize in India. Trop. Agric. Res. 8, 13–18.
Pereira M. G. and Lee M. 1995 Identification of genomic regions affecting plant height in sorghum and maize. Theor. Appl. Genet. 90, 380–388.
Prasanna B. M. and Hoisington D. 2003 Molecular breeding for maize improvement: an overview. Ind. J. Biotech. 2, 85–98.
Rao B. M., Shetty S. H. and Safeeulla K. M. 1984 Production of Peronosclerospora sorghi oospores in maize seeds and further studies on the seed-borne nature of the fungus. Ind. Phytopathol. 37, 278–283.
Raymundo A. D. 2000 Downy mildew of maize in Asia: new perspectives in resistance breeding. In Proceedings of the 7th Asian Regional Maize Workshop (ed. S. K. Vasal, C. F. Gonzalez and F. Xingming), pp. 277–284. PCARRD, Los Banos, Philippines.
Ribaut J. M. and Bertran F. J. 1999 Single large-scale marker-assisted selection (SLS-MAS). Mol. Breed. 5, 531–541.
Ribaut J. M. and Ragot M. 2007 Marker-assisted selection to improve drought adaptation in maize: the backcross approach, perspectives, limitations, and alternatives. J. Exp. Bot. 58, 351–360.
Ribaut J. M., Banzinger M., Betran J., Jiang C., Edmeades G. O., Dreher K. and Hoisington D. 2002 Use of molecular markers in plant breeding: drought tolerance improvement in tropical maize. In Quantitative genetics, genomics, and plant breeding (ed. M. S. Kang), pp. 85–99. CABI Publishing, Wallingford, UK.
Sabry A., Jeffers D., Vasal S. K., Frederiksen R. and Magill C. 2006 A region of maize chromosome 2 affects response to downy mildew pathogens. Theor. Appl. Genet. 113, 321–330.
Saghai-Maroof M. A., Soliman K. M., Jorgensen R. A. and Allard R. W. 1984 Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location and population dymnamics. Proc. Natl. Acad. Sci. USA 81, 8014–8018.
Searle S. R. 1971 Linear models. Wiley, New York, USA.
Simcox K. D., McMullen M. D. and Louie R. 1995 Co-segregation of the maize dwarf mosaic virus resistance gene, mmdm1, with the nucleolus organizer region in maize. Theor. Appl. Genet. 90, 341–346.
Singburaudom N. and Renfro B. L. 1982 Heritability of resistance in maize to sorghum downy mildew (Peronosclerospora sorghi (Weston and Uppal) C.G. Shaw). Crop Protec. 1, 323– 332.
Sharma R. C., De-Leon C. and Payak M. M. 1993 Diseases of maize in south and south-east Asia: problems and progress. Crop Protec. 12, 414–422.
Wang S., Basten C. J., Gaffney P. and Zeng Z. B. 2004 Windows QTL cartographer 2.0. user manual. Bioinformatics Research Centre, North Carolina State University, Raleigh, USA.
Williams R. J. 1984 Downy mildews of tropical cereals. Adv. Plant Pathol. 2, 1–103.
Xu S. 2003 Theoretical basis of the Beavis effect. Genetics 165, 2259–2268.
Zaitlin D., DeMars S. and Ma Y. 1993 Linkage of rhm, a recessive gene for resistance to southern corn leaf blight, to RFLP marker loci in maize (Zea mays L.) seedlings. Genome 36, 555–564.
Zeng Z. B. 1994 Precision mapping of quantitative trait loci. Genetics 136, 1457–1468.
Zwonitzer J. C., Coles N. D., Krakowsky M. D., Arellano C., Holland J. B., McMullen M. D. et al. 2010 Mapping resistance quantitative trait loci for three foliar diseases in a maize recombinant inbred line population—evidence for multiple disease resistance? Phytopathology 100, 72–79.
Acknowledgements
The financial support for this research was provided by the Department of Biotechnology, Ministry of Science and Technology, Government of India. Special thanks to Dr H. C. Prasanna, Senior Scientist (Plant Breeding), IIVR, Varanasi, for his critical comments on this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Lohithaswa H. C., Jyothi K., Sunil Kumar K. R., Puttaramanaik and Hittalmani S. 2015 Identification and introgression of QTLs implicated in resistance to sorghum downy mildew (Peronosclerospora sorghi (Weston and Uppal) C. G. Shaw) in maize through marker-assisted selection. J. Genet. 94, xx–xx]
Rights and permissions
About this article

Cite this article
LOHITHASWA, H.C., JYOTHI, K., SUNIL KUMAR, K.R. et al. Identification and introgression of QTLs implicated in resistance to sorghum downy mildew (Peronosclerospora sorghi (Weston and Uppal) C. G. Shaw) in maize through marker-assisted selection. J Genet 94, 741–748 (2015). https://doi.org/10.1007/s12041-015-0590-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-015-0590-1