
Abstract
Chickpea (Cicer arietinum L.) is the second most important cool season food legume cultivated in arid and semiarid regions of the world. The objective of the present study was to study variation for protein content in chickpea germplasm, and to find markers associated with it. A set of 187 genotypes comprising both international and exotic collections, and representing both desi and kabuli types with protein content ranging from 13.25% to 26.77% was used. Twenty-three SSR markers representing all eight linkage groups (LG) amplifying 153 loci were used for the analysis. Population structure analysis identified three subpopulations, and corresponding Q values of principal components were used to take care of population structure in the analysis which was performed using general linear and mixed linear models. Marker-trait association (MTA) analysis identified nine significant associations representing four QTLs in the entire population. Subpopulation analyses identified ten significant MTAs representing five QTLs, four of which were common with that of the entire population. Two most significant QTLs linked with markers TR26.205 and CaM1068.195 were present on LG3 and LG5. Gene ontology search identified 29 candidate genes in the region of significant MTAs on LG3. The present study will be helpful in concentrating on LG3 and LG5 for identification of closely linked markers for protein content in chickpea and for their use in molecular breeding programme for nutritional quality improvement.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chickpea (Cicer arietinum L.) is a cool season grain legume and is the major source of protein in majority of the vegetarian diets. Its protein content is ∼17–22% (Jukanti et al. 2012), which increases considerably after dehulling, i.e. it ranges from 25.3–28.9%. Chickpea protein is considered best among all legume proteins, and it also has good in vitro protein digestibility (IVPD). In addition to protein, it also contains important nutrients such as carbohydrates, minerals, vitamins, fats, fibers, lipids, oils, etc. It also offers medicinal and health benefits. It is a hypocholesterolaemic agent and also has a role in the control of diabetes, blood pressure and weight loss. It also has a role against cardiovascular and digestive diseases, and for some type of cancers (for a review, see Jukanti et al. 2012). It shows antifungal, antibacterial, antiinflammatory and antiulcerative properties. Besides its use as the main source of protein, it is consumed in many forms (Burstin et al. 2011). Chickpea has been grown for its good protein content and good protein quality, which is very less as compared with other important legume like soybean, which has 40–43% protein (Burstin et al. 2011). Moreover, it is generally observed that popular chickpea varieties grown by the farmers have moderate protein content. This is due to the general belief that the varieties with higher protein content have small seed size.
Tremendous progress has been made in the area of chickpea genomics in the past five years, and this has resulted in the generation of few thousand molecular markers (Choudhary et al. 2009; Hiremath et al. 2012; Kujur et al. 2013), several linkage maps (Nayak et al. 2010; Thudi et al. 2011; for a review, see Varshney et al. 2013a), identification of marker-trait associations (MTAs) / quantitative trait loci (QTLs) (Varshney et al. 2013a; Thudi et al. 2014a) and the draft genome sequence of chickpea (Varshney et al. 2013b; Jain et al. 2013). With this vast wealth of genomic resources now available, majority of the breeding attempts made in chickpea have been, and are being, focussed on improving yield, resistance to diseases like Ascochyta blight and Fusarium wilt (Varshney et al. 2014a) and on resistance to various abiotic stresses (Varshney et al. 2013c, 2014b; for a review, see Jha et al. 2014; Thudi et al. 2014b). However, there has been no impact of traditional breeding strategies in the improvement of the nutritional status of the chickpea. There are very limited breeding attempts which focussed on improving protein content and other nutritional components in this crop with the help of molecular markers. This offers a lot of scope to study an important trait like protein content in chickpea, by identifying MTAs/QTLs using molecular markers for their ultimate use in the marker-assisted breeding programmes.
Ideally, a mapping population derived from two contrasting parents is used for the identification of QTLs for the trait of interest. However, this way of QTL identification is time-consuming and, moreover, the QTLs identified are relevant for those breeding programmes where either or both of the parental genotypes involved in crossing are used (Gupta et al. 2014). On the contrary, the approach of association mapping, which relies on the principle of linkage-disequilibrium (LD) can help in identifying QTLs in a rapid way from a set of germplasm/breeding lines, which is more relevant to the breeding point of view (Gupta et al. 2014). Use of a diverse set of germplasm has been suggested to be a good material for the identification of QTLs for different quality traits (Breseghello and Sorrells 2006). Therefore, the present study was aimed at identification of the markers associated with protein content in a diverse set of chickpea genotypes using simple sequence repeat (SSR) markers.
Material and methods
Material include 187 chickpea genotypes, comprised of both international and exotic collections, as well as promising genotypes of the university, and represents both desi and kabuli types, including three popular varieties namely Vijay, Vishal and Digvijay. The list of genotypes used in the present study is provide in table 1 in electronic supplementary material at http://www.ias.ac.in/jgenet/. These genotypes were provided by the Pulses Improvement Project, Mahatma Phule Agricultural University, Rahuri, and were grown during the year 2012–2013. Each accession was grown on a single row of three metres. Approximately 30 seeds per row were sown with 45 cm × 10 cm spacing. Recommended package of practices for chickpea were followed.
Estimation of crude protein content from chickpea seeds
Estimation of crude protein content was done using NIR SpectraAlyzer (ZEUTEC, Rendsburg, Germany). To calibrate the protocol of this system, the crude protein content of 30 chickpea genotypes were first estimated by Kjeldahl method using Kelplus-Classic Dx KES 06L (Pelican Equipments, Chennai, India). These 30 genotypes represented both desi and kabuli types, with different seed coat colour. Some of the samples with lower and higher crude protein content were repeated for confirmation.
Genomic DNA isolation and PCR amplification using SSR markers
Genomic DNA was isolated from young leaflets of chickpea genotypes, following Dellaporta et al. (1983) with some modifications. A set of 23 SSR markers were used in this study (table 1). Markers were chosen in such a way that they represent all the eight linkage groups of chickpea. PCR amplification was carried out in a 5 μL reaction volume in GeneAmp\(^{{\circledR }}\) PCR System 9700 Thermal Cycler (Applied Biosystems, Foster City, USA), following Thudi et al. (2011). PCR products were checked for amplification on 1.2% agarose gel containing 0.5 μL/10 mL ethidium bromide (10 mg/mL), with a 100 bp DNA ladder, by running it at a constant voltage of 90 V for 25 min. The amplification was visualized under UV illumination using Uvi-Tech gel documentation system (DOL-008.XD, England). Subsequently, PCR products generated by four different fluorescence dye-labelled primers were pooled in equal volumes, and 1.0 μL each of FAM, VIC, NED and PET-labelled product (amplicon) were mixed with 7 μL of formamide (Applied Biosystems), 0.05 μL of the GeneScanTM 500 LIZ ®; Size Standard (Applied Biosystems), and 2.95 μL of distilled water. DNA fragments (amplicons) were denatured and size fractioned using capillary electrophoresis on an ABI 3730 DNA Genetic Analyzer (Applied Biosystems) at the Centre for Excellence in Genomics, International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), Patancheru, India.
Population structure analysis
Population structure was assessed using a set of 13 unlinked SSR markers, having least number of missing marker data points, and were chosen from all the eight linkage groups (table 1). The software package STRUCTURE 2.3 (Pritchard et al. 2000) was used with admixture model and the number of subpopulations (K), as 1 through 20 with the length of burn-in period and numbers of MCMC after burn-in period as 100,000 each. Numbers of subpopulations were decided based on the ad hoc criteria as suggested by Evanno et al. (2005). Principal component analysis (PCA) was also performed using software TASSEL 4.0 (Bradbury et al. 2007), to take into account the effect of population structure.
Association analysis
Identification of MTA/QTLs was carried out following the approach of association analysis using the software package TASSEL 4.0 (Bradbury et al. 2007). Association analysis was performed using general linear model (GLM) and mixed linear model (MLM) approaches, as implemented in the software package TASSEL 4.0. The 23 SSR markers used in the present study amplified a total of 153 loci, with the minimum of two loci each by markers CaM0787, CaM0881, CaM2036, to a maximum of 18 for the marker CaM1068 (table 1). After removing the markers with minor allele frequency (MAF) less than 1%, the total number of marker loci retained for association analysis were 130. To take into account the population structure, principal components (PCs) were obtained using TASSEL 4.0 and the corresponding Q values were used as covariates in GLM and MLM analyses. For MLM analysis, marker-based kinship matrix (K) obtained using TASSEL was used along with the Q matrix, to correct both family and population structure, and the phenotypic variation explained (R 2) by the marker is reported. The optimum levels of compression in combination with P3D for variance component estimation were used as criteria in MLM. Initially, analysis was done using the entire set of genotypes, and later it was extended to the subpopulations as well. As population structure analysis indicated presence of three subpopulations, association analysis was carried out with corresponding Q values of first three principal components (1st component, PC1; first two components, PC2; first three components, PC3) in GLM and MLM. In summary, analysis was carried out excluding population structure (naive), with Q matrix, with K only matrix, and with Q+K matrix. An MTA was considered robust when it was significant following majority of the approaches used. For the sake of convenience, MTA on the same linkage group are considered to be linked to the same QTL.
Identification of genes in the important regions of QTL
To identify the potential candidate genes present in the regions where significant MTAs/QTLs have been identified, the markers CaM1212 and CaM1515 that were mapped on to chickpea physical map (Varshney et al. 2014c), were mapped on the chickpea genome (Varshney et al. 2013b), and the corresponding UniProt IDs were retrieved. For functional characterization of the genes, the UniProt IDs of the genes were mapped onto UniProt KB database (http://www.uniprot.org/).
Results
Protein content in chickpea genotypes
In this study a wide range for crude protein content was reported in chickpea genotypes, and it ranged from 13.25% (IC305653) to 26.77% (EC442034). This distribution in trait value showed the average crude protein content of 20.16%, while most of the genotypes were having protein content around 18%. The three popular varieties used in this study, namely, Vijay, Digvijay and Vishal recorded 22.08%, 24.89% and 23.33% crude protein, respectively.
Population structure assessment
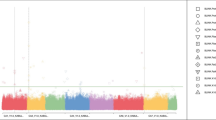
A set of 13 SSR markers representing all the eight linkage groups were used for the population structure assessment. Population structure analysis indicated presence of three subpopulations. PCA also separated the total genotypes into three components (figure 1). Majority of the indigenous genotypes were positioned to the left and towards the centre, while all the exotic collections (representing majority of the kabuli types) along with some indigenous types were positioned towards right. To rule out any confusion regarding number of subpopulations, association analysis was carried out using corresponding Q values of first three components.

Plot of PC1 drawn against PC2. Each circle (empty or filled) or plus represents an individual genotype in different subpopulations of the germplasm used for analysis.
Association analysis
When association analysis was carried out using GLM and MLM, by using different criteria, a total of 19 MTAs representing five QTLs having P<0.05 were detected in the entire population as well as in three subpopulations (tables 2 and 3). Three MTAs on LG1, two each on LG2, LG4 and LG5 and 10 on LG3 were detected for protein content in this study. Some of the associations detected in the entire population were common in individual subpopulations. Moreover, many of these associations may be linked with the same QTL, as some of them were present on the same linkage group. It is also likely that all of them were not true significant, as only few of them were actually significant following more than one approach, and also with MLM (tables 2 and 3). When the entire population was tested for association analysis, only two MTAs representing two QTLs associated with marker CaM1068.195 and TR26.205 for protein content were detected following all the criteria. These markers were mapped on LG5 and LG3, respectively (table 2). To identify the subpopulation specific QTLs, the analysis were also carried out in the three subpopulations, which were detected based on population structure study.
After studying the subpopulation 1 for association analysis, four putative MTAs were detected, three of which were on LG3 (table 3). Likewise, when subpopulation 2 was tested for association, six MTAs, three each on LG1 and LG3 associated with two QTLs were detected. This analysis was also extended to subpopulation 3. In this subpopulation, six MTAs associated with four putative QTLs were identified for crude protein content (table 3). Thus, a total of five QTLs were identified in subpopulations, four of which were common with those detected in the entire population. The amount of variation explained by these MTAs ranged from 2.4 to 5.1% in the entire population (table 2), while it ranged from 8.64 to 15.94% in the subpopulations (table 3).
Quantile-quantile (QQ) plots
Quantile-quantile (QQ) plot is a probability plot, which is a graphical method of comparing two probability distributions (observed versus expected), by plotting their quantiles against each other. The results obtained following various criteria of GLM and MLM are also explained using QQ plots (figure 2). It was observed that QQ plots obtained based on the results of MLM showed overfitting as compared to those obtained following GLM. The results obtained following GLM with first three principal components as covariates showed the expected distribution. This also supports the assumption ofpresence of three subpopulations in the present study.

QQ plots of the analysis for protein content in chickpea obtained by following various criteria: (a), naive (without population structure); (b), MLM with kinship only; (c), GLM with first PC taken as covariate; (d), MLM with first PC taken as covariate; (e), GLM with first two PCs taken as covariates; (f), MLM with first two PCs taken as covariates; (g), GLM with first three PCs taken as covariates; (h), MLM with first three PCs taken as covariates.
Candidate gene analysis in the important regions
The CaM series markers (CaM1122 and CaM1515) on LG3, which were found to be significant for protein content in the present study, were mapped on to the chickpea genome assembly, and an attempt was made to identify potential candidate genes in the region of these markers. The analysis has identified a total of 29 genes in this region (table 2 in electronic supplementary material). Further, based on Gene Ontology (GO) descriptions, these genes were functionally categorized as molecular function (17), cellular component (15) and biological process (14), some of which were common among each other (table 4).
Discussion
An effort has been made in this study to identify MTA for protein content in chickpea using a diverse set of genotypes. To our knowledge, this is the first such attempt to find the MTAs for protein content in chickpea, albeit with a small set of molecular markers. The wide range of protein content observed in the genotypes used in the present study justifies its use for association analysis. Use of diverse material from germplasm bank for the purpose of association mapping of quality traits has earlier been suggested (Breseghello and Sorrells 2006).
There was no definite relationship between the protein content and the type of chickpea (desi or kabuli), as well as between the colour of the seed and the protein content. This is against the general belief that kabuli types contain more protein compared to the desi types. For example, protein content in desi genotypes ranged from 13.26 to 26.06%, while in kabuli genotypes it ranged from 13.30 to 26.78%. These results are in agreement with those of Jukanti et al. (2012), who also did not find any consistency between the type of chickpea and protein content.
Population structure
Population structure was found to have little effect on the results of association analysis in the present study. The MTAs identified in the entire population following naive approach (without population structure), and those with various other criteria of GML considering population structure were almost the same. The only difference was with regard to marker H2B061.162, which was detected only through naive and not by other criteria. On the contrary, the MTAs on LG4 were detected by other criteria, and not following the naive (table 2). Moreover, the major QTLs linked with markers CaM1068.195 and TR26.205 were detected following all the criteria.
MTA analysis
In the present study, a total of 19 MTAs, representing five QTLs from five different LG were identified for protein content in the entire population as well as subpopulations of chickpea. Of the nine MTAs detected in the entire population, only six were found significant in the subpopulations with varying P values. Moreover, all these six were not common in all the subpopulations and one, two and three numbers of MTAs were common in subpopulations I (H2B061.162), II (TR56.253 and CaM1122.239) and III (CaM1068.195, TR26.205 and ICCM0249.142), respectively (tables 2 and 3). Association mapping is therefore sensitive in a way that change in genotype frequency arising due to change in the different sample sizes (in entire population and different subpopulations) may influence the power of detection of a QTL in the entire population. This is expected, as the P value tends to be low in the smaller set of genotypes. This is also evident from the smaller values of phenotypic variation explained by the MTAs explained in the entire population compared to the higher values explained by the MTAs identified in different subpopulations. Except for the marker CaM1515 in subpopulation I and marker GA11 in subpopulation II, other markers found significant in subpopulations were common with those found significant in the entire population (except for marker alleles). Generally, MLM is considered to be more robust as compared to GLM as chances of false positive associations tend to be more with GLM. However, the QQ plot showed that the results of MLM are overfitting and that of GLM and naive approach are more as expected. However, it should be noted that the approach of MLM may not always be rewarding, and the strategy of analysis may differ from trait to trait so that, for a given trait in a given set of genotypes, approach like naive or GLM may be more fruitful as has been reported by Zhao et al. (2011) for plant height in rice. It is, therefore, necessary that one should analyse the data using various criteria in association analysis, along with analysis in the subpopulations to find an appropriate way of identifying QTLs/MTAs in the given set of data (Gupta et al. 2014).
In our previous study using the same germplasm, it was observed that protein content is significantly positively correlated with 100-seed weight (R 2=0.28; significant at P<0.01) and two markers, one each on LG3 and LG5 were found to be significantly associated with 100-seed weight (unpublished results). In the present study, both these linkage groups were found to contain significant QTLs for protein content. Hundred-seed weight is the major component of yield in chickpea. Yield of the crop is always considered to be negatively correlated with its protein content. This has been considered as a limiting factor for simultaneous improvement in the protein content and yield in all the crops. However, positive correlation between protein content and the 100-seed weight as observed in the present study shows that simultaneous improvement for both the traits can be possible. Presence of significant QTLs for yield and other related traits on LG3 and LG5 have been reported in earlier studies in chickpea. For instance, Gowda et al. (2011) reported QTLs for yield in chickpea, which were associated with markers NCPGR52 and TA103x mapped on LG3 and LG5, respectively. Significant positive correlation of protein content has also been found with plant height (R 2=0.22; significant at P<0.01) in the present study. Gowda et al. (2011) identified a QTL on LG5 for plant height in chickpea. Similarly, Abbo et al. (2005) reported significant QTLs for beta carotene concentration in chickpea associated with markers TS19 and TR26. These markers were reported to be mapped on LG3 by Winter et al. (2000). These examples highlight the importance of LG3 and LG5 in chickpea.
Ideally, any genomewide association study should comprise of large numbers of markers and the genotypes. Although a small set of markers were used in the present study, care was taken so that they represent all the linkage groups. Moreover, the purpose of this study was to identify the potential MTAs in the given set of genotypes for a less studied trait like protein content and then focus on the important regions of the genome in future studies.
Candidate gene analysis
Of the 29 candidate genes identified based on GO annotation, gene-like protein-like malate synthase have been reported to be involved in carbohydrate metabolism pathway in soybean (Guex et al. 1995); whereas 6-phosphogluconate dehydrogenase is involved in carbohydrate degradation and pentose phosphate pathway, while SufE-like protein are involved in cofactor and iron–sulphur cluster biosynthesis, and it has been reported that its deficiency results in abnormal seed development and arrested embryo development in Arabidopsis (Xu and Moller 2006). These genes need to be studied further for their direct or indirect involvement in regulation of seed protein.
Conclusions
The present study has helped in identification of significant MTAs on LG3 and LG5, which have also been reported to contain important QTLs for various traits in different studies. This shows that LG3 and LG5 are potential candidate chromosomes for emphasizing future studies, with inclusion of additional markers from these chromosomes. Efforts are underway to develop biparental mapping populations using the genotypes having very low and high protein content, which can be used for high density mapping of the QTLs in future. This study also shows that association analysis can be effectively used to identify MTAs/QTLs for a trait-like protein content using a diverse set of genotypes even with a small set of markers. The significant markers identified in the present study can be used in marker-assisted breeding programme after validation.
References
Abbo S., Molina C., Jungmann R., Grusak M., Berkovitch Z., Reifen R. et al. 2005 Quantitative trait loci governing carotenoid concentration and weight in seeds of chickpea (Cicerarietinum L.) Theor. Appl. Genet. 111, 185–195.
Bradbury P. J., Zhang Z., Kroon D. E., Casstevens T. M., Ramdoss Y. and Buckler E. S. 2007 TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635.
Breseghello F. and Sorrells M. 2006 Association analysis as a strategy for improvement of quantitative traits in plants. Crop Sci. 46, 1323–1330.
Burstin J., Gallardo K., Mir R., Varshney R. and Duc G. 2011 Improving protein content and nutrition quality. In Biology and breeding of food legumes, (ed. A. Pratap and J. Kumar), pp. 314–328. CABI, Cambridge, USA.
Choudhary S., Sethy N. K., Shokeen B. and Bhatia S. 2009 Development of chickpea EST-SSR markers and analysis of allelic variation across related species. Theor. Appl. Genet. 118, 591–608.
Dellaporta S., Wood J. and Hick J. 1983 A plant DNA mini preparation. Plant Mol. Biol. Rep. 13, 19–21.
Evanno G., Regnaut S. and Goudet J. 2005 Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620.
Gowda S., Radhika P., Mhase L., Jamadagni B., Gupta V. and Kadoo N. 2011Mapping of QTLs governing agronomic and yield traits in chickpea. J. Appl. Genet. 52, 9–21.
Guex N., Henry H., Flach J., Richter H. and Widmer F. 1995 Glyoxysomal malate dehydrogenase and malate synthase from soybean cotyledons (Glycine max L.) enzyme association, antibody production and cDNA cloning. Planta 197, 369–375.
Gupta P. K., Kulwal P. L. and Jaiswal V. 2014 Association mapping in crop plants: Opportunities and challenges. Adv. Genet. 85, 109–147.
Hiremath P. J., Kumar A., Penmetsa R. V., Farmer A., Schlueter J. A., Chamarthi S. K. et al. 2012 Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol. J. 10, 716–732.
Jain M., Misra G., Patel R. K., Priya P., Jhanwar S., Khan A. W. et al. 2013 A draft genome sequence of the pulse crop chickpea (Cicerarietinum L.). Plant J. 74, 715–729.
Jha U. C., Chaturvedi S. K., Bohra A., Basu P. S., Khan M. S. and Barh D. 2014 Abiotic stresses, constraints and improvement strategies in chickpea. Plant Breed. 133, 163–178.
Jukanti A., Guar P., Gowda C. and Chibbar R. 2012 Nutritional quality and health benefits of chickpea. Br. J. Nutr. 108, S11– S26.
Kujur A., Bajaj D., Saxena M. S., Tripathi S., Upadhyaya H. D., Gowda C. L. et al. 2013 Functionally relevant microsatellite markers from chickpea transcription factor genes for efficientgenotyping applications and trait association mapping. DNA Res. 20, 355–374.
Nayak S. N., Zhu H., Varghese N., Datta S., Choi H. K., Horres R. et al. 2010 Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor. Appl. Genet. 120, 1415–1441.
Pritchard J., Stephens M. and Donnelly P. 2000 Inference of population structure using multi-locus genotype data. Genetics 155, 945–959.
Thudi M., Bohra A., Nayak S., Varghese N., Shah T., Penmetsa R. et al. 2011 Novel SSR markers from BAC-end sequences, DArT arrays and a comprehensive genetic map with 1,291 marker loci for chickpea (Cicerarietinum L.) PLoS One 6, e27275.
Thudi M., Upadhyaya H. D., Rathore A., Gaur P. M., Krishnamurthy L., Rookiwal M. et al. 2014a Genetic dissection of drought and heat tolerance in chickpea throughgenome-wide and candidate gene-based association mapping approaches. PLoS One 9, e96758.
Thudi M., Gaur P. M., Krishnamurthy L., Mir R. R., Kudapa H., Fikre A. et al. 2014b Genomics-assisted breeding for drought tolerance: A dream come true in chickpea. Funt. Plant Biol. (doi: 10.1071/FP13318).
Varshney R. K., Murali Mohan S., Gaur P. M., Gangarao N. V. P. R., Pandey M. K. et al. 2013a Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotech. Adv. 31, 1120–1134.
Varshney R., Song C., Saxena R., Azam S., Yu S., Sharpe A. et al. 2013b Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246.
Varshney R. K., Gaur P. M., Chamarthi S. K., Krishnamurthy L., Tripathi S., Kashiwagi J. et al. 2013c Fast-track introgression of “QTL-hotspot” for root traits and other drought tolerance traits in JG 11, an elite and leading variety of chickpea. Plant Genome (doi: 10.3835/plantgenome2013.07.0022).
Varshney R. K., Mohan S. M., Gaur P. M., Chamarthi S. K., Singh V. K., Srinivasan S. et al. 2014a Marker-assisted backcrossing to introgress resistance to fusarium wilt race 1 and ascochyta blightin C 214, an elite cultivar of chickpea. Plant Genome 7 (doi: 10.3835/plantgenome2013.10.0035).
Varshney R. K., Thudi M., Nayak S. N., Gaur P. M., Kashiwagi J., Krishnamurthy L. et al. 2014b Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.) Theor. Appl. Genet. 127, 445–462.
Varshney R. K., Mir R. R., Bhatia S., Thudi M., Hu Y., Azam S. et al. 2014c Integrated physical, genetic and genome map of chickpea (Cicer arietinum L.) Funct Integr. Genomics 14, 59–73.
Winter P., Benko-Iseppon A. M., Huttel B., Ratnaparkhe M., Tullu A., Sonnante G. et al. 2000 A linkage map of the chickpea (Cicer arietinum L.) genome, based on recombinant inbred lines from a C. arietinum × C. reticulatum cross: Localization of resistance genes for Fusarium wilt races 4 and 5. Theor. Appl. Genet. 101, 1155–1163.
Xu X. M. and Moller S. G. 2006 AtSufE is an essential activator of plastidic and mitochondrial desulfurases in Arabidopsis. EMBO J. 25, 900–909.
Zhao K., Tung C. W., Eizenga G. C., Wright M. H., Ali M. L., Price A. H. et al. 2011 Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2, 467.
Acknowledgements
Thanks are due to the Head, Department of Agricultural Process Engineering and Forage Breeding Scheme, MPAU, Rahuri, for providing the facilities for estimation of protein content. Thanks are also due to Aamir W. Khan, ICRISAT, for useful discussion on identifying candidate genes in the QTL region.
Author information
Authors and Affiliations
Corresponding author
Additional information
[Jadhav A. A., Rayate S. J., Mhase L. B., Thudi M., Chitikineni A., Harer P. N., Jadhav A. S., Varshney R. K. and Kulwal P. L. 2015 Marker-trait association study for protein content in chickpea (Cicer arietinum L.). J. Genet. 94, xx–xx]
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
JADHAV, A.A., RAYATE, S.J., MHASE, L.B. et al. Marker-trait association study for protein content in chickpea (Cicer arietinum L.). J Genet 94, 279–286 (2015). https://doi.org/10.1007/s12041-015-0529-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-015-0529-6