
Abstract
Quantitative trait locus (QTL) mapping is an important method in marker-assisted selection breeding. Many studies on the QTLs focus on cotton fibre yield and quality; however, most are conducted at the DNA level, which may reveal null QTLs. Hence, QTL mapping based on transcriptome maps at the cDNA level is often more reliable. In this study, an interspecific transcriptome map of allotetraploid cotton was developed based on an F2 population (Emian22 × 3-79) by amplifying cDNA using EST-SSRs. The map was constructed using cDNA obtained from developing fibres at five days post anthesis (DPA). A total of 1270 EST-SSRs were screened for polymorphisms between the mapping parents. The resulting transcriptome linkage map contained 242 markers that were distributed in 32 linkage groups (26 chromosomes). The full length of this map is 1938.72 cM with a mean marker distance of 8.01 cM. The functions of some ESTs have been annotated by exploring homologous sequences. Some markers were related to the differentiation and elongation of cotton fibre, while most were related to the basic metabolism. This study demonstrates that constructing a transcriptome linkage map by amplifying cDNAs using EST-SSRs is a simple and practical method as well as a powerful tool to map eQTLs for fibre quality and other traits in cotton.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cotton is one of the most important crops in the world, providing natural fibre used in many facets of daily life. The most extensively cultivated cotton species are the allotetraploid Gossypium hirsutum and G. barbadense, which account for ∼95 and 2% of total production worldwide (National Cotton Council, http://www.cotton.org). G. hirsutum and G. barbadense are known for their high yield and high fibre quality, respectively (Pang et al. 2012). The contrasting traits of two species make them ideal for studying the genetic basis of yield and fibre quality in cotton (Mei et al. 2004; Lacape et al. 2005, 2010; He et al. 2007; Yu et al. 2013). However, since yield and fibre quality in cotton are complex quantitative traits, studies on the genetic basis of these traits rely on genetic linkage maps.
The first interspecific linkage map was constructed with restriction fragment length polymorphism (RFLP) markers (Reinisch et al. 1994). However, a shortage of RFLP probes restricts worldwide use. Fortunately, the development of PCR-based markers, especially simple sequence repeats (SSRs), has increased rapidly. There are now 17,448 publicly available SSR markers in cotton (http://www.cottongen.org), which facilitate linkage map development and QTL mapping in cotton worldwide. EST-SSRs account for nearly half of all SSRs. Several high-density interspecific linkage maps (Rong et al. 2004; Yu et al. 2011; Zhao et al. 2012; Fang and Yu 2012) and intraspecific linkage maps have been developed in cotton. In addition, many QTL mapping studies for yield and fibre quality have been conducted with both interspecific (Mei et al. 2004; Lacape et al. 2005, 2010; He et al. 2007; Yu et al. 2013) and intraspecific populations (Ulloa et al. 2005; Zhang et al. 2005, 2009, 2012; Shen et al. 2006; Wang et al. 2006; Qin et al. 2008; Wu et al. 2009; He et al. 2011; Liu et al. 2012; Sun et al. 2012). These studies contribute much to the understanding of the complexity of genetic control of cotton yield and fibre quality.
Recently, the focus on QTL mapping has shifted to the transcriptional level in cotton. Liu et al. (2009) applied cDNA-amplified fragment length polymorphisms (AFLPs) to construct fibre transcriptome groups at the secondary cell wall (SCW) thickening stage, based on an interspecific backcross (BC1) of G. hirsutum ×G. barbadense. They mapped 78 transcript-derived fragments (TDFs) into eight transcriptome groups, and detected two significant QTLs, FS1 and FS2, which explained 16.08 and 15.87% of the fibre strength variance, respectively. Liu et al. (2011) constructed a transcriptome map based on cDNA-AFLPs, using an immortalized F2 (IF2) population of the cotton hybrid Xiangzamian 2 (G. hirsutum). A total of 302 TDFs were mapped onto 26 linkage groups, and 71 QTLs for yield and yield component traits were detected, based on four environments, with 13 QTLs identified in at least two environments. Claverie et al. (2012) used quantitative cDNA-AFLP to monitor variation in the expression level of cotton fibre elongation and secondary cell wall thickening transcripts in a population of interspecific G. hirsutum × G. barbadense recombinant inbred lines (RILs). Two-thirds of the normalized intensity ratios of TDFs were mapped between 1 and 6 eQTLs.
Genomewide transcriptome maps form the foundation of gene mapping and cloning as well as comparative genomics. Usually, transcriptome maps are constructed using cDNA-AFLPs since direct mapping of transcripts with the cDNA-AFLP technique is a rapid and practical method of direct mapping of expressed genes (Brugmans et al. 2002). Transcriptome maps have been successfully constructed in Arabidopsis and potato (Brugmans et al. 2002; Ritter et al. 2008) as well as in cotton (Liu et al. 2009, 2011; Claverie et al. 2012) by cDNA-AFLP. The cDNA-AFLP technique is high-throughput, however, the procedure is complex. cDNA-SRAP has been applied for the construction of a transcriptome map in B. oleracea (Li et al. 2003); this technique is similar to but simpler than cDNA-AFLP.
Compared to these two techniques, constructing transcriptome maps using EST-SSRs is also a practical method of directly mapping expressed genes. Although EST-SSRs are developed from ESTs, EST-SSRs are typically applied in DNA research as they are simpler and easier to work with. In fact, EST-SSRs can be directly used to amplify cDNA to construct SSR-based transcriptome linkage maps. There are many EST-SSRs in different plants, with more than 8000 EST-SSRs in cotton (http://www.cottongen.org). Thus, EST-SSRs are a valuable resource for constructing transcriptome maps. Generally, EST-SSRs were first applied in DNA-based genetic maps, implying that they have been mapped to chromosomes. When they are applied in transcriptome maps, the linkage groups can easily be assigned to corresponding chromosomes. However, most TDFs from cDNA-AFLPs and -SRAPs have not been mapped to chromosomes, since it is hard to assign them to chromosomes (Brugmans et al. 2002; Li et al. 2003; Ritter et al. 2008; Liu et al. 2009, 2011).
In this study, we utilized the cDNA samples obtained from developing fibres from an F2 population of Emian22 × 3-79 at five days post anthesis (DPA) as templates to explore the possibility that a transcriptome map could be constructed by amplifying cDNA using EST-SSRs. We have shown that this practical and simple method for constructing transcriptome linkage maps will facilitate eQTL mapping in cotton, which will help to analyse the genetic basis of economically desirable traits at the transcriptional level.
Materials and methods
The mapping parents were the G. hirsutum cultivar ‘Emian22’ and the G. barbadense accession ‘3-79’, which are described in the previous work with the BC1 mapping population (Yu et al. 2011). To construct a transcriptome linkage map, a new F2 population was developed. The F2 population (∼200 individuals) was planted in the experimental field of Huazhong Agriculture University, Wuhan, Hubei, China in 2009. However, only 69 plants were used to construct the map as it was possible to obtain enough samples for RNA extraction from these plants. In the previous study, it was found that the differences in developing fibres at five DPA were obvious between the mapping parents (Liu et al. 2013). Therefore, RNA was extracted from developing fibres at five DPA using the method described by Zhu et al. (2005). First-strand cDNA was synthesized using 3 μg of RNA from each sample, following the protocol provided with the Superscript®; III RT kit (Invitrogen, Carlsbad, USA).
Polymorphism screening
All the markers used in this study were obtained from the previously published BC1 linkage map (Yu et al. 2011), which have been mapped to chromosomes. Since the markers on the BC1 map included gSSRs and EST-SSRs, the EST-SSRs were directly used to screen for polymorphisms in the cDNA of developing fibres at five DPA; the sequences of the gSSRs were blasted against the cotton ESTs to identify SSRs derived from the transcribed sequences. Next, these SSRs were used to screen for polymorphisms.
PCR amplification and electrophoresis
Polymerase chain reaction (PCR) was conducted in 20 μL volume containing 25 ng DNA, 0.2 μmol L−1 forward primers, 0.2 μmol L−1 reverse primers, 1×buffer, 1.5 mmol L−1 MgCl2, 0.3 mmol L−1 dNTPs and 0.5 units of Taq polymerase. The PCR profile consisted of an initial denaturation at 94∘C for 2 min followed by 30 cycles of denaturation at 94∘C for 1 min, annealing at 55∘C for 1 min, and an extension at 72∘C for 1 min, with a final incubation at 72∘C for 10 min. First, the amplification products obtained with all of the primers were genotyped using the SSR analysis protocol on 6% denaturing polyacrylamide gels at room temperature (Lin et al. 2005). Next, the monomorphic PCR products were separated based on single-strand conformation polymorphisms (SSCPs) on 6% nondenaturing polyacrylamide gels at a constant 8 W for ∼16 h at 4∘C.
Transcriptome linkage map construction
After polymorphism screening, all the polymorphic markers were used to genotype the entire F2 population. If a primer detected multiple loci, letters (a, b, c...) were assigned to the loci according to the descending fragment size. The linkage map was constructed with JoinMap 3.0 (Stam 1993) using a logarithm of odds (LOD) threshold of 4.0 and a maximum recombination fraction of 0.4. Map distances were calculated in centi-Morgans (cM) using the Kosambi mapping function (Kosambi 1944). Linkage groups were assigned to the corresponding chromosomes using mapped SSRs (http://www.cottongen.org).
Functional annotation of SSR-ESTs
The SSR sequences were functionally annotated using Blast2GO (Conesa et al. 2005; Götz et al. 2008) with default parameters. First, blastx analysis was carried out with an E-value of 10−5 against the nr protein database in GenBank; and the sequences with >80% identity were retained. Next, GO-mapping, annotation analysis, InterPro Scan and InteProScan GOs were performed. Finally, GO-Slim (http://www.geneontology.org/GO.slims.shtml) was then carried out to acquire specific GO terms.
Results and discussion
Marker polymorphism
In this study, the mapping parents were the same as those used to construct the DNA-based linkage map (Yu et al. 2011). The EST-SSRs in the DNA-based linkage map were directly used to construct the transcriptome map. To map more markers, genomic SSRs in the map were blasted against the cotton ESTs to identify transcribed markers. Finally, 1270 transcript-derived SSRs were included in this study (table 1). The samples used for transcriptome map construction were cDNAs from developing fibres at five DPA, as it was confirmed that a great number of TDFs differed between the mapping parents at this stage (Liu et al. 2013). After polymorphism detection, only 303 primers (23.86%) showed polymorphism, 267 (21.02%) produced amplification products, but lacked polymorphism, and 700 (55.12%) did not amplify the target (table 1) The failed amplification is likely to be the reason as these SSRs could not target tissue-specific transcripts in developing fibres at five DPA.
Transcriptome linkage map construction
The cDNA samples obtained from the F2 population were genotyped with the 303 polymorphic primers; however, some primers generated ambiguous gel bands and were discarded in subsequent analyses. Subsequently, 244 primers produced clearly scorable gel bands with 279 polymorphic loci; among them, 58 (20.79%) were 3-79 dominant loci, 18 (6.45%) were Emian22 dominant, and 203 (72.76%) were codominant loci. Generally, SSRs are codominant. The corresponding genes of the codominant markers are all expressed in the parents. However, variations in the microsatellite region could occasionally cause alterations in the translated products, which could result in trait variations. The corresponding genes of dominant markers are only expressed in one parent, which could alter the gene network of the trait and result in trait variations.
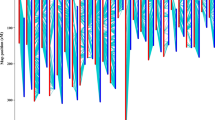
After linkage analyses, 242 loci were mapped into 32 linkage groups, with 37 remaining loci unmapped. The total length of the transcriptome linkage map was 1938.72 cM (table 2; figure 1). The longest linkage group was 132.74 cM with 15 loci (LG05/Chr05), while the shortest linkage group was 2.33 cM with two loci (LG31/Chr25); generally, the average length of a linkage group was 60.58 cM. The average marker interval distance was 8.01 cM, while it varied from 0.01 (LG16/Chr11) to 36.12 cM (LG11/Chr09). The AT genome included 18 linkage groups with a total of 114 loci; the total length was 1041.68 cM, and the average marker interval distance was 9.14 cM. Comparatively, the DT genome included 14 linkage groups with a total of 128 loci; the total length was 897.04 cM and the average marker interval distance was 7.01 cM. The genomewide distribution of EST-SSRs implied that the fibre development in cotton involves genes distributed throughout the entire genome.



Interspecific transcriptome linkage map of fibre development in cotton constructed using EST-SSRs.
We found that dominant markers were absent on Chr02, Chr04, Chr16, Chr17, Chr21 and Chr25 (table 3). Obviously, there were more chromosomes lacking dominant markers in the DT genome than in the AT genome; however, there were more dominant markers overall in the DT genome, indicating that the dominant markers were clustered on particular chromosomes of the DT genome, especially Chr15. The uneven distribution of EST-SSRs on the chromosomes implied that different chromosomes played different roles in developing fibres at five DPA. This result also provided evidence for the existence of tissue-specific transcriptomes.
This map is suitable for mapping eQTLs. Unfortunately, most of the cotton bolls were sampled for RNA extraction to construct the transcriptome map; thus, not enough bolls remained to phenotype the fibre quality. However, when transcriptome maps are constructed from permanent populations, it is feasible to map eQTL to understand the genetic basis of cotton fibre traits at the transcriptional level. We can also compare the eQTLs with previous QTLs based on DNA markers to uncover those reliable QTLs.
Functional annotation of SSR-ESTs
After blasting against the nr protein database, 53 of the 242 SSR-ESTs with >80% identity were functionally annotated, and they were classified into 10 categories (figure 2). Except for the 16% with unknown function, the top functions were transcription activity (15%) and transportation activity (14%). Some SSR-ESTs were involved in fibre differentiation (e.g. NAU3495), fibre initiation (e.g. NAU3496) and fibre elongation (e.g. MUSS250). These results were in accordance with the sampling phase (developing fibres at five DPA) as five DPA is the overlapping period of fuzz cell differentiation, lint initiation and elongation (Lee et al. 2007).

Functional annotation of mapped SSR-ESTs.
In conclusion, an EST-SSR-based interspecific transcriptome linkage map of fibre development in cotton was constructed. This innovative new tool for constructing transcriptome linkage maps can be easily used to assign linkage groups to chromosomes and facilitate eQTL mapping in plants.
References
Brugmans B., Fernandez del Carmen A., Bachem C. W., van Os H., van Eck H. J., and Visser R. G. 2002 A novel method for the construction of genomewide transcriptome maps. Plant J. 31, 211–222.
Claverie M., Souquet M., Jean J., Forestier-Chiron N., Lepitre V., Pré M., et al. 2012 cDNA-AFLP-based genetical genomics in cotton fibers. Theor. Appl. Genet. 124, 665–683.
Conesa A., Götz S., Garcia-Gomez J. M., Terol J., Talon M., and Robles M. 2005 Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676.
Fang D. D. and Yu J. Z. 2012 Addition of 455 microsatellite marker loci to the high-density Gossypium hirsutum TM-1 × G. barbadense 3-79 genetic map. J. Cotton Sci. 16, 229–248.
Götz S., García-Gómez J. M., Terol J., Williams T. D., Nueda M. J., Robles M., et al. 2008 High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435.
He D. H., Lin Z. X., Zhang X. L., Nie Y. C., Guo X. P., Zhang Y. X., and Li W. 2007 QTL mapping for economic traits based on a dense genetic map of cotton with PCR-based markers using the interspecific cross of Gossypium hirsutum × Gossypium barbadense. Euphytica 153, 181–197.
He X. H., Qin H., Hu Z., Zhang T., and Zhang Y. M. 2011 Mapping of epistatic quantitative trait loci in four-way crosses. Theor. Appl. Genet. 122, 33–48.
Kosambi D. D. 1944 The estimation of map distance from recombination values. Ann. Eugen. 12, 172–175.
Lacape J. M., Nguyen T. B., Courtois B., Belot J. L., Giband M., Gourlot J. P., et al. 2005 QTL analysis of cotton fiber quality using multiple Gossypium hirsutum × Gossypium barbadense backcross generations. Crop Sci. 45, 123–140.
Lacape J. M., Llewellyn D., Jacobs J., Arioli T., Becker D., Calhoun S., et al. 2010 Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum × G. barbadense RIL population. BMC Plant Biol. 10, 132.
Lee J. J., Woodward A. W., and Chen Z. J. 2007 Gene expression changes and early events in cotton fibre development. Ann. Bot. 100, 1391–1401.
Li G., Gao M., Yang B., and Quiros C. F. 2003 Gene for gene alignment between the Brassica and Arabidopsis genomes by direct transcriptome mapping. Theor. Appl. Genet. 107, 168–180.
Lin Z., He D., Zhang X., Nie Y., Guo X., Feng C., et al. 2005 Linkage map construction and mapping QTL for cotton fiber quality using SRAP, SSR and RAPD. Plant Breed. 124, 180–187.
Lin Z. X., Zhang Y. X., Zhang X. L., and Guo X. P. 2009 A high-density integrative linkage map for Gossypium hirsutum. Euphytica 166, 35–45.
Liu H. W., Wang X. F., Pan Y. X., Shi R. F., Zhang G. Y., and Ma Z. Y. 2009 Mining cotton fiber strength candidate genes based on transcriptome mapping. Chin. Sci. Bull. 54, 4651–4657.
Liu R., Wang B., Guo W., Wang L. and Zhang T. 2011 Differential gene expression and associated QTL mapping for cotton yield based on a cDNA-AFLP transcriptome map in an immortalized F2. Theor. Appl. Genet. 123, 439–454.
Liu R., Wang B., Guo W., Qin Y., Wang L., Zhang Y., and Zhang T. 2012 Quantitative trait loci mapping for yield and its components by using two immortalized populations of a heterotic hybrid in Gossypium hirsutum L. Mol. Breed. 29, 297–311.
Liu C., Yuan D., Zhang X., and Lin Z. 2013 Isolation, characterization and mapping of genes differentially expressed during fibre development between Gossypium hirsutum and G. barbadense by cDNA-SRAP. J. Genet. 92, 175–181.
Mei M., Syed N. H., Gao W., Thaxton P. M., Smith C. W., Stelly D. M., and Chen Z. J. 2004 Genetic mapping and QTL analysis of fiber-related traits in cotton (Gossypium). Theor. Appl. Genet. 108, 280–291.
Pang M., Percy R. G., Stewart J. Mc. D., Hughs E., and Zhang J. 2012 Comparative transcriptome analysis of Pima and Acala cotton during boll development using 454 pyrosequencing technology. Mol. Breed. 30, 1143–1153.
Qin H. D., Guo W. Z., Zhang Y. M., and Zhang T. Z. 2008 QTL mapping of yield and fiber traits based on a four-way cross population in Gossypium hirsutum L. Theor. Appl. Genet. 117, 883– 894.
Reinisch A. J., Dong J. M., Brubaker C. L., Stelly D. M., Wendel J. F., and Paterson A. H. 1994 A detailed RFLP map of cotton Gossypium hirsutum × Gossypium barbadense: chromosome organization and evolution in a disomic polyploid genome. Genetics 138, 829–847.
Ritter E., de Galarreta J. I. R., van Eck H. J., and Sánchez I. 2008 Construction of a potato transcriptome map based on the cDNA-AFLP technique. Theor. Appl. Genet. 116, 1003–1013.
Rong J. K., Abbey C., Bowers J. E., Brubaker C. L., Chang C., Peng W. C., et al. 2004 A 3347-locus genetic recombination map of sequence-tagged sites reveals features of genome organization, transmission and evolution of cotton (Gossypium). Genetics 166, 389–417.
Shen X. L., Zhang T. Z., Guo W. Z., Zhu X. F., and Zhang X. Y. 2006 Mapping fiber and yield QTLs with main, epistatic, and QTL × environment interaction effects in recombinant inbred lines of upland cotton. Crop Sci. 46, 61–66.
Stam P. 1993 Construction of integrated genetic linkage maps by means of a new computer package: join map. Plant J. 3, 739–744.
Sun F. D., Zhang J. H., Wang S. F., Gong W. K., Shi Y. Z., Liu A. Y., et al. 2012 QTL mapping for fiber quality traits across multiple generations and environments in upland cotton. Mol. Breed. 30, 569–582.
Ulloa M., Saha S., Jenkins J. N., Meredith W. R., McCarty J. C., and Stelly D. M. 2005 Chromosomal assignment of RFLP linkage groups harboring important QTLs on an intraspecific cotton (Gossypium hirsutum L.) joinmap. J. Hered. 96, 132–144.
Wang B. H., Guo W. Z., Zhu X. F., Wu Y. T., Huang N. T., and Zhang T. Z. 2006 QTL mapping of fiber quality in an elite hybrid derived-RIL population of upland cotton. Euphytica 152, 367–378.
Wu J., Gutierrez O. A., Jenkins J. N., McCarty J. C., and Zhu J. 2009 Quantitative analysis and QTL mapping for agronomic and fiber traits in an RI population of upland cotton. Euphytica 165, 231–245.
Yu Y., Yuan D. J., Liang S. G., Li X. M., Wang X. Q., Lin Z. X., et al. 2011 Genome structure of cotton revealed by a genome-wide SSR genetic map constructed from a BC1 population between Gossypium hirsutum and G. barbadense. BMC Genomics 12, 15.
Yu J., Zhang K., Li S., Yu S., Zhai H., Wu M., et al. 2013 Mapping quantitative trait loci for lint yield and fiber quality across environments in a Gossypium hirsutum × Gossypium barbadense backcross inbred line population. Theor. Appl. Genet. 126, 275–287.
Zhang Z. S., Xiao Y. H., Luo M., Li X. B., Luo X. Y., Hou L., et al. 2005 Construction of a genetic linkage map and QTL analysis of fiber-related traits in upland cotton (Gossypium hirsutum L.) Euphytica 144, 91–99.
Zhang Z. S., Hu M. C., Zhang J., Liu D. J., Zheng J., Zhang K., et al. 2009 Construction of a comprehensive PCR-based marker linkage map and QTL mapping for fiber quality traits in upland cotton (Gossypium hirsutum L). Mol. Breed. 24, 49–61.
Zhang K., Zhang J., Ma J., Tang S., Liu D., Teng Z., et al. 2012 Genetic mapping and quantitative trait locus analysis of fiber quality traits using a three-parent composite population in upland cotton (Gossypium hirsutum L.) Mol. Breed. 29, 335–348.
Zhao L., Lv Y., Cai C., Tong X., Chen X., Zhang W., et al. 2012 Toward allotetraploid cotton genome assembly: integration of a high-density molecular genetic linkage map with DNA sequence information. BMC Genomics 13, 539.
Zhu L. F., Tu L. L., Zhen F. C., Liu D. Q., and Zhang X. L. 2005 An improved simple protocol for isolation of high quality RNA from Gossypium spp. suitable for cDNA library construction. Acta Agromomica Sin. 31, 1657–1659.
Acknowledgement
This work was financially supported by the National Basic Research Program (no. 2011CB109303 and 2010CB126001).
Author information
Authors and Affiliations
Corresponding author
Additional information
[Liu C., Yuan D. and Lin Z. 2014 Construction of an EST-SSR-based interspecific transcriptome linkage map of fibre development in cotton. J. Genet. 93, xx–xx]
Rights and permissions
About this article

Cite this article
LIU, C., YUAN, D. & LIN, Z. Construction of an EST-SSR-based interspecific transcriptome linkage map of fibre development in cotton. J Genet 93, 689–697 (2014). https://doi.org/10.1007/s12041-014-0425-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-014-0425-5