
Abstract
Different reaction pathways of CO oxidation at the surface of trimetallic nanoclusters, Au(5-x-y)AgxCuy of varying compositions with 2 ≤ x + y ≤ 4 (x, y > 0) are investigated. The starting point of all pathways is the surface reaction between adsorbed CO and O2 via the Langmuir-Hinshelwood mechanism. The conventional reaction pathway leads to the formation of intermediate Au(5-x-y)AgxCuy.O* followed by a reaction with CO to release CO2. However, on these trimetallic clusters, the formation of the nanocluster-carbonate adduct is greatly facilitated, which is known to poison the catalytic surface because of its greater stability. Here, we propose a possible Eley-Rideal (ER) pathway leading to carbonate decomposition and catalytic surface regeneration. This ER-pathway involves interaction between nanocluster-carbonate adduct and CO in the gaseous state, forming Au(5-x-y)AgxCuy.O2COCO*, which decomposes to release CO2. The study suggests the best trimetallic clusters for CO oxidation are the ones where both reaction sites are Ag-sites.
Graphical abstract
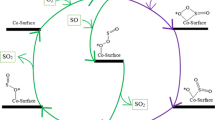
Different reaction pathways of CO oxidation at the surface of trimetallic nanoclusters, Au(5-x-y)AgxCuy, are investigated. These include the conventional LH-LH pathway and a newer alternative LH-ER pathway. The proposed LH-ER pathway involves an interaction between nanocluster-carbonate adduct and CO(g), forming Au(5-x-y)AgxCuy.O2COCO*, which decomposes to release CO2.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
With the continuous need to upgrade environment remediation technologies, significant focus has been on developing gold-nanocluster-based catalysts over the past several decades.1 CO is a toxic pollutant generated due to the incomplete oxidation of fossil fuels. The interest in gold catalysts for CO oxidation was generated following the report by Haruta et al., of exceptional reactivity of small nanosized gold catalysts (≈5 nm) dispersed on different transition metal oxides like α- Fe2O3 at temperatures as low as 200 K.2 Due to promising catalytic activity of gold nanoparticles at low temperatures, the use of gold-based catalysts in catalytic convertors installed in fossil fuel-powered vehicles can also provide a solution to cold-start emission problem. Cold-start emission is a known problem with conventional Pt and Pd-based catalytic converters.3 The gold nanoparticles are catalytically active only when particle size lies in the range between 1-5 nm; the bigger-sized particles or bulk gold are nonreactive.4,5,6 There are a large number of factors, such as charge effect of support, low coordination sites, strain, particle size, composition, and presence of quantum effect at the nano level that influence the catalytic activity of nanoparticles.7,8,9,10,11,12,13 As a result, a mechanistic investigation into catalytic activity of such systems requires a study from different perspectives.
A great contrast is observed in the catalytic properties of hydrogen chemisorbed silver clusters (AgnH) and pure silver clusters (Agn) for CO oxidation reaction, where it is found while the former is catalytically active, the latter is not.14 Addition of Na to Au20 nanocluster (NaAu20) results in two types of structures – tetrahedral and caged. Both of these structures are reported to have enhanced binding energies and substantially reduced activation energy barriers for CO oxidation.15 The studies on hydrogen-doped gold nanoclusters (Au(n-1)H) found the adsorption properties of the clusters to be the same as that of undoped gold clusters (Aun).16,17 These studies indicate the profound effect of the composition of nanoparticles on structural and thereby on their catalytic properties. Darby et al., report the structural difference between pure copper and pure gold nanoclusters. Further, replacing just one Au atom with Cu in a gold nanocluster significantly changes its structure.18
Nanoalloys as catalysts are interesting materials to explore as their catalytic capabilities can be fine-tuned with changes in the composition and position of dopants. In the case of Ag doping in Au13, an increase in co-adsorption energy and a decrease in the activation energy is reported due to enhanced electron density at shell atoms over the core.19 Zeng et al., in a systematic study of CO oxidation on Au18M (M = Ag, Cu, Pt, Pd) nanoclusters investigated in addition to conventional bi-molecular CO oxidation pathway, a tri-molecular reaction pathway in which co-adsorbed CO facilitates the dissociation of OCOO* intermediate.20 A similar but distinct alternative route for CO oxidation is also presented in the current investigation. Zeng et al., found doped clusters, especially with M = Ag, Cu, to be catalytically more active than parent gold nanocluster via a tri-molecular reaction mechanism. The nanocluster Au18M with M = Ag, Cu is found to be more reactive via the tri-molecular route due to the weaker d-π* back bonding in agreement with the Blyholder model.20 There are a number of studies that analyze the different aspects of catalytic activity of bi-/tri-metallic nanoclusters on the basis of local densities of states, electron localized function, and charge distribution to identify the catalytically most active composition of these nanoalloys.21,22,23,24,25,26 For example, dispersed nanoalloys of Au-Ag-Pd created by laser irradiation are investigated for catalytic activity, and these are found to be more effective catalysts for Heck coupling reactions as compared to monometallic or bimetallic nanoclusters.23 A uniquely designed Cu-Ag-Au trimetallic nano bowls having a cavity of few nanometers are found to exhibit maximum efficiency of 98.6% for reducing 4-Nitrophenol to 4-Aminophenol, which are reported to be 14, 23, and 43-fold more effective than the monometallic Au, Cu, and Ag systems, respectively.24. A comparative study of activation energy of rate-determining step of oxygen reduction reaction (ORR) reports trimetallic Pt-Co-Ni cluster as having higher catalytic performance over Pt containing bimetallic or pure Pt nanoclusters.25 The slightly weaker adsorption of reactant/intermediates on the surface of the catalyst is correlated with the better catalytic activity of these trimetallic nanoclusters. Kang et al., demonstrated the free valence electron centralization strategy to be an effective route for preparing ultrastable silver nanoclusters.26 The strategy involves stepwise preparation of nanoalloy of Ag with Cu and Au, which not only imparts higher thermal stability to these silver-based nanoclusters but is also shown to have improved catalytic performance. They reported a considerable reduction in reaction time from 5 h to 3 min upon employing trimetallic nanocluster Au1Ag16Cu12 as the catalyst for a multicomponent coupling reaction at high temperatures.26
In the current study, we investigate the effect of the composition of trimetallic Au-Ag-Cu nanocluster on activation energy barriers of CO oxidation reaction and also propose an alternative route for CO oxidation via decomposition of adsorbed carbonate species initiated by CO at the surface of these nanoclusters.
2 Computational details
All calculations are performed through Gaussian09 using density functional theory with PW91 exchange-correlation (xc) functional.27 For gold, silver, and copper atoms, relativistic effective core potential (ECP) LANL2TZ(f) is used, while for small atoms C and O 6-311+G(d) basis set is used. The LANL2TZ(f) is a LANL2 relativistic ECP of Hay and Wadt with a triple-zeta basis set supplemented with f polarization functions for treating the atom’s valence shell.28 The choice of ECP, basis set, and xc functional is justified and discussed by Saini et al.29 The bare nanoclusters are optimized for both doublet and quartet spin multiplicity, and nanoclusters with doublet multiplicity are found to be more stable. The geometries for various intermediates are also optimized for doublet spin multiplicity. No symmetry constraints are imposed in all calculations, and these calculations are spin unrestricted (UDFT). The structures are optimized using the keyword integral=ultrafine. The frequency analysis is carried out to verify the optimized geometries as minima or transition states. The intrinsic reaction coordinate (IRC) calculations are carried out to validate the corresponding transition state for a given set of reactants and products. All energies reported here include zero-point energy (ZPE) corrections.
3 Results and Discussion
3.1 Trimetallic clusters
The monometallic, bimetallic, and trimetallic nanoclusters have been optimized for different spin multiplicities. The clusters with doublet spin multiplicity are found to be the most stable. The binding energy per atom (B.E./atom) for these clusters is reported in Table 1 and compared with binding energy for monometallic clusters Au5, Ag5, Cu5, and bimetallic clusters Au3Ag2 and Au3Cu2. The binding energy is defined as the energy required to break the cluster into its constituent atoms. B.E. per atom is calculated using equation (A) given below. Here, E(Au5-x-yAgxCuy) is the energy of a cluster while E(M), M=Au, Ag, Cu is the energy of a single atom of Au/Ag/Cu in an unbound state.
As the silver atom is replaced by a copper atom in trimetallic cluster AuCuAg3, it is noted that B.E. per atom increases. A similar trend is observed as a gold atom in the trimetallic cluster AuCuAg3 replaces a silver atom. It can be interpreted from the table that, in general, replacement by Ag atom decreases B.E./atom while replacement by Cu increases B.E./atom. But in the case of trimetallic cluster Au3AgCu, the replacement of the gold atom by copper atom does not bring much change in B.E./atom for the cluster. Contrarily, B.E. per atom for Au2AgCu2 is more than that for AuAgCu3, albeit the increment is minor. This can be understood in terms of the strength of the bond between two atoms. The study of Table 2 shows that the strength of the Au-Cu bond is highest and least for the Ag-Ag bond. Through careful examination of the geometries of most stable trimetallic clusters (Figure 1), it is found that in Au2AgCu2, there are four Au-Cu bonds and one Ag-Cu bond, while in cluster AuAgCu3, the number of Au-Cu bonds reduces to two and there are two Ag-Cu bonds. This count explains why despite the replacement of Au by Cu in Au2AgCu2, the B.E. per atom for Au2AgCu2 (1.82 eV) is slightly greater than that for AuAgCu3 (1.81 eV). The count of different kinds of bonds in six trimetallic clusters is provided in the supplementary information (SI) Table S.1. The formation energy for these trimetallic clusters has also been calculated, and the results are detailed in SI (Table S.2). The formation energies for all six clusters are negative indicating the stability of these clusters.

Trimetallic nanoclusters of varying composition with important bond lengths (Å) and binding energy per atom (eV).
3.2 Reaction profile of CO oxidation on Au(5-x-y)AgxCuy clusters
Here, we discuss the reaction profile of CO oxidation at the surface of various trimetallic clusters with different compositions i.e., Au(5-x-y)AgxCuy where x, y varies between 1 to 3 with the restriction that 2 ≤ x + y ≤ 4. There are six such trimetallic clusters. CO oxidation catalysed by these clusters is a bimolecular surface reaction that can proceed either via the Langmuir-Hinshelwood mechanism (LH) or the Eley-Rideal mechanism (ER). In the present study, the initial point of all the reaction profiles is taken to be a cluster with CO and O2 adsorbed (LH mechanism) on adjacent three coordinated sites in top adsorption geometry. This very initial geometry is selected as we aimed to study the effect of the nature of the adsorption site (Au/Ag/Cu) on a given reaction profile, and secondly, the molecules adsorbed at these sites are oriented preferably for the reaction to proceed. The main steps involved in the CO oxidation on different compositions of trimetallic nanocluster considered in the current work are as follows.
The first step involves a surface reaction between adsorbed CO and O2, which is an activated process (Figure 2). In figures depicting the reaction profile, the chemical formula in the text implies that the species, e.g., CO, O2 and/or CO2, are infinitely separated from each other and from the cluster. Also, for all the transition states reported here, the coordinates, distance matrix, and a list of angles and dihedral angles are provided at the end of SI. Table 3 indicates the activation of O-O bond is maximum when O2 is adsorbed on a 3-coordinated Cu-site. Due to the least electronegativity of Cu among the three metals, the charge transfer is more from Cu to O2, leading to observed longest/weaker O-O bond among all six trimetallic clusters, which can account for the smallest barrier obtained for the surface reaction between CO* and O2* at the surface of Cu3AgAu. On the other five clusters, O-O activation is nearly of the same order and is supported by O-O bond length and charge on O2. The CO and O2 molecule adsorption studies on these six trimetallic clusters (Table S.3) indicate that C-atom forms the weakest bond with Ag, followed by Au and Cu. This can be explained based on s-d splitting. In the case of Ag, d levels lie deeper compared to Au and Cu. As a result, the strength of the Ag-CO bond is weaker due to the lesser availability of Ag d-orbitals for π back donation.31,32 Note that wherever the strength of interaction between the metal atom and C/O is discussed, it is supported by initial adsorption studies of CO and O2 carried out on these trimetallic clusters.

Reaction pathway for oxidation of first CO molecule by LH mechanism on the surface of Au3AgCu nanocluster. The probability of forming carbonate species by recapturing liberated CO2 is high due to the small energy barrier. The energies are reported in eV. For reaction profiles on remaining nanoclusters, refer to supplementary information.
Except for Au3AgCu, the surface reaction is a multistep reaction on the surface of trimetallic clusters. The multistep reaction facilitates CO oxidation. Table 3 tabulates the activation energy barrier involved in this surface reaction on different clusters. In this step, the interaction between adsorbed CO and O2 generates transiently CO2* and O* surface species. O* is stabilized in µ2 configuration, which facilities the desorption of CO2 from the nanocluster. After the desorption of CO2, two possibilities exist: (a) another CO molecule adsorbs on the cluster and interacts with O* surface species leading to the liberation of the second molecule of CO2, and the surface of nanocluster becomes available for the next catalytic cycle (Figure 3, LH-LH Pathway) or (b) the desorbing CO2 may be reabsorbed resulting in the formation of carbonate surface species that poisons the surface of nanocluster for the further oxidation reaction. The carbonate nanocluster adduct (IV in Figure 2) is highly stable relative to starting reactants (I in Figure 2 +CO+ O2). This is a common observation for the reaction profile on all six trimetallic nanoclusters. In our current study, we have further explored the possibility of a decomposition reaction involving Structure IV and CO which has been discussed in section 3.2.2. This pathway is depicted as an LH-ER pathway as after the initial LH step (Figure 2), the reaction between structure IV and CO takes place via ER mechanism. To clearly state, the LH-LH pathway consists of steps (i) and (iii) of equation (B), while the LH-ER pathway consists of steps (i), (ii), and (iv) of equation (B). Section 3.2.1 below discusses the possibility (a), Figure 3 with reaction proceeding via conventional LH-LH pathway.

Reaction pathway for oxidation of the second molecule of CO by LH mechanism on the surface of Au3AgCu nanocluster. A high barrier of 0.99 eV is involved. The reported energies are in eV.
3.2.1 Reaction of Au(5-x-y)AgxCuy.O* with CO
To explore the surface reaction between Au(5-x-y)AgxCuy.O* and CO via LH mechanism, the adsorption study of CO on both 3-coordinated sites of Au(5-x-y)AgxCuy.O* has been carried out, and the most stable adduct Au(5-x-y)AgxCuy.O*.CO* is selected for further reaction mechanism study. The reaction pathway for Au3AgCu is presented in Figure 3. The reaction profiles are presented in the supplementary information for the rest of the nanoclusters. Table 4 lists the activation energy barrier involved in this surface reaction on different trimetallic clusters. The activation energy for the reaction is least when both the 3-coordinated sites involved in the reaction are Ag sites which is justified as interaction energy for CO and O on the Ag site is lowest among all three metal sites – Au, Ag, and Cu. The orientation of CO* with respect to O* also significantly affects the activation energy barrier. Table 4 shows that the larger is the C-M1-O bond angle greater is the energy barrier. It is the most for Au3AgCu, where the bond angle is 174.2°, and the barrier is 0.99 eV, followed by Cu2Au2Ag with a bond angle of 170.3°. In both clusters, one of the 3-coordinated sites is the Au site. For other clusters, the orientation is better suited for the surface reaction, which is also reflected in lower values of activation energy.
For cluster Cu3AgAu both reaction sites are Cu sites, and the angle C-M1-O is 153.8°. This angle is lower than the corresponding angle for Cu2Au2Ag, but the activation energy barrier for surface reaction on Cu3AgAu surface is only 0.01 eV less than on Cu2Au2Ag. This is explained by the stronger bonding interaction between Cu and O, as evident from the shortest bond length. The stronger the M1-O and M2-O interactions, the more energy would be required for a surface reaction between adsorbed CO and O, where O makes a new bond with the carbon of CO and breaks away completely/partially from metal sites. Once this surface reaction completes, the next step is the desorption of the product, that is, CO2, from the surface of the metal cluster, making it once again available for the next catalytic cycle. A significant activation energy barrier is involved in the desorption of CO2 from the surface of Cu3AgAu, which is expected as the interactions of C and O with Cu are stronger. An additional barrier of 0.35 eV is reported in this case (Figure S.16). Another point to be noted here is that Cu3AgAu.CO2* is considerably more stable than Cu3AgAu, similar to surface energetics of trimetallic clusters, Au3AgCu (Figure 3) and Cu2Ag2Au (Figure S.24). For the remaining three trimetallic clusters, an alternative pathway exists in which CO2 is desorbed immediately after completion of the surface reaction.
3.2.2 Decomposition of cluster-carbonate adduct
It is widely reported in the literature that the formation of carbonate on the surface of a nanocluster affects the catalytic activity of the CO oxidation reaction. In the present study, we explore the new aspect of the decomposition of the cluster-carbonate adduct in the presence of CO. The reaction between carbonate-nanocluster adduct and CO is an activated process. For the clusters, the activation energy required is less if the reaction site is (Ag, Ag) or (Au, Ag) [Refer Table 5 and 6].
However, if reaction site is Cu-site, the activation energy required for the reaction between carbonate-nanocluster adduct and CO is considerably higher which is because of strong interaction between Cu and O. Further, once Au(5-x-y)AgxCuy.O2COCO* is formed, the decomposition may proceed by two alternate routes – the one which involves dihedral angle rotation about C-O-C-O by nearly 180 degrees and second without any dihedral angle rotation. The activation energy required for decomposition is higher for the route without dihedral angle rotation (Refer to discussion in SI). Tables 5 and 6 present the activation energy barriers involved for Route1 (with dihedral angle rotation) and Route2 (without dihedral angle rotation) for all six trimetallic nanoclusters, respectively. The reaction profile via Route1 at the surface of Au3AgCu is shown in Figure 4, while the reaction profile via Route2 on the same cluster is presented in Figure 5. The reaction profiles for the rest of the trimetallic nanoclusters are provided in supplementary information.

An alternative decomposition reaction pathway by ER mechanism involving dihedral angle rotation (Route1).

An alternative decomposition reaction pathway by ER mechanism without dihedral angle rotation (Route2).
Again, in the clusters where at least one of the reaction sites is Cu, the activation energy is greater for Route2 (without dihedral angle rotation) and highest when both the reaction sites are Cu sites. For Route1 where after the formation of Au(5-x-y)AgxCuy.O2COCO*, the next step is dihedral angle rotation which is as well an activated process, but the barrier for rotation is nearly the same for all trimetallic clusters and is of the order of 0.24 eV. However, as with Route2, even for Route1, the sum of all activation barriers for the decomposition of carbonate on the surface of the trimetallic cluster with Cu-site as one of the reaction sites is higher. Also, when both the reaction sites are Cu-sites, the CO2 molecule does not desorb from the surface. As per Sabatier principle, the best catalyst for a reaction is one that has optimal interaction with reactants and products that is neither too strong nor too weak. The clusters with Cu as reaction sites are not good catalysts for CO oxidation reaction as Cu interacts strongly with the reactants O2 and CO as well with product CO2.
Another important observation is the presence of two different kinds of intermediates (Figure 6) during the decomposition of Au(5-x-y)AgxCuy.O2COCO* adduct on nanoclusters where one of the reaction sites is Cu-site. The decomposition of Au(5-x-y)AgxCuy.O2COCO* adduct leads to the release of two molecules of CO2. The release of the second CO2 molecule upon decomposition of Au(5-x-y)AgxCuy.O2COCO* adduct may either be directly from intermediate A, B or it may be that firstly intermediate A transforms to intermediate B followed by the release of CO2. The release of CO2 from intermediate B may or may not occur depending on the strength of interaction between the metal site and C. The study shows that if the intermediate involved is of type A (Cu2Ag2Au, Figure S.25 and Figure S.26), the release of CO2 is energetically more favourable compared to if the intermediate is of type B (Cu2Au2Ag, Figure S.21, and S.22) is involved. For clusters Cu3AgAu (Figure S.17 and Figure S.18) with reaction site as (Cu, Cu), because of stronger interactions between Cu and C, the transformation of intermediate A to B is facilitated, which increases the energetic cost for the release of CO2 molecule and also for the regeneration of catalytically active sites. On the other hand, for trimetallic clusters with reaction sites (Au, Ag); (Ag, Ag), and (Cu, Ag) as one the reaction site is Ag site which has weaker interactions with C, the release of CO2 takes place either directly from intermediate A or from a structure with bonding characteristics similar to intermediate A.

Two types of possible intermediates involved in the release of second molecule of CO2 via Route1 or Route2.
The carbonate decomposition via ER mechanism through Route1 (with dihedral angle rotation) is most favourable on the surface of Au3AgCu, Au2Ag2Cu, and Ag3AuCu where at least one reaction site is Ag-site.
4 Conclusions
CO oxidation at the surface of different trimetallic clusters can occur through several pathways, albeit the starting point is the surface reaction between adsorbed CO and O2 via the Langmuir-Hinshelwood mechanism. This surface reaction leads to the formation of Au(5-x-y)AgxCuy.O* in µ2-configuration, and one molecule of CO2 is released. From this point onwards, the study presents several possible ways for the release of subsequent CO2 molecules. Broadly, it can be summarized that two main pathways exist for CO oxidation at the surface of trimetallic clusters: the LH-LH pathway and the LH-ER pathway. Both CO2 molecules are released following surface reaction via the LH mechanism in the LH-LH pathway. In the LH-ER pathway, the initial step occurs via LH mechanism, followed by carbonate formation and further decomposition via ER pathway. ER-pathway involves an interaction between nanocluster-carbonate adduct and CO in the gaseous state leading to the formation of Au(5-x-y)AgxCuy.O2COCO*, which decomposes to release two molecules of CO2.
The ER mechanism may happen either via Route1 or Route2. Route1 is where an additional step, that is, dihedral angle rotation, is involved that considerably lowers the activation energy required for decomposition reaction in comparison to Route2. Table 7 details the total activation energy barrier involved for these different pathways. The study of Table 7 suggests the best trimetallic cluster for CO oxidation is the one where both the reaction sites are Ag-sites. The trimetallic clusters Au2Ag2Cu and Ag3AuCu are better suited for CO oxidation. Initially, both CO2 molecules may be generated at the surface of these trimetallic clusters via the LH-LH pathway. However, when the concentration of CO2 is considerable, the chances of carbonate formation will be higher, and hence the possibility of the reaction proceeding via the LH-ER pathway will be more. It is to be noted that for clusters where even if, one of the reaction sites is Cu-site, the total energy barrier is higher for both the LH-LH pathway and LH-ER pathway making Cu3AuAg, Cu2Au2Ag, and Cu2AuAg2 trimetallic clusters less suited for the reaction. This is mainly because of stronger interactions of the Cu-site with the reactants (CO, O2) and product (CO2).
Also, our study on CO oxidation at the surface of the Au5 cluster shows that the chances of the formation of carbonate are negligible here as the activation energy required for its formation is quite high, i.e., 0.97 eV.29 Therefore, at the surface of Au5 the reaction proceeds via the LH-LH pathway involving a total energy barrier of 0.64 eV (0.50 eV + 0.14 eV). To conclude, it is suggested that due to the ease of formation of carbonate species at the surface of trimetallic clusters, the oxidation of CO has equal chances of proceeding via the LH-LH and LH-ER pathway, as discussed here. A similar study on the surface of AuxAgy and AuxCuy bimetallic clusters has also been carried out.30
References
Okumura M, Fujitani T, Huang J and Ishida T 2015 A Career in Catalysis: Masatake Haruta ACS Catal. 5 4699
Haruta M, Kobayashi T, Sano H and Yamada N 1987 Novel Gold Catalysts for the Oxidation of Carbon Monoxide at a Temperature far Below 0°C Chem. Lett. 16 405
Liu C, Tan Y, Lin S, Li H, Wu X, Li L, Pei Y and Zeng X C 2013 CO self-promoting oxidation on nanosized gold clusters: Triangular Au3 active site and CO induced O-O scission J. Am. Chem. Soc. 135 2583
Lyalin A and Taketsugu T 2010 Reactant-promoted oxygen dissociation on gold clusters J. Phys. Chem. Lett. 1 1752
Crawley J W M, Gow I E, Lawes N, Kowalec I, Kabalan L, Catlow C R A, Logsdail A J, Taylor S H, Dummer N F and Hutchings G J 2022 Heterogeneous Trimetallic Nanoparticles as Catalysts Chem. Rev. 122 6795
Huang W, Zhai H J and Wang L S 2010 Probing the interactions of O2 with small gold cluster anions (Aun−, n = 1−7) Chemisorption vs physisorption J. Am. Chem. Soc. 132 4344
Stamatakis M, Christiansen M A, Vlachos D G and Mpourmpakis G 2012 Multiscale modeling reveals poisoning mechanisms of MgO-supported Au clusters in CO oxidation Nano Lett. 12 3621
Lopez N and Nørskov J K 2002 Catalytic CO oxidation by a gold nanoparticle: A density functional study J. Am. Chem. Soc. 124 11262
An H, Kwon S, Ha H, Kim H Y and Lee H M 2016 Reactive structural motifs of Au nanoclusters for oxygen activation and subsequent CO oxidation J. Phys. Chem. C 120 9292
Nikbin N, Mpourmpakis G and Vlachos D G 2011 A combined DFT and statistical mechanics study for the CO oxidation on the Au10-1 cluster J. Phys. Chem. C 115 20192
Nikbin N, Austin N, Vlachos D G, Stamatkis M and Mpourmpakis G 2014 Catalysis at the sub-nanoscale: complex CO oxidation chemistry on a few Au atoms Catal. Sci. Technol. 5 134
Li Ma, Melander M, Weckman T, Lassonen K and Akola J 2016 CO Oxidation on the Au15Cu15 Cluster and Role of Valencies in The MgO(100) Support J. Phys. Chem. C 120 26747
Yadav J and Saini S 2020 Atop adsorption of oxygen on small sized gold clusters: Analysis of size and site reactivity from restructuring perspective Comput. Theor. Chem. 1191 113014
Manzoor D and Pal S 2015 Reactivity and Catalytic Activity of Hydrogen Atom Chemisorbed Silver Clusters J. Phys. Chem. A 119 6162
Molina L M and Hammer B 2005 The activity of the tetrahedral Au20 cluster: Charging and impurity effects J. Catal. 233 399
Megha, Kamal C, Mondal K, Ghanty T K and Banerjee A 2019 Remarkable Structural effect on Gold-Hydrogen Analogy in Hydrogen-Doped Gold Cluster J. Phys. Chem. A 123 1973
Megha, Kamal C, Mondal K, Ghanty T K and Banerjee A 2022 Gold-Hydrogen Analogy in Small-Sized Hydrogen-Doped Gold Clusters Revisited ChemPhyChem 18 e202200261
Darby S, Johnston R, Mortimer-Jones T and Roberts C 2002 Theoretical study Of Cu-Au Nanoalloy clusters using a genetic algorithm J. Chem. Phys. 116 1536
Ma W and Chen F 2013 CO oxidation on the Ag-doped Au nanoparticles Catal. Lett. 143 84
Zeng W, Tang J, Wang P and Pei Y 2016 Density functional theory (DFT) studies of CO oxidation reaction on M13 and Au18M clusters (M = Au, Ag, Cu, Pt and Pd): the role of co-adsorbed CO molecule RSC Adv. 6 55867
Li H J and Ho J J 2012 Theoretical calculations on the oxidation of CO on Au55, Ag13Au42, Au13Ag42, and Ag55 clusters of nanometer size J. Phys. Chem. C 116 13196
Zhang Y and He X 2017 Reaction mechanisms of CO oxidation on cationic, neutral, and anionic X-O-Cu (X = Au, Ag) clusters Chem. Phys. Lett. 686 116
Venkatesan P and Santhanalakshmi J 2012 Synthesis and Characterization of Surfactant Stabilized Trimetallic Au-Ag-Pd Nanoparticles for Heck Coupling Reaction Phys. Chem. 2 12
Haldar K K, Tanwar S, Biswas R, Sen T and Latinen J 2019 Noble copper-silver-gold trimetallic nanobowls: An efficient catalyst J. Colloid Interface Sci. 556 140
Zhang W, Zhu J, Cheng D and Zeng X C 2020 PtCoNi Alloy Nanoclusters for Synergistic Catalytic Oxygen Reduction Reaction ACS Appl. Nano Mater. 3 2536
Kang X, Abroshan H, Wang S and Zhu M 2019 Free Valence Electron Centralization Strategy for Preparing Ultrastable Nanoclusters and Their Catalytic Application Inorg. Chem. 58 11000
Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, M Robb M A, Cheeseman J R, Scalmani G, Barone V, Mennucci B, Petersson G A, Nakatsuji H, Caricato M, Li X, Hratchian H P, Izmaylov A F, Bloino J, Zheng G, Sonnenberg J L, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J A, Jr., Peralta J E, Ogliaro F, Bearpark M, Heyd J J, Brothers E, Kudin K N, Staroverov V N, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant J C, Iyengar S S, Tomasi J, Cossi M, Rega N, Millam J M, Klene M, Knox J E, Cross J B, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann R E, Yazyev O, Austin A J, Cammi R, Pomelli C, Ochterski J W, Martin R L, Morokuma K, Zakrzewski V G, Voth G A, Salvador P, Dannenberg J J, Dapprich S, Daniels A D, Farkas Ö, Foresman J B, Ortiz J V, Cioslowski J and Fox D J 2009 Gaussian, Inc., Wallingford CT
Roy L E, Hay P J and Martin R L 2008 Revised Basis set for LANL Effective Core potentials J. Chem. Theory Comput. 4 1029
Saini S, Yadav J and Parkash J 2019 Mechanistic investigation into CO oxidation catalyzed by Au5 gold cluster AIP Conf. Proc. 2142 19002
Yadav J 2021 Theoretical and Computational studies of CO oxidation catalyzed by noble metal nanocluster Ph.D. Thesis Kurukshetra University Kurukshetra India p. 102 and 107
Albert K, Neyman K M, Pacchioni G and Rolsch N 1996 Electronic and Geometric Structure of Bimetallic Clusters: Density Functional Calculations on [M4{Fe(CO)4}4]4- (M = Cu, Ag, Au) and [Ag13{Fe(CO)4}8]n- (n = 0–5) Inorg. Chem. 35 7370
Popolan DM, Neossler M, Mitric R, Bernhardt T M and Bonacic-Koutecky V 2011 Tuning Cluster Reactivity by Charge State and Composition: Experimental and Theoretical Investigation of CO Binding Energies to AgnAum+/- (n + m = 3) J. Phys. Chem. A 115 951
Acknowledgements
SS acknowledges Kurukshetra University, Kurukshetra, for providing financial support (Seed-Money Grant No. DPA-I/32/22/MRP/2358-2500 dated 15-03-2022). JP acknowledges UGC, Delhi, for Senior Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Parkash, J., Saini, S. An alternative decomposition reaction pathway for CO oxidation at Au5-x-yAgxCuy (2 ≤ x + y ≤ 4) nanoclusters. J Chem Sci 135, 94 (2023). https://doi.org/10.1007/s12039-023-02212-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-023-02212-y