
Abstract
The present study deals with a computational investigation on the role of dithiocarbamate (DTC) anions in the stabilization of σ-aromatic trinuclear mono-cationic metal clusters (M = Cu, Ag and Au). Electrostatic potential, aromaticity, binding energy, thermodynamical parameters and nature of bonding are estimated. Nucleus independent chemical shifts (NICS) and their variants such as NICStotal and FiPC-NICS are employed to calculate aromaticity. The nature of bonding is assessed by the quantum theory of atoms-in-molecules (QT-AIM) and NBO methods. The charge density map in the complex has been assessed by molecular electrostatic potential analysis. Comparison of complexation properties of DTC ligand to common monodentate ligands (pyrazolates, NHC, pyridine, furan and isoxazole) explored in past reveal that DTC anions are more efficient in stabilizing metal complexes.
Graphic abstract
Synopsis. A computational approach is undertaken to investigate complexation properties of dithiocarbamate (DTC) anions with electron-deficient M3+ (M = Cu, Ag & Au) clusters. DTC ligands are found to be efficient chelators as compared to commonly used monodentate ligands to bind and stabilize such unstable trinuclear mono-cationic metal clusters

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The family of dithiocarbamate acids and their anionic salts constitute an important class of molecular species with multiple useful properties and are extensively used in agriculture, medicine and the rubber industry.1,2,3 Their anions are known as dithiocarbamate anions (R2NCSS-, Figure 1), are easily obtained from the reaction of carbon disulphide with secondary amines in presence of a base.4,5,6 Ammonia reacts with carbon disulfide in alcohol or ester solvent to give ammonium dithiocarbamate salt.7 DTC anions are planar in structure and sterically less demanding. These anions act as bidentate ligands with both sulphur atoms available to chelate with metal centres.8 Based on binding energy values, DTC anions show strong affinity towards electron-deficient centres and therefore these are found suitable to stabilize higher oxidation states of transition metals.9 DTC anions are therefore used as efficient chelators to remove heavy metals like Pb, Zn and Cd from polluted water.10 Coordination chemistry of complexation of DTC anion with transition metals, main group metals and lanthanides has been previously explored in detail.11,12,13 The unique and diversified role profile of DTC anions is attributed majorly to the existence of its stable resonance forms.14 Similarly, in a recent mechanistic study on the conversion of monosulfiram (used in the treatment of scabies) into disulfiram (used in the treatment of alcoholism), we found DTC radicals also play an important role.15

Optimized structure of M3+ metal clusters, Dithiocarbamate ligand and bonding modes in M3+ DTC complexes on M06-2X/def2-TZVP level.
In recent years, there has been a surge in synthesis, isolation and exploration of properties of all-metal inorganic clusters. Boldyrev et al., pioneered in synthesizing such all-metal aromatic compounds e.g. Al42- and related clusters which are found to obey classical rules of aromaticity.16 Due to this inherent existence of aromatic character, these clusters are comparatively more stable and exhibit useful chemical properties.17 The classical concept of Huckel’s aromaticity18 i.e., (4n+2) π-electron rule which has established itself as a powerful tool for organic molecules is now extended to σ bonded metal clusters and cages too and such systems are known as σ-aromatic.19 Such inorganic systems obey most of the parameters which could essentially be called as aromaticity criterion.20
Inorganic metal clusters made of group 11 coinage metals such as Cu3+, Ag3+ and Au3+ (Figure 1) have been synthesized lately.21,22,23 These metal clusters contain 3-center 2-electron bond and can be considered as isolobal analogues of extensively studied σ-aromatic species H3+ and Li3+.24 Metal-metal bond is found to be shorter and stronger in the case of gold clusters because of a shorter covalent radius of gold atoms on the virtue of relativistic and correlation effects.25,26 This shortening of bond length results in a special aurophilic interaction that has comparable energy as that of a hydrogen bond and is more profound when bond lengths are less than 3.6 Å, which is equivalent to its van der walls radii.27 These cyclic trinuclear complexes (CTCs) have found applications as luminescent materials and in supramolecular chemistry.28,29 Such triatomic mono-cationic clusters contain two free electrons which are available for delocalization and thereby follow (4n+2) e- rule which applies to classical π-aromatic molecules and therefore these are classified as σ-aromatic species. On the virtue of this special aromatic nature and stability, these M3+ clusters are used extensively in supramolecular design. Due to the presence of positive charge and therefore high electron affinity (EA), M3+ clusters need to be stabilized with suitable ligands. Any potential ligand to be used for this purpose needs to be electron-rich and nucleophilic in nature.
Pyrazolates have been found to bind M3+ clusters very efficiently and the resultant clusters are studied experimentally in detail.30,31 Other five and six-membered heterocyclic ligands such as triazolates, amidazolates, pyridiniates and their substituted derivatives have also been utilized.30,32,33 A theoretical attempt has been recently made by Frenking et al., to decipher bonding properties of such CTCs.34 Chattaraj et al., have computationally explored some other ligands such as NHC, furan, isoxazole etc., as suitable stabilizing ligands.35 Emphasising the fact that M3+ clusters have a net positive charge, it becomes intuitive to explore its binding abilities with an anionic ligand. It is anticipated that such an interaction will lead to the formation of a charge-neutral species which is expected to have far greater stability.
DTC anions are found to coordinate with a large number of electron-deficient species.8,9,10 DTC ligands have a unique property of bridging at 1, 3 centres and thus forming a five-membered ring with little ring strain (Figure 2). DTC anions have been effectively employed as a sensitizer in designing lanthanide complexes resulting in strong photoluminescence.13 Yang et al., have observed that ligands having rigid planar structure show enhanced lanthanide luminescence.36 DTC anions are structurally planar and rigid which makes them suitable in designing luminescent M3+ based complexes. The parent molecules of DTC anions i.e., thiuram compounds or carbon disulphide and amines are relatively cheaper and commercially easily available. Owing to their similarity to molecular systems employed in the past, these clusters could prove to be efficient agents for trapping noble gases too.24

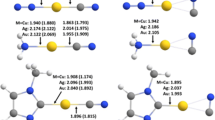
Optimized geometries of M3+ DMTC conformers obtained at M06-2X/def2-TZVP level of theory with important bond lengths (in Å). P = Cu, Q = Ag, R = Au, A = DMTC, B = DETC*, 1=> Both S atoms attached to single metal center, 2=> S atoms attached to two different metal centers. (* = used later).
This is the first time a computational study has been done on an open chain bidentate anionic ligand with 1, 3 chelating centres as a suitable candidate for coordination with M3+ clusters. To the best of our belief, DTC ligands have never been employed to coordinate with electron-deficient poly-metallic clusters.
In the present study we have tried to gain insights into the complexation properties such as stability, change in aromaticity and nature of bonding of Cu3+, Ag3+ and Au3+ metal clusters with two DTC ligands dimethyldithiocarbamate (DMTC, Me2NCSS-) and diethyldithiocarbamate (DETC, Et2NCSS-). We have tried to put in efforts to compare the suitability of DTC anions with other ligands such as pyrazolates, NHC, pyridine, furan and isoxazole which have been explored experimentally or computationally in the past.
2 Computational methods
All studies in this work have been performed on Gaussian 09 suite of programs37 except topological studies38 which have been done using Multiwfn39 program. All structures have been optimized in the gaseous phase by density functional theory (DFT) method using M06-2X functional in conjunction with def2-TZVP basis set. Basis set superposition error (BSSE) corrected binding energies have been obtained using the counterpoise method as proposed by Boys and Bernardi.40 Harmonic vibrational frequency studies have been performed to make sure that all obtained optimized geometries are true minima. Thermodynamic parameters such as enthalpy change and free energy change have been obtained from statistical parameters derived from vibrational analysis. Nucleus independent chemical shifts (NICS)41 values have been obtained from NMR calculations using Gauge independent atomic orbital (GIAO) method42 performed at M06-2X/def2-TZVP level of theory. Other NICS variants are obtained from different components of the NICS tensor. Quantum theory of atoms-in-molecules (QT-AIM) analysis has been done using Multiwfn software to identify the nature of critical points.43
Conceptual DFT parameters like electron affinity (A), hardness (η) and electrophilicity (ω) have also been discussed. For an N-electron system with total energy E and external potential v, chemical potential (μ), electronegativity (χ)44 and hardness (η)45 can be defined as:
Using a finite difference approximation, χ and η can be rewritten as:
Using Koopmans’ theorem,46 electron affinity and hardness47 of metal clusters and metal complexes can be expressed as:
Electrophilicity (ω) is defined by Parr et al.,48 as:
Change in enthalpy and change in free energy from Gaussian calculations are obtained from the following equations:
Where ε0 is electronic energy, Hcorr is thermal correction to enthalpy and Gcorr is thermal correction to Gibbs free energy.
3 Results and discussion
3.1 Geometry
Geometries of all three M3+ (M = Cu, Ag, Au) clusters and two dithiocarbamate (DMTC and DETC) anions were individually optimized and confirmed as energy minimum structures with no imaginary frequencies found. The structure of the DTC anion shows a C-N bond length of around 1.37 Å which is in between a typical carbon-nitrogen single bond and a double bond. This intermediate character of C-N bond indicates conjugation between lone pair on nitrogen and negative charge on either sulfur atoms. In the case of CTCs, M-M bond lengths are in order Cu-Cu < Au-Au < Ag-Ag. This anomaly is expected because of the compact first d shell in copper and the relativistic effect found in the case of gold.49 Devoid of these factors silver clusters stand apart from the other two metals of the group. Upon conformational analysis of M3+ DTC complex, two stable conformers were obtained in each case (Figure 2). For instance, in case of interaction of Cu3+ and DMTC, we obtained two conformers PA1 and PA2 (Notation explained under Figure 2 caption). In PA1 both sulfur centers of DTC anion are bonded to a single Cu atom (three centered bonding) while in PA2 the two sulfur centers are attached separately to two different Cu atoms (four centered bonding). The geometry of M3+ rings has changed from an equilateral triangle to an isosceles triangle in all six conformers where two M-M bonds have different bond length than that of the third M-M bond as can be seen in Figure 2.
Similar changes in geometry have been observed for the rest of the conformers. Some important geometrical parameters have been given in Table 1. A negative deviation from the original value of 124.1° for S-C-S bond angle was observed for PA1, QA1 and RA1 while PA2, QA2 and RA2 showed a positive deviation depending on the strain in the resultant ring.
The high amount of ring and angle strain is expected on account of constrained geometry in three centered complexes (PA1, QA1 and RA1). On the other hand, four centered complexes contain a five-membered ring and are less strained comparatively. This can be exemplified by considering the case of Au3+ bound complex with DMTC ligand where a comparison of ring strain and therefore the stability of the complex can be made in terms of S-C-S bond angle. Free DMTC, RA1 and RA2 have S-C-S bond angles of 124.1°, 118.8° and 131.0° respectively. RA2 is therefore much more geometrically relaxed than RA1. Such geometry dependent stability is directly correlated with thermochemical changes that occurred during the process and the same has been discussed later.
Nitrogen-Carbon (R2N-CS2) bond length changes from 1.37 Å to 1.33-1.34 Å for different conformers. There appears a shift of electron density from NR2 region to CS2- region due to efficient conjugation. NR2 group shows an electron-donating character in these complexes and thus helps in charge transfer from DTC ligand to M3+ clusters. A decreased N-C bond length essentially signifies an enhanced double bond character of the bond and hence greater rigidity in the complex. This gain in structural rigidity makes DTC ligands highly suitable for designing luminescent complexes with metal clusters.36 Averaged metal-sulfur bond lengths show a trend in order Cu-S < Au-S < Ag-S and are shorter in the case of PA2, QA2 and RA2 as compared to PA1, QA1, and RA1 respectively. This result is due to a greater extent of bonding in four centered complexes as compared to three centered complexes and a shorter Au-S bond depicts higher affinity in the case of a gold cluster. Averaged carbon-sulfur (R2NC-S2) bond length remains the same for all conformers at around 1.72 Å that is only slightly higher than 1.70 Å, which is observed in the case of isolated DMTC anion. M3+ molecular plane is found to be coplanar with DTC molecular plane in all conformers. Similar geometrical changes were encountered upon complexation of M3+ with DETC ligand and are given in Supplementary Information (Table S1).
3.2 Binding energy
Binding energy is expected to be large on account of ionic interaction and significant charge transfer. BSSE corrected binding energies have been obtained for all conformers at M06-2X/def2-TZVP level and are listed in Table 2. For a given ligand, Au3+ complexes are found to have the highest binding energies followed by Cu3+ and Ag3+ complexes respectively. Similar trends have been observed for M-S bonds (M = Cu, Ag, Au) in binary alloys in studies done by Pakiari et al.50 Change of alkyl group in DTC ligand is found to have a negligible effect as DETC bound complexes are found to have similar energies as that of DMTC bound complexes. This clearly rules out any role of the size of alkyl group attached to nitrogen on overall interaction. Similarly, we tried to put a phenyl ring instead of alkyl group and observed a decrease in binding energy values. This is expected because of the delocalization of lone pair of nitrogen on phenyl ring and its electron density donation towards sulfur atoms get reduced. Out of the two binding modes obtained each for Cu3+, Ag3+ and Au3+ bound complexes, the four centered bonded conformers (i.e. PA2, QA2 and RA2) have higher binding energies as compared to the three centered bonded conformers (i.e. PA1, QA1 and RA1). This is on account of different ring strain and extent of charge transfer in respective conformers.
Binding energy values are supplemented by enthalpy change (ΔrH0) and free energy change (ΔrG0) values based on free ions complexation scheme (M3+ + DTC− → M3DTC), which also follow a similar trend. ΔrH0 and ΔrG0 values have been obtained from the vibrational analysis of the respective complexes and constituent ionic fragments in the gas phase at 298.15K and 101.325 kPa as given in equation 9 and 10.51 The effect of ring strain is noticeably visible while comparing two bonding modes where the formation of four centered bonded complexes is much more exothermic than those of complexes with three centered bonding. For example, the formation of RA2 which contains a five-membered ring is more exothermic than RA1 which contains a four-membered ring by around 15 kcal/mol. An attempt has been made to obtain BSSE corrected dissociation energies assuming that the complex breaks into two constituent radicals as R2NCSS· and M3· (Table 2). This approach avoids overemphasis on the electrostatic attraction between oppositely charged ions and provides insights into the stabilities of different conformers.
To consider the influence of counter ions during complexation, we have tried to obtain thermodynamical parameters for a general scheme of complex formation taking Cu+ and Cl- as counter ions (M3Cl + CuDTC → M3DTC + CuCl). It is observed that although the scale of values has changed but the relative differences in magnitudes of enthalpy and free energy change for different conformers remain similar to that of the earlier scheme in which counter ions are ignored (Table S2 (a)). An attempt was made to compare the efficacy of DTC ligands towards stabilizing M3+ clusters. With reference to an earlier study,35 dissociation energy values reveal that DTC ligands have comparable or better binding abilities towards Au3+ clusters (Table S2(b)).
3.3 Population analysis
Natural Bond Orbitals method has been applied to get insights about charge transfer from ligand to the metal cluster. Substantial charge transfer is observed in all cases. NBO charges on constituent ions and the complexes are collected in Table 3.

Dimethyl Dithiocarbamate ligand with atomic labels.
Charge density at donor sulfur atoms is substantially reduced and enhanced at acceptor metal centers after complexation. These values clearly reflect a high amount of charge transfer from DTC ligand to M3+ clusters. Among all three M3+ DTC clusters, the highest amount of charge transfer is observed for the gold cluster followed by copper and silver clusters. This is reflected from the NBO charges on S and Au atoms in RA1 and RA2 complexes (Table 3). Three centered bonded conformers (PA1, QA1 and RA1) show more change in charge density accumulation than those of four centered bonded conformers (PA2, QA2, and RA2). Out of all complexes, the most significant charge reduction is observed for RA1. The gold atom which is bonded to sulfur atoms shows a huge change in NBO charge from 0.333 to 0.016, while sulfur atoms also show a similar change from − 0.406 to − 0.165. This exceptional charge transfer is the reason behind the enhanced stability of RA1 as compared to other complexes. As noticed earlier, the same trend is observed in binding energies and thermodynamical parameters.
3.4 Molecular electrostatic potential surface
An effort has been made to obtain electrostatic potential (ESP) maps for different conformers. The key reason behind the stabilization of M3+ cations after complexation with DTC ligands is because of charge neutralization and subsequent redistribution of charge density from electron-rich atoms to electron-deficient metal centers. Apart from the evident donation of electron density from sulfur atoms, DTC ligands have an additional electron-donating moiety in form of NR2 group. Lone pair on nitrogen is in conjugation with a negative charge on sulfur atoms and when needed, nitrogen center pushes more electron density towards sulfur atoms. Earlier we noticed a shortening of CN bond and a decrease in the population at N center upon complexation. This phenomenon can be clearly seen from the ESP diagram of complexes where the NR2 region has turned electron deficient (Figure 4). Comparing with the results obtained from binding energies and thermodynamic calculations, it can be inferred from ESP diagrams that the complexes which show a more uniform distribution of charge density are more stable and the ones in which charge accumulation still remains are less stable. Four centered bonded complexes show decreased charge density at nitrogen center and show the enhanced distribution of charge all over the complex. While three centered complexes show good charge transfer but the distribution of charge density is less uniform comparatively.

Molecular Electrostatic Potential mapped on 0.002 au isovlaue surface for DMTC bonded complexes.
3.5 Aromaticity
Aromaticity has classically been interpreted under three major criteria: geometrical, energetic and magnetic. Planar shape, extended conjugation, high resonance energy and high diamagnetic susceptibility have been some of the important parameters. Talking about magnetic criteria in relation to cyclic inorganic compounds, the nucleus independent chemical shifts technique has been used quite successfully as this is highly suitable to study aromatic properties of non-conjugated and non-carbon containing sigma bonded inorganic molecules. Schleyer has popularized the technique in last two decades and it has been on the list of nearly all computational organic chemists.
NICS is defined as negative of the absolute chemical shifts at the geometrical center or at a point in space preferably on the line passing through the center and perpendicular to the molecular plane. High negative values indicate aromaticity while positive values indicate anti-aromaticity.52 Non-aromatic molecules are known to exhibit near-zero NICS values. NICS is a tensor quantity which can be further resolved in three different components (NICStotal = -1/3(σxx + σyy + σzz)) where σxx, σyy and σzz are isotropic shielding constants along mutually perpendicular x, y and z directions respectively. The plot of NICSout of plane component (−1/3 σzz) vs NICSin plane component (−1/3(σxx + σyy)) allows us to classify aromatic properties of a molecular system.53 This approach is popularly called as free of in plane component NICS or FiPC-NICS and it is based on the fact that in plane components are more sensitive towards induced magnetic field.54
In our study, we calculated NICStotal values at the points starting from center of the M3+ cluster and on the z axis at an interval of 0.5Å. These values have been plotted in Figure 5. The change in aromaticity upon complexation has been observed carefully. σ-aromaticity in these molecular systems is shown by NICStotal values which are highly negative near ring center and gradually decrease to zero at a distance of 5Å. In all cases, NICS value is found to decrease after complex formation. This is on account of the loss of regular triangle structure and henceforth decrease in circular ring current. The only aberration is found in the case of Au3+ cluster where NICS value has increased a little from its original values for RA1. For a given ligand, the NICStotal values for these three metal clusters follow the order Ag ~ Cu < Au. Among DMTC and DETC bound M3+ complexes hardly any difference was observed. Again, it starts decreasing and shows near-zero values after reaching distance of 5Å.

NICStotal vs. distance plot of M3+ clusters and DTC bound M3+ complexes.

Location of Bond Critical Points on different complexes and important parameters associated with them.
NICSout of plane component is highly sensitive towards the aromatic character of the cyclic rings and this is exemplified by the fact that Au3+ and its complexes show most negative values and hence are more aromatic in nature. For four centered complexes, the rate of fall of NICS out of plane values is higher than three centered complexes and bare clusters. This observation reaffirms the fact that more is the strength of the bonding and resulting change in geometry, more will be the rate of drop in the out of plane NICS component. NICSzz component is often considered to be a better index to study π-aromaticity in organic compounds. Nevertheless, we tried to obtain additional information out of NICSzz plots for our systems too. We found that NICSzz values show high negative values near the ring center, becomes gradually positive on moving upwards and reach its peak value in 1Å–2Å range thereafter it starts diminishing as expected. FiPC-NICS and NICSzz plots are given in Supplementary Information (Figure S1).
3.6 Quantum theory of atoms-in-molecules (QT-AIM) analysis
Atoms in molecules theory, proposed by Bader in the early 1990s is based on the topology of electron density (ρ) in a molecule.38 All points in space where the electron density is maximum or minimum i.e. Gradient (∇ρ) of electron density is equal to zero, are called critical points (CP). Double derivative i.e. Laplacian (∇2ρ) tells about the nature of the CP. ∇2ρ can be written in form of a symmetric matrix known as Hessian of ρ. Based on rank (ω) and signature (σ), different CPs are represented as (ω, σ). There are four defined critical points named as nuclear critical points {NCP (3,-3)}, bond critical points {BCP (3,−1)}, ring critical points {RCP (3,1)} and cage critical points {CCP (3,3)}.43 Existence of a bond critical point (BCP) is a necessary and sufficient condition for the existence of a bond or interaction. A BCP signifies the accumulation of electron density between two atoms thus suggesting a possible interaction. Magnitude and sign of electron density, its Laplacian and energy density at the BCP tells about the nature and strength of bonding.
In this study, BCPs have been found at all expected binding sites which confirms the existence of bonds between the electron-rich sulfur atom and metal centers. Locations of BCPs and different topological parameters related to these critical points have been shown in Figure 5. ρ values are found to be positive and in the range 0.05–0.10, while ∇2ρ values are also positive and in the range 0.13–0.20. Other parameters being considered are local kinetic energy density (G), local potential energy density (V) and the sum of both called total energy density (H) which are given in Supplementary Information (Table S3 (a)). High positive values of electron density and high negative values of total energy density (H) are favorable conditions for a stable bond to be formed.55 Laplacian values are reliable only for simple organic molecules containing covalent bonds. Considerable electron density and negative H is observed for all complexes. As is evident from these values, most favorable values are obtained for gold complexes which again supports our assumption that DTC binds more suitably with the gold cluster as compared to copper and silver clusters.
An attempt has been made to compare the binding ability of DMTC ligand with Au3+ with some of the previously studied ligands in light of QT-AIM.35 Numerical values of ρ, ∇2ρ and H are listed in Table S3 (b) (Supplementary Information) to compare bonding properties of different ligands. These values indicate that bonding in DTC bound complexes is favorable and is comparable with NHC bound M3+ complex. Considering negative values of total energy density (H) as the decisive parameter, DTC is found to be a better ligand or with comparable ability as of other candidates.
3.7 Conceptual DFT parameters
Electron affinity (EA) of a chemical species is a measure of its electron deficiency. M3+ cationic clusters have high electron affinity and hence get attracted to electron-rich DTC anions. As evident from the significant decrease in EA values of different complexes as compared to M3+, DTC acts as an excellent nucleophile towards stabilizing M3+ clusters (Table S4 (a), Supplementary Information). Even though a significant decrease in EA is observed, the resulting complexes are found to be softer than bare clusters on account of decreased HOMO-LUMO gap. Decreased hardness is crucial for making the resultant cluster suitable for further binding with other ligands such as noble gases and other small gaseous molecules. Similar trends were found for DETC bonded complexes too and values are given in Supplementary Information (Table S4 (b)). An advanced conceptual DFT parameter, electrophilicity proposed by Parr et al.,48 has also been utilized to gain further insights. Electrophilicity is considered to be a measure of reactivity of a system towards attracting electrons from an electron-rich species. A big decline in electrophilicity values after complexation is testimony to the excellent ability of DTC ligand to donate electrons and form a strong bond with M3+ clusters.
4 Conclusions
DTC ligands are found highly suitable for stabilizing M3+ clusters owing to a considerable affinity between the two. Out of all three metal clusters, DTC ligands interact with Au3+ more favorably as compared to Cu3+ and Ag3+ as evident from binding energy values as well as thermochemical data. Electrostatic potential maps explain the transfer and distribution of charge density before and after complexation. A significant shift in charge density is observed at NR2 as well as CS2 region of the DTC ligand. Aromaticity properties have also been discussed and it was noted that a noticeable shift in aromatic character occurs after complexation on account of geometrical changes. NBO and QT-AIM studies indicate that significant charge transfer occurs from ligand to metal cluster and the bonds formed are very strong. A huge drop in values of electron affinity and electrophilicity is the testimony of an excellent interaction. Three centered complexes are less stable than four centered complexes because of high ring strain. The exceptional stability of gold complexes is due to aurophilic interaction and could be partly attributed to an increase in aromaticity which is not observed in the other two cases.
References
Halls D J 1969 The properties of dithiocarbamates: A review Mikrochim Acta 57 62
Nieuwenhuizen P J, Reedijk J, Duin M V and McGill W J 1997 Thiuram- and dithiocarbamate-accelerated sulfur vulcanization from the chemist’s perspectives; Methods, materials and mechanisms reviewed Rub. Chem. Technol. 70 368
Sunderman F W 1979 Efficacy of sodium diethyldithiocarbamate (dithiocarb) in acute nickel carbonyl poisoning Ann Clin. Lab. Sci. 9 1
Schubart R 2000 Dithiocarbamic Acid and Derivatives. In Ullmann's Encyclopedia of Industrial Chemistry, (Ed.). 11 495
Hogarth G 2005 In Progress in Inorganic Chemistry K D Karlin (Ed.) (New Jersey: John Wiley) p. 1978
Azizi N, Aryanasab F, Torkiyan L, Ziyaei A and Saidi M R 2006 One-pot synthesis of dithiocarbamates accelerated in water J. Org. Chem. 71 3634
Mathes R A 1950 In Inorganic Syntheses vol. III Ludwig F Audrieth (Ed.) (New York: McGraw-Hill Book Company, Inc.) p. 48
Steggerda J J, Cras J A and Willemse J 1981 Reactions of complexes of dithiocarbamate and related ligands Recl. Rev. Prog. Curr. Res. 100 41
Hogarth G 2012 Metal-dithiocarbamate complexes: chemistry and biological activity Mini-Rev. Med. Chem. 12 1202
Abu-El-Halawa R and Zabin S A 2017 Removal efficiency of Pb, Cd, Cu and Zn from polluted water using dithiocarbamate ligands J. Taibah Univ. Sci. 11 57
Heard P J 2005 In Progress in Inorganic Chemistry K D Karlin (Ed.) (New Jersey: Wiley) p. 1
Boncher W L, Regulacio M D and Stoll S L 2010 Thermolysis of lanthanide dithiocarbamate complexes J. Solid State Chem. 183 52
Regulacio M D, Pablico M H, Vasquez J A, Myers P N, Gentry S, Prushan M, et al. 2008 Luminescence of Ln(III) dithiocarbamate complexes (Ln = La, Pr, Sm, Eu, Gd, Tb, Dy) Inorg. Chem. 47 1512
Coucouvanis D 1979 In Progress in Inorganic Chemistry Vol. 26 S J Lippard (Ed.) (New York: Wiley)
Singh V K, Gupta S and Gupta A 2019 Exploring the mechanism of conversion of monosulfiram into disulfiram Ind J. Chem. Sect. A 58 429
Li X, Kuznetsov A E, Zhang H F, Boldyrev A I and Wang L S 2001 Observation of all-metal aromatic molecules Science 291 859
Popov I A, Starikova A A, Steglenko D V and Boldyrev A I 2018 Usefulness of the σ-aromaticity and σ-antiaromaticity concepts for clusters and solid-state compounds Chem. A Eur. J. 24 292
Huckel E 1931 Quantentheoretische Beitrage zum Benzolproblem Zeitschrift für Phys. 70 204
Zubarev D Y, Averkiev B B, Zhai H J, Wang L S and Boldyrev A I 2008 Aromaticity and antiaromaticity in transition-metal systems Phys. Chem. Chem. Phys. 10 257
Krygowski T M, Cyranski M K, Czarnocki Z, Hafelinger G and Katritzky A R 2000 Aromaticity: A theoretical concept of immense practical importance Tetrahedron 56 1783
Burini A, Mohamed A A and Fackler J P 2003 Cyclic trinuclear gold(I) compounds: Synthesis, structures and supramolecular acid-base π-stacks Comments Inorg. Chem. 24 253
Robilotto T J, Bacsa J, Gray T G and Sadighi J P 2012 Synthesis of a trigold monocation: An isolobal analogue of [H3]+ Angew Chemie Int. Ed. 51 12133
Shayeghi A, Johnston R L, Rayner D M, Schäfer R and Fielicke A 2015 The nature of bonding between argon and mixed gold-silver trimers Angew Chemie Int. Ed. 54 10675
Chakraborty A, Giri S and Chattaraj P K 2010 Trapping of noble gases (He-Kr) by the aromatic H3+ and Li3+ species: A conceptual DFT approach New J. Chem. 34 1936
Desclaux J P and Pyykkö P 1976 Dirac-Fock one centre calculations. The molecules CuH, AgH and AuH including p-type symmetry functions Chem. Phys. Lett. 39 300
Shorygin P P and Burshtein K Y 1991 Conjugation and the periodic system of the elements Russ. Chem. Rev. 60 1
Pathaneni S S and Desiraju G R 1993 Database analysis of Au ⋯ Au interactions J. Chem. Soc. Dalt. Trans. 319
Omary M A, Rawashdeh-Omary M A, Gonser M W A, Oussama Elbjeirami, Grimes T and Cundari T R 2005 Metal Effect on the Supramolecular Structure, Photophysics, and Acid-Base Character of Trinuclear Pyrazolato Coinage Metal Complexes Inorg Chem. 44 8200
Hou L, Shi W J, Wang Y Y, Wang H H, Cui L, Chen P X and Shi Q Z 2011 Trinuclear-based copper(I) pyrazolate polymers: Effect of trimer π-acid· · ·halide/pseudohalide interactions on the supramolecular structure and phosphorescence Inorg. Chem. 50 261
Mohamed A A, Burini A and Fackler J P 2005 Mixed-metal triangular trinuclear complexes: Dimers of gold - silver mixed-metal complexes from Gold(I) carbeniates and Silver(I) 3,5-diphenylpyrazolates J. Am. Chem. Soc. 127 5012
Zhang J P and Kitagawa S 2008 Supramolecular isomerism, framework flexibility, unsaturated metal center, and porous property of Ag(I)/Cu(I) 3,3’,5,5’-Tetrametyl-4,4’-Bipyrazolate J. Am. Chem. Soc. 130 907
Halcrow M A 2009 Pyrazoles and pyrazolides—flexible synthons in self-assembly Dalton Trans. 2059
Bovio B, Bonati F and Banditelli G 1984 X-ray crystal structure of tris [µ-3,5-bis(trifluoromethyl)pyrazolato-N, N’] trigold(I), a compound containing an inorganic nine-membered ring Inorg. Chim. Acta 87 25
Caramori G F, Piccoli R M, Segala M, Munoz-Castro A, Guajardo-Maturana R, Andrada D M and Frenking G 2015 Cyclic trinuclear copper(I), silver(I), and gold(I) complexes: A theoretical insight Dalton Trans. 44 377
Pan S, Saha R, Mandal S and Chattaraj P K 2016 σ-Aromatic cyclic M3+ (M = Cu, Ag, Au) clusters and their complexation with dimethyl imidazol-2-ylidene, pyridine, isoxazole, furan, noble gases and carbon monoxide Phys. Chem. Chem. Phys. 18 11661
Yang Y S, Gong M L, Li Y Y, Lei H Y and Wu S L 1994 Effects of the structure of ligands and their Ln3+ complexes on the luminescence of the central Ln3+ ions J. Alloys Compd. 208 112
Gaussian 09, Revision D.01, Frisch M J et al. 2009 Gaussian, Inc., Wallingford CT
Bader R F W 1985 Atoms in molecules Acc. Chem. Res. 18 9
Lu T and Chen F 2012 Multiwfn: A multifunctional wavefunction analyzer J. Comput. Chem. 33 580
Boys S F and Bernardi F 1970 The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors Mol. Phys. 19 553
Schleyer P V R, Maerker C, Dransfeld A, Jiao H and Hommes N J R V E 1996 Nucleus-independent chemical shifts: A simple and efficient aromaticity probe J. Am. Chem. Soc. 118 6317
Wolinski K, Hinton J F and Pulay P 1990 Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations J. Am. Chem. Soc. 112 8251
Kumar P S V, Raghavendra V and Subramanian V 2016 Bader’s theory of atoms in molecules (AIM) and its applications to chemical bonding J. Chem. Sci. 128 1527
Parr R G, Donnelly R A, Levy M and Palke W E 1978 Electronegativity: The density functional viewpoint J. Chem. Phys. 68 3801
Parr R G and Pearson R G 1983 Absolute hardness: companion parameter to absolute electronegativity J. Am. Chem. Soc. 105 7512
Koopmans T 1934 Über die zuordnung von wellenfunktionen und eigenwerten zu den einzelnen elektronen eines atoms Physica 1 104
Pearson R G 1986 Absolute electronegativity and hardness correlated with molecular orbital theory Proc Natl. Acad. Sci. 83 8440
Parr R G, Szentpaly L V and Liu S 1999 Electrophilicity Index J. Am. Chem. Soc. 121 1922
Yan S, Chen J and Wang R 2008 Towards anomalous diffusion with nonlinear interactions for hamiltonian chaotic systems Physica A 387 1786
Pakiari A H and Jamshidi Z 2010 Nature and strength of M-S bonds (M = Au, Ag, and Cu) in binary alloy gold clusters J. Phys. Chem. A 114 9212
Ochterski J W 2000 Thermochemistry in Gaussian Gaussian Inc. 1–19
Chen Z, Wannere C S, Corminboeuf C, Puchta R and Schleyer P V R 2005 Nucleus-independent chemical shifts (NICS) as an aromaticity criterion Chem. Rev. 105 3842
Stanger A 2006 Nucleus-independent chemical shifts (NICS): Distance dependence and revised criteria for aromaticity and antiaromaticity J. Org. Chem. 71 883
Torres-Vega J J, Vasquez-Espinal A, Caballero J, Valenzuela M L, Alvarez-Thon L, Osorio E and Tiznado W 2014 Minimizing the risk of reporting false aromaticity and antiaromaticity in inorganic heterocycles following magnetic criteria Inorg. Chem. 53 3579
Cremer D and Kraka E 1984 Chemical bonds without bonding electron density — does the difference electron-density analysis suffice for a description of the chemical Bond? Angew. Chemie Int. Ed. Eng. 23 627
Acknowledgements
VKS thanks CSIR, New Delhi for a junior research fellowship. The authors are thankful to reviewers for valuable suggestions.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Singh, V.K., Shrivastava, A. & Gupta, A. Complexation of Mono-anionic Bidentate Ligand Dithiocarbamate with σ-Aromatic M3+ Clusters: A DFT Study. J Chem Sci 133, 33 (2021). https://doi.org/10.1007/s12039-021-01895-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-021-01895-5