
Abstract
An unprecedented metal-, halogen- and solvent-free, MSAA-promoted S-carbonylation of thiols with feedstock acids has been developed. This new transformation provides an efficient and atom-economic strategy for the synthesis of thioesters in a single operation from readily available and inexpensive starting materials. The reaction avoids the use of expensive and hazardous coupling reagents, bases and generates water as the only by-product, thus making this chemical synthetic process more viable, environment-friendly and contributing towards sustainable chemistry.
Graphic abstract

An efficient and novel strategy was established for dehydrative nucleophilic substitution reaction of feedstock acids with thiols, which systematically unravels the feasibility and practicality of thioester formation in a step- and atom-economical fashion.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
In the modern era of chemistry, undisputed attention has been placed towards solvent-less technology due to the poisonous, expensive and volatile nature of many organic solvents, particularly chlorinated hydrocarbons.1,2,3 With the emergence of the concept of novelty, selectivity, and effectiveness, the synthetic chemists have been very attentive in rendering the required reactions in metal-, halogen-, and solvent-free conditions to lead the goal of triple bottom-line benefits of environmental, economic, and social improvements.4 Recently, the desire of greener, hazard-free, waste-free, and energy-efficient sustainable synthetic routes has increased for bond-forming steps which are fundamental to the pharmaceutical industries.
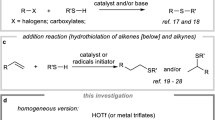
Among the widespread category of reactions, thioesterification has seen as a significant example of such fundamental transformation. There exists and continues to be a significant amount of progress in this reaction because thioesters represent excellent building blocks for chemical biology and organic synthesis.5,6,7,8 Thioesters act as an important class of acyl donors and are protagonists in the biosynthesis of many natural polyketide antibiotics, including erythromycin, enterobactin, vancomycin, penicillin, and bacitracin biochemistry.5,9,10 These air-stable acyl donors are tolerant to column purification on silica gel and therefore easier to handle than the corresponding acyl halides. Examples of applications of thioesters in organic synthesis are illustrated in Scheme 1. Fukuyama et al., established a chemoselective protocol to access the ketones from thioesters using Pd-catalysis and a zinc aryl or alkyl species.11 Different procedures have also been developed for the synthesis of acetylene ketones, esters, amides, and acylsilanes from thiol esters.12,13,14 Furthermore, it is possible to reduce this carboxylic acid derivative selectively to either alcohol or aldehyde depending on the conditions used while leaving other functional groups such as esters and amides intact.15,16 Sekiya and Lawesson demonstrated the successful replacement of the oxygen atom of thioester by sulfur or CCl2 group.17,18 In addition, benzodithioate products can be exploited as an attractive source for the synthesis of different sulfur containing heterocycles. Furthermore, it is possible to synthesize 2-methylthio-1,3-oxazoles from both aryl and alkyl thioesters using N-(ethoxycarbonylmethyl)-iminodithiocarbonate.19

Thioesters as precursors in organic synthesis.
In consideration of their relevant role in biological systems and their synthetic versatility, preparation of thioesters is still an urgent need. Despite the broad range of described methods reported for their synthesis, the acylation of thiols or thiolate anions by reaction with carboxylic acid derivatives, namely acid anhydrides and acyl halides in organic solvent20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36 is probably the most widely used. Moreover, thioesters have been synthesized by direct reaction of carboxylic acids with thiols and in the presence of diverse reaction conditions and catalysts.37,38,39,40,41 Although most of these approaches provide efficient access to thioesters, they suffer from the use of corrosive reagents, harsh reaction conditions, expensive catalysts or reagents which could have detrimental environmental effects, unfriendly organic solvents and long reaction time. There are many drawbacks associated with the use of moisture sensitive acyl chlorides and anhydrides for S-acylation, e.g. reaction of thiols with acyl chlorides can be highly exothermic and this transformation produces an equal amount of non-eco-friendly halide ion when acyl halides were used.42,43,44,45,46 Therefore, to address the aforementioned challenging issues, we were eager to develop an environmentally benign reaction course, based on readily available inexpensive and eco-friendly precursors and organic reagents for the synthesis of diverse thioesters.
Methanesulfonic acid (MSA) is a low molecular weight compound, derived from biomass and commonly used as a strong acid.47,48,49 MSA is considered to be attractive from a sustainability point of view and it has been used to catalyse a wide variety of transformations and as a solvent for rearrangement and condensation reactions. Suitable industrial processes have already been disclosed for the biodegradation of MSA, would be beneficial for waste steam processing.50 Eaton’s reagent (P2O5/MSA) has been used in intermolecular acylation reaction.51,52,53,54,55 The use of MSA as a solvent for similar reactions promoted by alumina or graphite has also been detailed.56,57,58 Methanesulfonic anhydride (MSAA), the anhydride of MSA, has been used as an excellent reagent for the improvement in Friedel-Crafts acylation reaction.59 MSAA is readily available, inexpensive, eco-friendly, bench stable, non-hygroscopic reagent and easy to handle. However, to the best of our knowledge, MSAA has never been explored for S-carbonylation of thiols leading to thioesters. Intrigued by the aforementioned attractive assets of MSAA and our ongoing work on metal-free catalysis,60,61,62,63,64,65 in the present paper, we report a MSAA-promoted direct thioester formation from feedstock acids and thiols under solvent-free conditions.
2 Experimental
2.1 General information
All chemicals were purchased at the highest purity grade and used for solvent-free protocol without further purification. All syntheses were performed in standard glassware without any special precautions taken for the removal of moisture or air. Merck precoated 0.25 mm silica gel plates (60F-254) were used to perform the analytical TLC. Visualization was achieved with shortwave UV light. Column chromatography was carried out with silica gel (100–200 mesh) using EtOAc/hexanes. NMR spectra were recorded in CDCl3 and using TMS as internal standard on JEOL ECX-400-II. Chemical shifts of 1H NMR spectra were given in parts per million with respect to TMS and the coupling constant J was measured in Hz. The signals from solvent CDCl3, 7.26 and 77.0 ppm, are set as the reference peaks in 1H NMR and 13C NMR spectra, respectively. Melting points were recorded on a Perfit melting point instrument and are uncorrected. The following abbreviations were used to describe the multiplicities: s = singlet, d = doublet, t = triplet, m = multiplet.
2.2 General procedure for the synthesis of thioesters 3
To a mixture of carboxylic acid 1 (0.5 mmol) and thiol 2 (0.6 mmol), was added methanesulfonic anhydride (0.65 mmol, 1.3 equiv.) The reaction was heated to 80 °C for 4 h, after which the mixture was placed under high vacuum at room temperature. The reaction mixture was diluted with ether (10 mL) and the slurry was kept at 0 °C for 20 min. The ether solution was concentrated and the residue was subjected to silica gel column chromatography, using ethyl acetate and hexanes (5:95) as eluent to afford the pure product 3.
2.3 Characterization data
2.3.1 S-p-Tolyl 4-nitrobenzothiolate (3aa):66
Yield: 131 mg (96%) as light yellow solid; M.p.: 101–103 °C; 1H NMR (400 MHz, CDCl3): δ 8.34 (d, J = 8.4 Hz, 2H), 8.17 (d, J = 8.8 Hz, 2H), 7.46 (s, 1H), 7.39 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 2.42 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.4, 150.7, 141.3, 140.6, 134.91, 130.5, 128.6, 124.1, 122.6, 21.5 ppm.
2.3.2 S-Phenyl 4-nitrobenzothiolate (3ab):67
Yield: 124 mg (96%) as light yellow solid; MP: 141–143 °C; 1H NMR (400 MHz, CDCl3): δ 8.35 (d, J = 8.8 Hz, 2H), 8.18 (d, J = 8.8 Hz, 2H), 7.54–7.44 (m, 5H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.0, 150.7, 141.3, 135.0, 130.2, 129.6, 128.6, 126.2, 124.1 ppm.
2.3.3 S-4-Chlorophenyl 4-nitrobenzothiolate (3ac):68
Yield: 136 mg (93%) as light yellow solid; M.p.: 75–77 °C; 1H NMR (400 MHz, CDCl3): δ 8.35 (d, J = 8.8 Hz, 2H), 8.17 (d, J = 8.4 Hz, 2H), 7.46 (t, J = 11.2 Hz, 4H) ppm; 13C NMR (100 MHz, CDCl3): δ 188.5, 150.8, 141.1, 136.7, 136.2, 129.9, 128.6, 124.6, 124.2 ppm.
2.3.4 S-4-Bromophenyl 4-nitrobenzothiolate (3ad):65
Yield: 150 mg (89%) as light yellow solid; M.p.: 74–76 °C; 1H NMR (400 MHz, CDCl3): δ 8.35 (d, J = 8.0 Hz, 2H), 8.17 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ 188.3, 150.9, 141.0, 136.4, 132.8, 128.6, 125.3, 125.0, 124.2 ppm.
2.3.5 S-p-Tolylbenzothiolate (3ba):69
Yield: 105 mg (92%) as white solid; M.p.: 73–75 °C; 1H NMR (400 MHz, CDCl3): δ 8.0 (d, J = 7.6 Hz, 2H), 7.58 (t, J = 7.6 Hz, 1H), 7.48–7.37 (m, 4H), 7.25 (d, J = 8.4 Hz, 2H), 2.38 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.8, 139.9, 136.9, 135.1, 133.7, 130.2, 128.8, 127.6, 123.8, 29.5 ppm.
2.3.6 S-Phenyl benzothiolate (3bb):70
Yield: 96 mg (90%) as white solid; M.p.: 51–53 °C; 1H NMR (400 MHz, CDCl3): δ 8.20 (d, J = 7.6 Hz, 1H), 8.01 (d, J = 7.6 Hz, 1H), 7.64–7.57 (m, 1H), 7.51–7.42 (m, 6H), 7.28–7.19 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.3, 136.7, 135.2, 133.8, 129.6, 129.3, 128.8, 127.6, 127.4 ppm.
2.3.7 S-4-Chlorophenyl benzothiolate (3bc):71
Yield: 106 mg (85%) as white solid; MP: 73–75 °C; 1HNMR (400 MHz, CDCl3): δ 8.02 (d, J = 8.0 Hz, 2H), 7.62 (t, J = 7.2 Hz, 1H), 7.52–7.44 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.7, 136.4, 136.1, 134.0, 129.6, 128.9, 127.6, 125.9 ppm.
2.3.8 S-4-Bromophenyl benzothiolate (3bd):72
Yield: 125 mg (85%) as white solid; M.p.: 70–71 °C; 1H NMR (400 MHz, CDCl3): δ 8.01 (d, J = 7.6 Hz, 2H), 7.64–7.58 (m, 3H), 7.50 (t, J = 7.6 Hz, 2H), 7.38 (d, J = 7.6 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.6, 136.6, 136.4, 134.0, 132.6, 128.9, 127.6, 126.5, 124.4 ppm.
2.3.9 S-Napthalen-2-yl benzothiolate (3be):73
Yield: 116 mg (88%) as white solid; M.p.: 101–103 °C; 1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 7.2 Hz, 2H), 7.93–7.85 (m, 3H), 7.65–7.49 (m, 6H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.6, 136.7, 135.1, 133.8, 133.7, 133.5, 131.5, 128.9, 128.9, 128.1, 127.9, 127.6, 127.3, 126.6, 124.8 ppm.
2.3.10 S-p-Tolyl 3-methylbenzothiolate (3ca):74
Yield: 108 mg (89%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.84–7.81 (m, 2H), 7.44–7.34 (m, 4H), 7.28–7.26 (m, 2H), 2.43 (s, 3H), 2.41 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.8, 139.8, 138.7, 136.8, 135.1, 134.4, 130.2, 128.7, 128.0, 124.8, 124.0, 21.5, 21.4 ppm.
2.3.11 S-Phenyl 3-methylbenzothiolate (3cb):71
Yield: 101 mg (89%) as white solid; M.p.: 95–97 °C; 1H NMR (400 MHz, CDCl3): δ 7.83–7.81 (m, 1H), 7.52–7.34 (m, 6H), 7.28–7.19 (m, 1H), 2.42 (s, 3H), 2.41 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.3, 138.8 136.8, 135.2, 134.5, 129.6, 129.3, 128.7, 128.0, 128.0, 124.8, 21.4 ppm.
2.3.12 S-4-Chlorophenyl 3-methylbenzothiolate (3cc):65
Yield: 105 mg (83%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.83–7.81 (m, 2H), 7.44–7.36 (m, 6H), 2.44 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.8, 138.8, 136.5, 136.4, 136.0, 134.7, 129.6, 128.8, 128.0, 126.1, 124.8, 21.4 ppm.
2.3.13 S-Naphthalen-2-yl 3-methylbenzothiolate (3ce):65
Yield: 115 mg (83%) as white solid; M.p.: 95–97 °C; 1H NMR (400 MHz, CDCl3): δ 8.06 (s, 1H), 7.94–7.85 (m, 5H), 7.58–7.38 (m, 5H), 2.45 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.6, 141.6, 138.8, 136.8, 135.1, 134.6, 133.7, 133.5, 131.5, 128.9, 128.1, 127.9, 127.3, 126.6, 126.6, 124.9, 124.9, 21.5 ppm.
2.3.14 S-p-Tolyl 4-methoxybenzothiolate (3da):71
Yield: 107 mg (91%) as light yellow solid; M.p.: 61–63 °C; 1H NMR (400 MHz, CDCl3): δ 8.0 (d, J = 9.2 Hz, 2H), 7.39 (d, J = 10.0 Hz, 2H), 7.27–7.25 (m, 2H), 6.95 (d, J = 9.2 Hz, 2H), 3.88 (s, 3H), 2.40 (s, 3H) ppm; 13CNMR (100 MHz, CDCl3): δ 189.1, 164.0, 139.7, 135.2, 130.13, 129.8, 129.6, 124.1, 114.0, 55.6, 21.5 ppm.
2.3.15 S-Phenyl 4-methoxybenzothiolate (3db):70
Yield: 110 mg (90%) as white solid; M.p.: 92–94 °C; 1H NMR (400 MHz, CDCl3): δ 8.16 (d, J = 8.8 Hz, 1H), 8.01 (d, J = 8.8 Hz, 1H), 7.52–7.40 (m, 3H), 7.28–7.19 (m, 2H), 6.97 (t, J = 10.0 Hz, 2H), 3.90 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 188.7, 135.3, 132.4, 129.8, 129.52, 129.3, 125.8, 121.9, 113.9, 55.6 ppm.
2.3.16 S-4-Chlorophenyl 4-methoxybenzothiolate (3dc):71
Yield: 121 mg (87%) as white solid; M.p.: 93–95 °C; 1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 8.8 Hz, 2H), 7.43 (t, J = 9.6 Hz, 4H), 6.96 (d, J= 8.8 Hz, 2H), 3.89 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 188.1, 164.2, 136.5, 135.9, 129.8, 129.5, 129.2, 126.2, 114.1, 55.7 ppm.
2.3.17 S-4-Bromophenyl 4-methoxybenzothiolate (3dd):71
Yield: 126 mg (78%) as white solid; M.p.: 102–104 °C; 1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 8.8 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.0 Hz, 2H), 6.96 (d, J= 8.4 Hz, 2H), 3.89 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 187.9, 164.2, 136.7, 132.4, 129.9, 129.2, 126.9, 124.2, 114.1, 55.7 ppm.
2.3.18 S-Naphthalen-2-yl 4-methoxybenzothiolate (3de):65
Yield: 93 mg (75%) as white solid; M.p.: 123–125 °C; 1H NMR (400 MHz, CDCl3): δ 8.06 (s, 1H), 7.94–7.85 (m, 4H), 7.58–7.38 (m, 5H), 7.23–7.17 (m, 1H), 2.45 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 188.9, 164.1, 135.1, 133.7, 133.5, 131.7, 129.8, 129.5, 128.8, 128.1, 127.9, 127.2, 126.6, 125.1, 114.0, 55.7 ppm.
2.3.19 S-p-Tolylethanethiolate (3ea):75
Yield: 69 mg (83%) as yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.29 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 8.0 Hz, 2H), 2.40 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 194.8, 139.8, 134.5, 130.1, 124.5, 30.2, 21.4 ppm.
2.3.20 S-Phenyl ethanethiolate (3eb):76
Yield: 61 mg (80%) as yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.42 (m, 5H), 2.43 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 194.2, 134.5, 129.5, 129.3, 128.0, 30.3 ppm.
2.3.21 S-4-Chlorophenyl ethanethiolate (3ec):76
Yield: 73 mg (78%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.42–7.26 (m, 4H), 2.42 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 193.5, 135.9, 135.7, 129.5, 126.4, 30.3 ppm.
2.3.22 S-4-Bromophenyl ethanethiolate (3ed):75
Yield: 90 mg (78%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.52 (d, J= 8.0 Hz, 2H), 7.26–7.24 (m, 2H), 2.41 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 193.4, 136.0, 132.5, 127.0, 124.2, 30.3 ppm.
2.3.23 S-Naphthalen-2-yl ethanethiolate (3ee):75
Yield: 81 mg (80%) as white solid; M.p.: 105–107 °C; 1H NMR (400 MHz, CDCl3): δ 7.98–7.95 (m, 1H), 7.88–7.73 (m, 4H), 7.53–7.51 (m, 1H), 7.46–7.44 (m, 1H), 2.46 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 194.5, 134.4, 133.6, 133.4, 130.98, 128.9, 128.1, 127.9, 127.3, 126.7, 125.3, 30.4 ppm.
2.3.24 S-Benzyl 4-nitrobenzothiolate (3af):77
Yield: 132 mg (97%) as light yellow solid; M.p.: 80–82 °C; 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 8.8 Hz, 2H), 8.09 (d, J = 8.8 Hz, 2H), 7.37–7.24 (m, 5H) 4.34 (s, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.9, 150.5, 141.4, 136.6, 129.1, 128.8, 128.3, 127.7, 124.0, 34.0 ppm.
2.3.25 S-Benzyl benzothiolate (3bf):71
Yield: 108 mg (95%) as white solid; M.p.: 36–38 °C; 1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 7.6 Hz, 2H), 7.56 (t, J = 7.6 Hz, 1H), 7.45–7.38 (m, 4H), 7.34–7.24 (m, 3H) 4.34 (s, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ 191.4, 137.6, 136.9, 133.6, 129.1, 128.8, 128.8, 127.5, 127.4, 33.5 ppm.
2.3.26 S-Benzyl 3-methylbenzothiolate (3cf):77
Yield: 109 mg (90%) as white solid; M.p.: 81–83 °C; 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 6.8 Hz, 2H), 7.38–7.22 (m, 7H), 4.31 (s, 2H), 2.39 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 191.5, 138.6, 137.6, 136.9, 134.3, 129.1, 128.7, 128.6, 127.9, 127.4, 124.6, 33.4, 21.4 ppm.
2.3.27 S-Benzyl 4-methoxybenzothiolate (3df):78
Yield: 107 mg (92%) as white solid; M.p.: 53–55 °C; 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 8.8 Hz, 2H), 7.38–7.22 (m, 5H), 6.91 (d, J = 8.8 Hz, 2H), 4.30 (s, 2H), 3.84 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.9, 163.9, 137.9, 129.7, 129.6, 129.1, 128.7, 127.3, 113.9, 55.6, 33.3 ppm.
2.3.28 S-Benzyl ethanethiolate (3ef):75
Yield: 73 mg (88%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.31–7.20 (m, 5H), 4.10 (s, 2H), 2.33 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 195.3, 137.7, 128.9, 128.7, 127.4, 35.5, 30.4 ppm.
2.3.29 S-Cyclohexyl 4-nitrobenzothiolate (3ag):79
Yield: 125 mg (94%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 8.4 Hz, 2H), 8.09 (d, J = 8.4 Hz, 2H), 3.81–3.71 (m, 1H), 2.04–1.96 (m, 2H), 1.77–1.74 (m, 2H), 1.65–1.59 (m, 2H), 1.53–1.45 (m, 2H), 1.37–1.30 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.4, 150.4, 142.1, 128.2, 123.9, 43.5, 33.0, 26.0, 25.6 ppm.
2.3.30 S-Cyclohexylbenzothiolate (3bg):78
Yield: 102 mg (93%) as yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 7.2 Hz, 2H), 7.53 (t, J = 7.6 Hz, 1H), 7.42 (t, J = 8.0 Hz, 2H), 3.76–3.70 (m, 1H), 2.04–2.01 (m, 2H), 1.76–1.73 (m, 3H), 1.63–1.60 (m, 2H), 1.56–1.43 (m, 2H), 1.35–1.28 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ 191.9, 137.5, 133.2, 128.6, 127.2, 42.6, 33.2, 26.1, 25.7 ppm.
2.3.31 S-Cyclohexyl3-methylbenzothiolate (3cg):65
Yield: 104 mg (89%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.77–7.73 (m, 2H), 7.36–7.19 (m, 2H), 3.78–3.70 (m, 1H), 2.46–2.38 (m, 3H), 2.07–2.00 (m, 2H), 1.82–1.73 (m, 2H), 1.68–1.56 (m, 2H), 1.40–1.30 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ 192.1, 138.4, 137.6, 133.9, 128.5, 127.7, 124.4, 42.5, 33.3, 26.1, 25.7, 21.4 ppm.
2.3.32 S-Cyclohexyl 4-methoxybenzothiolate (3dg):41
Yield: 113 mg (90%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 3.82 (s, 3H), 3.71–3.64 (m, 1H), 2.02–1.99 (m, 2H), 1.80–1.71 (m, 2H), 1.62–1.46 (m, 5H), 1.44–1.28 (m, 1H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.4, 163.6, 130.4, 129.3, 113.7, 55.5, 42.4, 33.4, 26.1, 25.7 ppm.
2.3.33 Methyl 3-(4-nitrobenzoylthio)propanoate (3ah):65
Yield: 121 mg (90%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 8.4 Hz, 2H), 8.09 (d, J = 8.4 Hz, 2H), 3.71 (s, 3H), 3.35 (t, J = 6.8 Hz, 2H), 2.75 (t, J = 6.8 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.1, 172.0, 150.6, 141.4, 128.3, 124.0, 52.1, 33.9, 24.6 ppm.
2.3.34 Methyl 3-(benzoylthio)propanoate (3bh):65
Yield: 106 mg (95%) as colourless oil; 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 7.2 Hz, 2H), 7.55 (t, J = 7.6 Hz, 1H), 7.42 (t, J = 7.6 Hz, 2H), 3.69 (s, 3H), 3.30 (t, J = 6.8 Hz, 2H), 2.72 (t, J = 7.2 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ 191.52, 172.22, 136.87, 133.57, 128.70, 127.29, 51.93, 34.33, 24.09 ppm.
2.3.35 Methyl 3-(3-methylbenzoylthio)propanoate (3ch):65
Yield: 105 mg (88%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.71 (d, J = 6.4 Hz, 2H), 7.34–7.13 (m, 2H), 3.67 (s, 3H), 3.26 (t, J = 7.2 Hz, 2H), 2.69 (t, J= 7.2 Hz, 2H), 2.36 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 191.7, 172.3, 138.6, 136.9, 134.4, 128.6, 127.8, 124.5, 52.0, 34.4, 24.1, 21.4 ppm.
2.3.36 Methyl 3-(4-methoxybenzoylthio)propanoate (3dh):80
Yield: 95 mg (85%) as light yellow oil; 1H NMR (400 MHz, CDCl3): δ 7.90 (d, J = 8.8 Hz, 2H), 6.89 (d, J= 8.8 Hz, 2H), 3.83 (s, 3H), 3.68 (s, 3H), 3.27 (t, J = 7.2 Hz, 2H), 2.70 (t, J = 6.8 Hz, 2H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.9, 173.2, 163.9, 129.8, 129.5, 113.8, 55.6, 51.9, 34.5, 24.0 ppm.
2.3.37 S-o-Tolylbenzothiolate (3ib):66
Yield: 106 mg (93%) as colorless oil; 1H NMR (400 MHz, CDCl3): δ 8.05 (d, J = 7.6 Hz, 2H), 7.61 (t, J = 7.2 Hz, 1H), 7.49 (t, J = 7.2 Hz, 4H), 7.40 –7.36 (m, 2H), 2.40 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 190.5, 139.2, 136.8, 135.8, 133.7, 132.2, 130.5, 129.2, 128.8, 127.6, 127.1, 21.4 ppm.
2.3.38 S-o-Tolylethanethiolate (3ie):81
Yield: 70 mg (84%) as colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.30–7.26 (m, 1H), 7.22–7.19 (m, 3H), 2.38 (s, 3H), 2.34 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 194.5, 139.2, 136.2, 131.6, 130.4, 129.1, 127.7, 30.3, 21.4 ppm.
2.3.39 S-m-Tolylbenzothiolate (3jb):66
Yield: 102 mg (90%) as colorless oil; 1H NMR (400 MHz, CDCl3): δ 8.08 (d, J = 7.6 Hz, 2H), 7.62 (t, J = 7.6 Hz, 1H), 7.50 (t, J = 7.6 Hz, 3H), 7.42–7.38 (m, 2H), 7.30–7.26 (m, 1H), 2.43 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 189.7, 142.8, 136.9, 136.5, 133.7, 130.9, 130.3, 128.8, 127.6, 126.9, 126.8, 20.9 ppm.
2.3.40 S-m-Tolylethanethiolate (3je):82
Yield: 72 mg (87%) as colorless oil; 1H NMR (400 MHz, CDCl3): δ 7.40 (d, J = 8.0 Hz, 1H), 7.36–7.31 (m, 2H), 7.24–7.20 (m, 1H), 2.42 (s, 3H), 2.36 (s, 3H) ppm; 13C NMR (100 MHz, CDCl3): δ 193.9, 142.0, 137.0, 130.8, 130.2, 127.5, 126.7, 30.3, 20.8 ppm.
3 Results and Discussion
Initially, 4-nitrobenzoic acid (1a) and 4-methylthiophenol (2a) were chosen as model substrates to screen the optimal conditions, and the results are summarized in Table 1. In our first attempt, trifluoroacetic anhydride (TFAA) was used for the preparation of thioester. Delightedly, 90% yield of the desired thiol ester 3aa was obtained, when the reaction was carried out in acetonitrile at 80 °C (entry 1). While this fluorinated reagent was beneficial, it suffered from a few drawbacks such as the mass of waste associated with stoichiometric reagent and the process for the disposal of fluorinated waste streams found to be difficult. The reaction was sensitive to solvent: it appeared limited to aprotic solvents in a model scheme (entries 2 and 3), and very low yield of product was observed with the use of tetrahydrofuran (THF) under otherwise identical conditions (entry 3) but there was no formation of even traces of thiolate in solvent-free case (entry 4). Afterwards, we began to consider how to improve this reaction by avoiding the use of halogenated reagents. We reasoned that a stronger acid than TFAA may extend the reaction in a green and economic fashion. Thus, we sought a strong acid, with a low molecular weight, and preferably not halogenated to aid waste treatment. This consideration led us to use methanesulfonic anhydride (MSAA). We began the work by screening conditions similar to those used with TFAA. Gratifyingly, the desired product 3aa was obtained in excellent yield when the reaction was carried out under the influence of MSAA in dichloromethane (entry 5). Switching the solvent to acetonitrile did not affect the rate of reaction and the yield of the product (entry 6). The reaction worked well under neat conditions and the reactivity of precursors 1a and 2a was preserved (entry 7). Probably, the reaction of MSAA with thiol under the influence of MSAA may be proceeding via the in situ generation of mixed anhydride species which releases methanesulfonic acid. This acid could be strong enough to facilitate the subsequent reaction with thiol leading to thioester. Then this process was examined to check the impact on reaction time and yield by varying the quantity of MSAA. Decreasing the loading of MSAA from 1.3 to 1.0 equiv., resulted in a somewhat diminished rate of the reaction and the yield of 3aa (entries 8 and 9). This screening led to the identification that stoichiometric amount of MSAA is necessary to furnish the product 3aa in excellent yield. It is noteworthy that variation in the yield of product was observed while changing the reaction temperature. Excellent amount of product was formed at 80 °C (entry 4); lowering the reaction temperature up to 40 °C gave the product in reduced yield (entry 10). Even longer reaction time could not improve the yield of the product (entry 11), yet the reaction at 100 °C did not exhibit obvious promotion in the rate of reaction and the yield of 3aa (entry 12) with respect to the results of entry 7. The reaction failed to promote the carbonylation of 1a in the absence of MSAA, indicating the important role of this anhydride in the direct synthesis of thioester (entry 13). Thus the exclusive tuning of 1a and 2a with MSAA at 80 °C provided the best conditions for further studies.

After optimizing the reaction conditions (Table 1, entry 7), we investigated the substrate scope and generality for this novel thioester-forming protocol. For this purpose, the reactions of acids 1 with aryl thiols 2a–2e were first studied, and the results are collected in Table 2. Thiophenol and its derivatives with electron-donating and withdrawing substituents at the para-position of the phenyl group, when treated with 1a, were transformed into the corresponding products 3aa–3ad with very good to excellent yields. The reaction of 1a with bulky substrate 2-naphthol (2e) furnished the thiolate 3ae in 85% yield. Similarly, benzoic acid (1b) reacted smoothly with 2a–2e, and afforded the target products 3ba–3be in high yields. Encouraged by these results, benzoic acids bearing electron-donating substituents 1c and 1d were next investigated to check the efficacy of the present reaction system. These substrates responded nicely in the current protocol to furnish the target compounds 3ca, 3cb, 3cc, 3ce and 3da–3de in good to excellent yields. Remarkably, aliphatic acid 1e could also provide the aryl alkyl thioesters 3ea–3ee in good yields ranging from 78–83%.
With the promising results obtained in the case of aryl thiols, we turned our attention towards alkyl thiols to broaden the substrate scope of this elegant methodology. As revealed in Table 3, the developed process once again showed good functional group compatibility. Carboxylic acids possessing electron-withdrawing groups displayed high reactivity with benzylmercaptan (2f) under the optimized conditions and generated the product 3af in excellent yield. Benzoic acid and its derivatives bearing m-Me and p-OMe substituents (1b, 1c and 1d) served as powerful aroyl surrogates when treated with thiol
2f and furnished the S-aroylated products 3bf, 3cf and 3df in high yields. Moreover, it was possible to expand the scope of this carbonylation process to acetic acid and the product 3ef was seen in a very good yield. As expected, benzoic acids 1a–1d worked well with cyclohexanethiol (2g) and afforded their corresponding products 3ag–3dg in 90–94% isolated yields.
Further, synthetically attractive ester functionalized thiol 2h, acts as an ideal substrate with 1a–1d in this protocol and furnished the alkyl thioesters 3ah–3dh in very good yields.
With the above positive results, structurally diverse thiocresols were explored as well to examine their substituent effect at different positions of phenyl ring on product outcome, and the representative results are presented in Figure 1. Thiocresols such as ortho-thiocresol (2i) and meta-thiocresol (2j) were found to be ideal reaction partners with benzoic acid (2b) and acetic acid (2e) under similar reaction conditions affording the corresponding products 3ib, 3ie, 3jb as well as 3je in good to excellent yields. The above observations explicate that thiophenols with the methyl group in ortho-, meta- or para-position showed no significant difference in the yields. Compared to the 92% yield of 3ib might be due to the steric effect of the methyl group on the ortho-position and similarly in the case of 3ea and 3ie.

Scope of thiocresols: Reaction conditions: feedstock acid 1 (0.5 mmol), thiocresol 2i/2j (0.6 mmol), MSAA (0.65 mmol), 80 °C, 4 h.
Finally, in order to demonstrate the synthetic utility of developed process; the reaction of 4-nitrobenzoic acid (1a) and 4-methylthiophenol (2f) as starting materials was conducted on a 5 mmol scale under similar reaction conditions. The product S-benzyl 4-nitrobenzothiolate (3af) was isolated in a quite good amount. This confirmed the efficacy of the present solvent-free track for the gram-scale preparation of thioesters (Scheme 2).

Gram-scale synthesis of thioester 3af.
All these results described above indicated that in situ generated mixed anhydride mediates the coupling with thiols which is the key step for the success of this dehydrative thioesterification process. Thus, the present methodology for the carbonylation of thiols under neat conditions is highly versatile and holds the potential for the synthesis of a large variety of thioesters.
On the basis of the above experimental results and the previous literature report,59 a plausible mechanism for this transformation is proposed in Scheme 3. The in situ generation of mixed anhydride intermediate 4 from carboxylic acid 1 and MSAA triggers the reaction by the release of methanesulfonate and activated complex 5 of carboxylic acid 1. The ensuing intermolecular attack of thiol 2 toward 5 and subsequent elimination of methanesulfonate liberates the thioester product 3. The methanesulfonate combines with proton to give the by-product methanesulfonic acid (6).

Plausible reaction mechanism.
4 Conclusions
In summary, we have established an efficient and novel strategy for dehydrative nucleophilic substitution reaction of feedstock acids with thiols, which systematically unravels the feasibility and practicality of thioester formation in a step- and atom-economical fashion. The successful implementation of this C–S bond-forming strategy relies on the in situ generation of mixed anhydride intermediate from carboxylic acid and cheap and easily handled MSAA which was an initial starting point to drive this reaction. The reaction can be run at gram-scale. Moreover, the power of this sustainable paradigm for the synthesis of thioesters has been fully exemplified by the tolerance of various functionalities that can serve as useful synthetic handles for subsequent chemical manipulation. We believe that this metal-, halogen- and solvent-free approach for the synthesis of thioesters will generate broad applications among practitioners of synthetic, pharmaceutical and industrial chemistry. Exploring the utility of MSAA to construct other useful products is currently underway in our laboratory.
References
Khour C B, Darth, C B, Lalnger A and Daves M E 1994 Studies on the catalytic oxidation of alkanes and alkenes by titanium silicates J. Catal. 149 195
Andrade C K Z and Alves L M 2005 Environmentally benign solvents in organic synthesis: Current topics Curr. Org. Chem. 9 195
Aparicio S and Alcalde R 2009 The green solvent ethyl lactate: an experimental and theoretical characterization Green Chem. 11 65
Elkington J 1994 Towards the sustainable corporation: win-win-win sustainable development Calif. Manag. Rev. 36 90
Staunton J and Weissman K J 2001 Polyketide biosynthesis: A millennium review Nat. Prod. Rep. 18 380
Yang H, Li H, Wittenberg R, Egi M, Huang W and Liebeskind L S 2007 Ambient temperature synthesis of high enantiopurity N-protected peptidyl ketones by peptidyl thiol ester-boronic acid cross-coupling J. Am. Chem. Soc. 129 1132
Crich D and Sharma I 2009 Epimerization-free block synthesis of peptides from thioacids and amines with the Sanger and Mukaiyama reagents Angew. Chem. Int. Ed. 48:2355
Kunchithapatham K, Eichman C C and Stambuli J P 2011 Synthesis of diaryl ketones via a phosphine-free Fukuyama reaction Chem. Commun. 47 12679
Keating T A and Walsh C T 1999 Initiation, elongation, and termination strategies in polyketide and polypeptide antibiotic biosynthesis Curr. Opin. Chem. Biol. 3 598
Khosla C, Tang Y, Chen A Y, Schnarr N A and Cane D E 2007 Structure and mechanism of the 6-deoxyerythronolide B synthase Annu. Rev. Biochem. 76 195
Tokuyama H, Yokoshima S, Yamashita T and Fukuyama T 1998 A novel ketone synthesis by a palladium-catalyzed reaction of thiol esters and organozinc reagents Tetrahedron Lett. 39 3189
Ueda M, Seki K and Imai Y 1981 S- and N-acyl derivatives of 2-mercaptobenzoxazole: New, highly reactive acylating agents for synthesis of amides and esters Synthesis 12 991
Mehta V P, Sharma A and Eycken E V 2008 The first palladium-catalyzed desulfitative sonogashira-type cross-coupling of (hetero)aryl thioethers with terminal alkynes Org. Lett. 10 1147
Azuma H, Okano K and Tokuyama H 2011 Synthesis of acylsilanes by palladium-catalyzed cross-coupling reaction of thiol esters and silylzinc chlorides Chem. Lett. 40 959
Kotsuki H, Yoshimura N, Ushio Y, Ohtsuka T and Ochi M 1986 Facile reduction of benzenethiol esters under mild conditions with zinc borohydride Chem. Lett. 15 1003
Fukuyama T, Lin S C and Li L 1990 Facile reduction of ethyl thiol esters to aldehydes: application to a total synthesis of (+)-neothramycin A methyl ether J. Am. Chem. Soc. 112 7050
Nagashima E, Suzuki K and Sekiya M 1983 Reactions of 2,2-dichlorovinyl and 2,2-dichlorovinylidene sulfides with butyllithium Chem. Pharm. Bull. 31 3306
Yousif N M, Pedersen U, Yde B and Lawesson S O 1984 Studies on organophosphorus compounds. XLVIII. Synthesis of dithioesters from phosphorus- and sulfur-containing reagents and carboxylic acids and their derivatives Tetrahedron 40 2663
Alvarez-Ibarra C, Mendoza M, Orellana G and Quiroga M L 1989 A novel method of synthesis of 2-(methylthio)-1,3-oxazoles Synthesis 7 560
Steglich W and Höfle G 1969 N,N-Dimethyl-4-pyridinamine, a very effective acylation catalyst Angew. Chem. Int. Ed. Engl. 8 981
Vedejs E, Bennett N S Conn L M, Diver S T, Gingras M, Lin S, Oliver P A and Peterson M J 1993 Tributylphosphine-catalyzed acylations of alcohols: scope and related reactions J. Org. Chem. 58 7286
Ishihara K, Kubota M, Kurihara H and Yamamoto H 1996 scandium trifluoromethanesulfonate as an extremely active Lewis acid catalyst in acylation of alcohols with acid anhydrides and mixed anhydrides J. Org. Chem. 61 4560
Ishihara K, Kubota M and Yamamoto H 1996 A new scandium complex as an extremely active acylation catalyst Synlett 39 265
Procopiou P A, Baugh S P D, Flack S S and Inglis G G A 1998 An extremely powerful acylation reaction of alcohols with acid anhydrides catalyzed by trimethylsilyl trifluoromethanesulfonate J. Org. Chem. 63 2342
Saravanan P and Singh V K 1999 An efficient method for acylation reactions Tetrahedron Lett. 40 2611
Pansare S V, Malusare M G, Pansare S V, Malusare M G and Rai A N 2000 Magnesium bromide catalyzed acylation of alcohols Synth. Commun. 30 2587
Derdau V and Snieckus V 2001 Condensation of laterally lithiated o-methyl and o-ethyl benzamides with imines mediated by (-)-sparteine. Enantioselective synthesis of tetrahydroisoquinolin-1-ones J. Org. Chem. 66 1992
Nakae Y, Kusaki I and Sato T 2001 Lithium perchlorate catalyzed acetylation of alcohols under mild reaction conditions Synlett 10 1584
Orita A, Tanahashi C, Kakuda A and Otera J 2001 Highly powerful and practical acylation of alcohols with acid anhydride catalyzed by Bi(OTf)3 J. Org. Chem. 66 8926
Kumar P, Pandey R K, Bodas M S, Dagade S P, Dongare M K and Ramaswamy A V 2002 Acylation of alcohols, thiols, and amines with carboxylic acids catalyzed by yttria-zirconia-based lewis acid J. Mol. Catal A Chem. 181 207
Shah S T A, Khan K M, Heinricha A M and Voelter W 2002 An alternative approach towards the syntheses of thioethers and thioesters using CsF-Celite in acetonitrile Tetrahedron Lett. 43 828
Chakraborti A K and Gulhane R 2003 Indium(III) chloride as a new, highly efficient, and versatile catalyst for acylation of phenols, thiols, alcohols, and amines. Tetrahedron Lett. 44 6749
Hao Z, Xi W, Wang P and Cai M 2009 Ruthenium(III) chloride catalyzed acylation of alcohols, phenols, and thiols in room temperature ionic liquids Molecules 14 3528
Basu B, Paul S and Nanda A K 2010 Silica-promoted facile synthesis of thioesters and thioethers: A highly efficient, reusable and environmentally safe solid support Green Chem. 12 767
Werner K, Robert K H, Christina S, Fritz H, Christine S, Hannelore D and Jurgen S 2012 A single-cell NMR membrane transport assay Eur. J. Chem. 9 2501
Prajapti S K, Nagarsenkar A and Babu B N 2014 Tris(pentafluorophenyl)borane catalyzed acylation of alcohols, phenols, amines, and thiophenols under solvent-free condition Tetrahedron Lett. 55 1784
Sucheta K, Reddy G S R, Ravi D and Rao N R 1994 A novel, general route to the synthesis of carboxylic acid esters and thiol esters Tetrahedron Lett. 35 4415
Bandgar B P and Pandit S S 2004 A novel and direct synthesis of thiolesters using cyanuric chloride under mild conditions J. Sulfur Chem. 25 343
Roy H N, Sarker A K and Al Mamun A H 2010 Rapid and regiospecific phenylthiolation of some organic acids catalyzed by AlCl3 in the presence of excess anhydrous ZnCl2 Synth. Commun. 40 2158
Lara R G, Rodrigues D C, Samue R, Mendes Panatieri R B, Jacob R G, Alves D, Lenardáo E J and Perin G 2011 Synthesis of thiol esters by the reaction of ricinoleic acid with thiols under solvent-free conditions Synth. Commun. 41 2974
El-Azab A S, Abdel-Aziz and A A.-M 2012 An efficient synthesis of thioesters via TFA-catalyzed reaction of carboxylic acid and thiols: remarkably facile C-S bond formation Phosphorus, Sulfur Silicon Relat. Elem. 187 1046
Katritzky A R, He H Y and Suzuki K 2000 N-acylbenzotriazoles: neutral acylating reagents for the preparation of primary, secondary, and tertiary amides J. Org. Chem. 65 8210
Prasad H S, Srinivasa G R, Gowda D C 2005 Convenient, cost-effective, and mild method for the N-acetylation of anilines and secondary amines Synth. Commun. 35 1189
Taylor J E, Jones M D, Williams J M J and Bull S D 2012 N-acyl DBN tetraphenylborate salts as N-acylating agents J. Org. Chem. 77 2808
Taylor J E, Williams J M J, Bull S D 2012 N-Acyl 1,5-diazabicyclo[4.3.0] non-5-ene (DBN) tetraphenylborate salts as O-acylating agents Tetrahedron Lett. 53 4074
Chikkulapalli A, Aavula S K, Mona R N P, Karthikeyan C, Kumar V C, Sulur M G and Sumathi S 2015 Convenient N-acetylation of amines in N,N-dimethylacetamide with N,N-carbonyldiimidazole Tetrahedron Lett. 56 3799
Gernon M D, Wu M, Buszta T and Janney P 1999 Environmental benefits of methanesulfonic acid. Comparative properties and advantages Green Chem. 1 127
Jamshad M, Murrell J C and Fülöp V 2007 Purification and crystallization of the hydroxylase component of the methanesulfonate monooxygenase from Methylosulfonomonas methylovora strain M2 Protein Expr. Purif. 52 472
Susperregui N, Delcroix D, Martin-Vaca B, Bourissou D and Maron, L 2010 Ring-opening polymerization of ε-caprolactone catalyzed by sulfonic acids: Computational evidence for bifunctional activation J. Org. Chem. 75 6581
Boyle R and Venkataramani E S 1995 Biodegradation of methanesulfonic acid PCT Int. Appl. WO 9521135 (CAN 123:207938)
Stott P E, Bradshaw J S, Parish W W and Copper J W 1980 Modified crown ether catalysts. 2. Synthesis of alkanoyl-, aroyl-, α-hydroxyalkyl- and alkylbenzo and alkylcyclohexano crown ethers J. Org. Chem. 45 4716
Kelly T R and Ghoshal M 1985 Expeditious synthesis of resistomycin J. Am. Chem. Soc. 107 3879
Eck G, Julia M, Pfeiffer B and Rolando C 1985 Access to the spiro hydrindandione ring system of fredericamycin A through a Friedel-Crafts reaction Tetrahedron Lett. 26 4723
Li J J, Mitchell L H and Dow R L 2010 Thyroid receptor agonists for the treatment of androgenetic alopecia Biorg. Med. Chem. Lett. 20 306
Wu Z, Guo W, Lian G and Yu B 2010 Synthesis of mangiferin, isomangiferin, and homomangiferin J. Org. Chem. 75 5725
Sharghi H and Kaboudon B J 1998 Alumina in methanesulfonic acid (AMA) as a new efficient reagent for direct acylation of phenol derivatives and Fries rearrangement. a convenient synthesis of o-hydroxyarylketones J. Chem. Res., Synop. 10 628
Sharghi H and Hosseini-Sarvari M 2004 Simple and improved procedure for the regioselective acylation of aromatic ethers with carboxylic acids on the surface of graphite in the presence of methanesulfonic acid Synthesis 13 2165
Sharghi H, Hosseini-Sarvari M and Eskandari R 2006 Direct acylation of phenol and naphthol derivatives in a mixture of graphite and methanesulfonic acid Synthesis 12 2047
Wilkinson M C 2011 “Greener” Friedel-Crafts acylations: a metal- and halogen-free methodology Org. Lett 13 2232
Choudhary G, Peddinti R K 2011 An expeditious, highly efficient, catalyst-free and solvent-free synthesis of nitroamines and nitrosulfides by Michael addition Green Chem. 13 276
Parumala S K R and Peddinti R K 2015 Iodine catalyzed cross-dehydrogenative C–S coupling by C(sp2–H bond activation: direct access to aryl sulfides from aryl thiols Green Chem. 17 4068
Singh P and Peddinti R K 2017 Waste-free swift synthesis of symmetrical and unsymmetrical diarylmethyl thioethers from diaryl carbinols Synthesis 49 3633
Singh P, Singh U P and Peddinti R K 2017 PTSA-catalyzed functionalization of hydroquinones with benzhydryl alcohols in water Tetrahedron Lett. 58 2813
Singh P and Peddinti R K 2017 Metal-free alkyl(aryl) transfer–aromatization–alkylation domino approach: facile synthesis of branched hydroquinones from p-quinols and diaryl carbinols ChemistrySelect 2 3622
Singh P and Peddinti R K 2017 Harnessing the catalytic behaviour of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP): An expeditious synthesis of thioesters Tetrahedron Lett. 58 1875
Singh S and Yadav L D S 2012 The direct thioesterification of aldehydes with disulfides via NHC-catalyzed carbonyl umpolung strategy Tetrahedron Lett. 53 5136
Dan W, Deng H, Chen J, Ding J and Wu H 2010 A new odorless one-pot synthesis of thioesters and selenoesters promoted by Rongalite® Tetrahedron 66 7384
Feng J, Lv M-F, Lu G-P and Cai C 2015 Direct oxidative coupling of thiols and benzylic ethers via C(sp3)–H activation and C–O cleavage to lead thioesters Org. Biomol. Chem. 13 677
Rong G, Mao J, Liu D, Yan H, Zheng Y and Chen J 2015 Formation of C(sp2)–S bonds through decarboxylation of α-oxocarboxylic acids with disulfides or thiophenols RSC Adv. 5 26461
Zhu X, Shi Y, Mao H, Cheng Y and Zhu C 2013 Tetraethylammonium bromide-catalyzed oxidative thioesterification of aldehydes and alcohols Adv. Synth. Catal. 355 3558
Ali W, Guin S, Rout S K, Gogoi A and Patel B K 2014 Thioesterification of alkylbenzenes with thiols via copper-catalyzed cross-dehydrogenative coupling without a directing group Adv. Synth. Catal. 356 3099
He C, Qian X and Sun P 2014 Syntheses of thiol and selenol esters by oxidative coupling reaction of aldehydes with RYYR (Y = S, Se) under metal-free conditions Org. Biomol. Chem. 12 6072
Shakoor S M A, Choudhary S, Bajaj K, Muthyala M K, Kumar A and Sakhuja R 2015 Imidazolium-supported benzotriazole: an efficient and recoverable activating reagent for amide, ester and thioester bond formation in water RSC Adv. 5 82199
Yan K, Yang D, Wei W, Zhao J, Shuai Y, Tian L and Wang H 2015 Catalyst-free direct decarboxylative coupling of α-keto acids with thiols: A facile access to thioesters Org. Biomol. Chem. 13 7323
Pijper T C, Robertus J, Browne W R and Feringa B L 2015 Mild Ti-mediated transformation of t-butyl thio-ethers into thio-acetates Org. Biomol. Chem. 13 265
Kashyap B and Phukan P 2013 A new ferrocene-based bulky pyridine as an efficient reusable homogeneous catalyst RSC Adv. 3 15327
Katritzky A R, Shestopalov A A and Suzuki K 2004 A new convenient preparation of thiol esters utilizing N-acylbenzotriazoles Synthesis 11 1806
Uno T, Inokuma T and Takemoto Y 2012 NHC-catalyzed thioesterification of aldehydes by external redox activation Chem. Commun. 48 1901
Iranpoor N, Firouzabadi H, Khalili D and Motevalli S 2008 Easily prepared azopyridines as potent and recyclable reagents for facile esterification reactions. An efficient modified mitsunobu reaction J. Org. Chem. 73 4882
Swain S P, Chou Y-L and Hou D-R 2015 Thioesterifications free of activating agent and thiol: A three-component reaction of carboxylic acids, thioureas, and michael acceptors Adv. Synth. Catal. 357 2644
Lai C and Backes B J 2007 Efficient preparation of S-aryl thioacetates from aryl halides and potassium thioacetate Tetrahedron Lett. 48 3033
Petrillo G, Novi M, Garbarino G and Filiberti M 1989 The reaction between arenediazonium tetrafluoroborates and alkaline thiocarboxylates in DMSO: A convenient access to aryl thiolesters and other aromatic sulfur derivatives Tetrahedron 45 7411
Acknowledgements
P.S. thanks UGC (New Delhi) for a research fellowship.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Singh, P., Peddinti, R.K. Methanesulfonic anhydride-promoted sustainable synthesis of thioesters from feedstock acids and thiols. J Chem Sci 133, 20 (2021). https://doi.org/10.1007/s12039-020-01871-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-020-01871-5