
Abstract
A series of five new donor–acceptor molecules based on imidazo-anthraquinone-amines have been synthesized and characterized. The structure-property relationship in these molecules is systematically examined by absorption-emission spectroscopy, cyclic voltammetry and theoretical studies. Optical properties of these molecules have been studied in solvents of varying polarity as well as in neat solid film and found to be affected by the nature of triarylamine substituent with broad absorption windows, strong charge transfer transitions (425–502 nm), high molar extinction coefficients and emission in green light (500–568 nm). Electrochemical data indicated that the dyes possess relatively low-lying LUMO values (\(-3.18\) to \(-3.42\) eV) while TGA studies revealed good thermal stability. The donor-acceptor architecture and HOMO–LUMO energies were further rationalized using DFT calculations. Experimental studies along with theoretical calculations suggest that these compounds have potential to be used as n-type materials in optoelectronic devices.
GRAPHICAL ABSTRACT
Imidazoanthraquinone–amine derivatives with donor–acceptor molecular architecture were synthesized using Buchwald–Hartwig amination reaction featuring ICT absorption and low lying LUMO energy level (\(-3.18\) to \(-3.42\) eV). These are comparable with well-known n-type materials for their applications in organic electronics.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Triarylamine-containing heteroaromatics are widely investigated and applied as electro-optical materials in applications such as organic light emitting diodes (OLEDs), [1] organic field effect transistors (OFETs),[2] organic photovoltaic cells (OPVs),[3] nonlinear optical devices[4] and in chemo-sensing applications. [5] The triarylamine unit is a well-known substituent used for the synthesis of diverse organic molecules for applications in organic electronics because of its ability to alter their optoelectronic properties due to its ease of oxidation of nitrogen centre and its ability to transport charge carriers via radical cation species with high stability. [6] The introduction of electron-rich triarylamine unit into the electron-deficient core molecule creates an electron donor-acceptor (D–A) type of molecular architecture, which tunes the absorption and emission properties of the molecule through intramolecular charge transfer transitions (ICT) and extends the conjugation in the molecular structure. Further, the presence of bulkier polyaromatic groups such as carbazole, fluoranthene, pyrene, etc., around the trigonal amine nitrogen often hinders the aggregation of molecules in the solid state and found to enhance thermal stability. [7] \(\uppi \)-conjugated small organic molecules with D–A architecture is currently of interest in optoelectronic devices because of their well-defined structures, ease of purification, reliable reproducibility and better solubility in most organic solvents. [8] A variety of molecules based on D–A approach derived from electron-rich triarylamine unit and electron-deficient core such as borane-amine,[9] quinoxaline-fluorine, [10] triazole-amine,[11] bathophenanthroline-amine,[12] acridone-amine, [13] tribenzophenazine-amine [14] and oxadiazole-carbozole [15] have been synthesized and used for applications in organic electronics.
Imidazoanthraquinone derivatives attracted a lot of research attention because of their successful biological application as anticancer[16] drugs and as ion sensors. [17]In our recent publication, we have explored imidazoanthraquinone aryl derivatives for application in organic electronics. [18] However, imidazoanthraquinone-triarylamines are not yet synthesized and explored for their ability to accept electron in connection with n-type organic semiconductor application. Keeping the above mentioned benefits of triarylamines and the D–A architecture therein in mind, we report the synthesis of new D–A molecules based on 1H–anthra[1,2–d]imidazole–6,11–dione substituted with triarylamines, 1–5 (Figure 1) and their detailed photophysical, electrochemical and thermal studies.

Molecular structure of imidazoanthraquinone-amine derivatives.
The D–A architecture and HOMO-LUMO energies in context with n–type materials were further rationalized using DFT calculations. Imidazoanthraquinone acts as typical electron-accepting unit while the donor unit comprises of various triarylamine units. Incorporation of triarylamines into the imidazoanthraquinone backbone is expected to affect the optoelectronic properties effectively. Morpholine amine was used to see the effect of the rigid cyclic aliphatic amine on the opto-electrochemical properties of Imidazoanthraquinone. Further, the studies also explore the effect of electron donating (\(-\hbox {OCH}_{3}\)) and withdrawing (\(-\hbox {NO}_{2})\) substituents on the triarylamine moiety on the photophysical, electrochemical and thermal properties such as ICT, HOMO–LUMO energy, and band gap.
2 Experimental
2.1 Materials and methods
All the reagents were purchased from commercial sources (Sigma Aldrich and Alfa Aesar) and were used without any further purification. The organic solvents were of analytical or spectroscopic grade and were dried and freshly distilled using the standard procedures whenever anhydrous solvents were required. \(^{1}\)H–NMR (500 MHz) spectra were recorded on a Varian 600 MHz Ultrashield spectrometer with tetramethylsilane (TMS) as internal reference and residual \(\hbox {CHCl}_{3}\) in \(\hbox {CDCl}_{3 }\) as reference and \(^{13}\)C–NMR (75 MHz) spectra were recorded on a Bruker Avance II 300 MHz Ultrashield spectrometer with tetramethylsilane (TMS) as internal reference and residual \(\hbox {CHCl}_{3}\) in \(\hbox {CDCl}_{3}\) and DMSO–d\(_{6}\) as reference. Fourier transform infrared (FT-IR) spectra were recorded on a Perkin Elmer Frontier 91579. Mass spectrometric measurements were recorded using MALDI-TOF (Bruker). Melting points of the products were determined by differential scanning calorimetry. Elemental analyses were obtained on Elemental Analyser-EURO Vector Italy EA-3000. Cyclic voltammetry and differential pulse voltammetry were carried out on a computer controlled Palmsens[3] potentiostat/galvanostat. Typically, a three-electrode cell equipped with a glassy carbon as working electrode, Ag/AgCl (non-aqueous) as reference electrode and platinum (Pt) wire as counter electrode were employed. The measurements were carried at room temperature in anhydrous acetonitrile with tetrabutylammonium hexafluorophosphate solution (0.1 M) as supporting electrolyte with a scan rate of \(100\,\hbox {mVs}^{-1}\). The potential of Ag/AgCl reference electrode was calibrated by using ferrocene/ferrocenium redox couple. Absorption and fluorescence data were acquired using \(\sim 2\,\times \,10^{-6}\) M solutions of Std and 1–5. UV-visible spectra were recorded on SHIMADZU U.V-A114548 and fluorescence studies were done on Horiba Fluorolog 3 at room temperature. The fluorescence quantum yields (\(\Phi _{\mathrm{F}})\) were calculated relative to fluorescein (\(\Phi _{\mathrm{F}}=0.79\) in 0.1 M NaOH). The neat solid films of compounds 2–6 were prepared by using a spin coater (Holmarc HO-TH-05) at 1000 rpm for 2 min using-\(\sim -6\,\hbox {mg}\,\hbox {mL}^{-1}\) of sample in chloroform. The area of the neat solid film is \(2\,\times \,2~\hbox {cm}^{2 }\) and approximately around 100-200 \(\upmu \hbox {L}\) of the solution was used for coating. The AFM topographical images were recorded on a Bruker Dimension Icon AFM instrument. Thermal studies were performed on \(\hbox {Stare}^{\mathrm{e}}\) system Metler Toledo TGA 1 with a heating rate of \(10\,{^{\circ }}\hbox {C}\,\hbox {min}^{-1}\) under nitrogen atmosphere. Density functional theoretical (DFT) calculations were performed using Gaussian 03 software package using B3LYP as exchange-correlation functional at 6-311 G basis set.
2.2 Synthesis
The Std molecule (2-phenyl-anthra[1,2-d]imidazole- 6,11-dione) and 2-(4-bromo) phenyl -anthra[1,2-d]imidazole-6,11-dione (6) were synthesized according to the reported procedure and confirmed by their reported melting point. [16]\(^\mathrm{b}\), [17]\(^\mathrm{d}\) A mixture of 1,2-diaminoanthraquinone (3 g, 12.6 mmol) and 4-bromobenzaldehyde (2.78 g, 15 mmol) was heated in nitrobenzene (20 mL) at \(140\,{^{\circ }}\hbox {C}\) for 18 h. Then the mixture was cooled to room temperature and hexane was added to obtain a solid precipitate which was washed several times with hexane. The crude solid thus obtained was purified by column chromatography using hexane/chloroform to obtain a bright yellow solid. Yield: 4.53 g (90%); M.p. \(303\,{^{\circ }}\hbox {C}\). [16]\(^\mathrm{b}\), [17]\(^\mathrm{d}\)

Synthesis of imidazoanthraquinone-amine derivatives 1–5.
2.2.1 General method for the synthesis of compounds 1-5
In a three-necked round bottom flask equipped with a reflux condenser and argon inlet and outlet ports, 2-(4-bromo)phenyl-anthra[1,2-d]imidazole-6,11-dione (6) (1.0mmol) and diarylamine (1.2 mmol) were dissolved in anhydrous toluene (20 mL) under argon atmosphere. The palladium catalyst [\(\hbox {Pd}_{2}\hbox {(dba)}_{3}\)] (5–8 mol%), 2-dicyclohexylphoshpino-2’,6’-dimethyl biphenyl (SPhos) (10–15 mol%) and sodium-t-butoxide (3.1 mol) were added to the reaction mixture. The reaction mixture was thoroughly stirred under argon atmosphere while the temperature was slowly raised to \(100\, {^{\circ }}\hbox {C}\). Reaction mixture was stirred at this temperature for 24 h. Reaction mixture was cooled to room temperature and extracted with chloroform (3 \(\times \) 50 mL) followed by water wash (3 \(\times \) 50 mL). All the organic layers were combined and dried over anhydrous \(\hbox {Na}_{2}\hbox {SO}_{4}\) and evaporated to get the crude product which was further purified by silica gel column chromatography.
2.2.2 2-(4-(diphenylamino)phenyl)-1H-anthra[1,2-d]imidazole-6,11-dione (1)
A mixture of 6 (0.40 g, 1 mmol) and diphenylamine (0.20 g, 1.2 mmol) were reacted in toluene as mentioned in the general method. The crude solid thus obtained was purified by column chromatography using hexane/chloroform to obtain a bright yellow solid. Yield: 0.27 g (56%); M.p. : \(295\,{^{\circ }}\hbox {C}\); FT-IR (KBr, \(v_{\mathrm{max}}/\hbox {cm}^{-1})\): 3390.18, 1656.45, 1589.56, 1472.69, 1289.71, 1006.84, 834.99, 715.43, 593.32, 510.83; \(^{1}\)H–NMR (600 MHz, \(\hbox {CDCl}_{3}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \), ppm: 7.15 (t,2H, ArH, \(J=5.4~\hbox {Hz}\)), 7.19 (d, 4H, ArH, \(J=7.8~\hbox {Hz}\)), 7.34 (d,4H, ArH, \(J=2.4~\hbox {Hz}\)), 7.73 (d, 2H, ArH, \(J=8.4~\hbox {Hz}\)), 7.83 (m, 2H, ArH), 8.05 (2H, ArH, d, \(J=7.8~\hbox {Hz}\)), 8.14 (1H, ArH, d, \(J=8.4~\hbox {Hz}\)), 8.26 (d, ArH,1H, \(J=8.4~\hbox {Hz}\)), 8.30 (t,1H,ArH, \(J=9.0~\hbox {Hz}\)), 8.36 (d, 1H,ArH, \(J=9.0~\hbox {Hz}\)), 11.32 (s, 1H, -NH); \(^{13}\)C–NMR (75 MHz, DMSO–d\(_{6}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \), ppm: 111.30, 114.50, 121.13, 122.68, 124.70, 125.28, 126.20, 126.39, 126.75, 127.83, 128.06, 129.32, 131.02, 131.08, 131.76, 131.96, 132.34, 133.34, 134.45, 141.47, 151.01, 182.38, 185.49; MALDI-TOF: mass calcd. for \(\hbox {C}_{33}\hbox {H}_{21}\hbox {N}_{3}\hbox {O}_{2}\) [\(\hbox {M}^{+}\)]: 491.54; found: 491.54; anal. Calcd. for \(\hbox {C}_{33}\hbox {H}_{21}\hbox {N}_{3}\hbox {O}_{2}\): C, 80.64; H, 4.31; N, 8.55% Found: C, 80.45; H, 4.22; N, 8.36%.
2.2.3 2-(4-(naphthalen-1-yl(phenyl)amino)phenyl)-1H-anthra[1,2-d]imidazole-6,11-dione (2)
A mixture of 6 (0.40 g, 1 mmol) and N–phenylnapathylamine (0.26 g, 1.2 mmol) were reacted in toluene as mentioned in the general method. The crude solid thus obtained was purified by column chromatography using hexane/chloroform to obtain a bright yellow solid. Yield: 0.29 g (54%); M.p.: \(244\,{^{\circ }}\hbox {C}\); FT-IR (KBr, \(v_{\mathrm{max}}/\hbox {cm}^{-1})\): 3599.72, 3391.24, 1659.07, 1589.87, 1474.29, 1295.20, 1008.33, 798.92, 715.04, 597.72; \(^{1}\)H–NMR (\(600\hbox { MHz},\hbox { CDCl}_{3}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \) ppm: 7.04 (d, 1H, ArH, \(J=9.0~\hbox {Hz}\)), 7.09 (t, 1H, ArH, \(J=7.8~\hbox {Hz}\)), 7.24 (d, 1H, ArH, \(J=7.8~\hbox {Hz}\)), 7.32 (t,1H, ArH, \(J=7.8~\hbox {Hz}\)), 7.41 (t, 2H, ArH, \(J=7.2~\hbox {Hz}\)), 7.52 (m, ArH, 2H), 7.76 (d, 2H, ArH, \(J=8.4~\hbox {Hz}\)), 7.83 (m, 4H, ArH), 7.94 (m, ArH, 1H), 8.07 (d, 2H, ArH, \(J=8.4~\hbox {Hz}\)), 8.16 (d, 1H, ArH, \(J=8.4~\hbox {Hz}\)), 8.23 (d,1H, ArH, \(J=8.4~\hbox {Hz}\)), 8.28 (d,1H, ArH, \(J=8.4~\hbox {Hz}\)), 8.30 (d,1H,ArH, \(J=9.0~\hbox {Hz}\)), 8.38 (d,1H,ArH, \(J=9.0~\hbox {Hz}\)), 11.34 (1H, s, -NH); \(^{13}\)C–NMR (75 MHz, \(\hbox {CDCl}_{3}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \), ppm: 115.17, 120.00, 119.38, 122.24, 132.03, 123.83, 124.13, 125.70, 126.45, 127.06, 127.51, 127.62, 128.39, 128.50, 128.64, 129.23, 129.56, 129.78, 130.98, 131.30, 132.61, 132.78, 133.27, 133.44, 133.72, 134.12, 134.42, 135.21, 135.36, 141.32, 142.24, 181.35, 185.28; MALDI-TOF: mass calcd. for \(\hbox {C}_{37}\hbox {H}_{23}\hbox {N}_{3}\hbox {O}_{2}\) [\(\hbox {M}^{+}\)]: 541.60; found: 542.45; anal. Calcd. for \(\hbox {C}_{37}\hbox {H}_{23}\hbox {N}_{3}\hbox {O}_{2}\): C, 82.05; H, 4.28; N, 7.76% Found: C, 81.89; H, 4.32; N, 7.64%.
2.2.4 2-(4-(bis(4-methoxyphenyl)amino)phenyl)-1H-anthra[1,2-d]imidazole-6,11-dione (3)
A mixture of 6 (0.40 g, 1 mmol) and bis(4-methoxyphenyl)amine (0.27 g, 1.2 mmol) were reacted in toluene as mentioned in the general method. The crude solid thus obtained was purified by column chromatography using hexane/chloroform to obtain a dark red solid. Yield: 0.34 g (62%); M.p.: \(197\,{^{\circ }}\hbox {C}\); FT-IR (KBr, \(v_{\mathrm{max}}/\hbox {cm}^{-1})\): 2849.41, 1661.79, 1582.76, 1470.89, 1280.89, 1031.76, 826.67, 715.34, 575.59; \(^{1}\)H–NMR (600 MHz, \(\hbox {CDCl}_{3}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \) ppm: 3.83 (s, 6H, \(-\hbox {OCH}_{3})\), 6.90 (d, 4H, ArH, \(J=9.0~\hbox {Hz}\)), 6.99 (d, 2H, ArH, \(J=8.4~\hbox {Hz}\)), 7.14 (d, 4H, ArH, \(J=8.4~\hbox {Hz}\)), 7.80 (m, ArH, 2H), 7.93 (d, 2H, ArH, \(J=8.4~\hbox {Hz}\)), 8.05 (d, 1H, ArH, \(J=8.4~\hbox {Hz}\)), 8.21 (d,1H, ArH, \(J=8.4~\hbox {Hz}\)), 8.27 (t, 1H, ArH, \(J=8.4~\hbox {Hz}\)), 8.34 (d,1H, ArH, \(J=8.4~\hbox {Hz}\)), 11.14 (s,1H, -NH); \(^{13}\)C–NMR (75 MHz, \(\hbox {CDCl}_{3}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \), ppm: 55.51, 115.02, 117.51, 118.93, 118.66, 122.06, 124.70, 126.44, 127.55, 127.67, 127.91, 128.15, 128.49, 133.28, 133.49, 133.63, 134.13, 134.32, 139.43, 151.71, 156.95, 157.09, 157.09, 182.57, 185.27; MALDI-TOF: mass calcd. for \(\hbox {C}_{35}\hbox {H}_{25}\hbox {N}_{3}\hbox {O}_{4}\) [\(\hbox {M}^{+}\)]: 551.59; found: 551.26; anal. Calcd. for \(\hbox {C}_{35}\hbox {H}_{25}\hbox {N}_{3}\hbox {O}_{4}\): C, 79.21; H, 4.57; N, 7.62% Found: C, 78.89; H, 4.51; N, 7.91%.
2.2.5 2-(4-((4-nitrophenyl)(phenyl)amino)phenyl)-1H-anthra[1,2-d]imidazole-6,11-dione (4)
A mixture of 6 (0.40 g, 1 mmol) and 4–nitro–N–phenylaniline (0.25 g, 1.2 mmol) were reacted in toluene as mentioned in general method. The crude solid thus obtained was purified by column chromatography using hexane/chloroform to obtain a deep yellow solid. Yield: 0.22 g (43%); M.p.: \(281\,{^{\circ }}\hbox {C}\); FT-IR (KBr, \(v_{\mathrm{max}}/\hbox {cm}^{-1})\): 3374.84, 1640.14, 1572.78, 1453.19, 1272.57, 987.21, 814.84, 710.85, 582.26, 491.54; \(^{1}\)H–NMR (600 MHz, \(\hbox {CDCl}_{3}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \) ppm: 7.10 (d, 2H, ArH, \(J=9.0~\hbox {Hz}\)), 7.22 (d, 2H, ArH, \(J=8.4~\hbox {Hz}\)), 7.29 (t, 2H, ArH, \(J=7.2~\hbox {Hz}\)), 7.32 (d, 2H,ArH, \(J=8.4~\hbox {Hz}\)), 7.43 (t, 2H, ArH, \(J=7.2~\hbox {Hz}\)), 7.73 (d, 2H, ArH, \(J=8.4~\hbox {Hz}\)), 7.82 (s, ArH, 2H), 8.12 (m, 3H, ArH), 8.29 (d, 1H, ArH, \(J=9.0~\hbox {Hz}\)), 8.36 (d,1H, ArH, \(J=9.0~\hbox {Hz}\)), 11.31 (s,1H, -NH); \(^{13}\)C–NMR (75 MHz, \(\hbox {CDCl}_{3}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \), ppm: 114.72, 117.21, 118.09, 118.36, 121.76, 124.40, 126.14, 127.25, 127.37, 127.61, 127.85, 128.19, 128.68, 132.27, 133.19, 133.33, 133.83, 134.02, 139.13, 140.64, 149.50, 151.41, 156.65, 156.79, 182.27, 184.97; MALDI-TOF: mass calcd. for \(\hbox {C}_{33}\hbox {H}_{20}\hbox {N}_{4}\hbox {O}_{4}\) [\(\hbox {M}^{+}\)]: 536.54; found: 537.62; anal. Calcd. for \(\hbox {C}_{33}\hbox {H}_{20}\hbox {N}_{4}\hbox {O}_{4}\): C, 73.87; H, 3.76; N, 10.44% Found: C, 73.54; H, 3.81; N, 10.73%.
2.2.6 2-(4-morpholinophenyl)-1H-anthra[1,2-d]imidazole-6,11-dione (5)
A mixture of 6 (0.40 g, 1 mmol) and morpholine (0.10 g, 1.2 mmol) were reacted in toluene as mentioned in the general method. The crude solid thus obtained was purified by column chromatography using hexane/chloroform to obtain a deep red solid. Yield: 0.26 g (65%); M.p.: \(314\,{^{\circ }}\hbox {C}\); FT-IR (KBr, \(v_{\mathrm{max}}/\hbox {cm}^{-1})\): 3435.61, 2850.57, 1666.20, 1490.33, 1249.78, 1292.25, 1121.33, 817.21, 717.58, 575.75; \(^{1}\)H–NMR (600 MHz, \(\hbox {CDCl}_{3}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \) ppm: 3.33 (t, 4H, ArH, \(J=4.8~\hbox {Hz}\), -\(\hbox {NCH}_{2})\), 3.89 (t,4H, ArH, \(J=5.4~\hbox {Hz}\), \(-\hbox {OCH}_{2})\), 7.03 (d, 2H,ArH, \(J=9.0~\hbox {Hz}\)), 7.80 (m,2H, ArH,), 8.06 (m, 3H, ArH,), 8.21 (d, 1H, ArH, \(J=8.4~\hbox {Hz}\)), 8.25 (d, 1H, ArH, \(J=9.0~\hbox {Hz}\)), 8.34 (d, 1H, ArH, \(J=9.0~\hbox {Hz}\)), 11.18 (s,1H, -NH); \(^{13}\)C–NMR (75 MHz, \(\hbox {CDCl}_{3}\), \(25\,{^{\circ }}\hbox {C}\)) \(\delta \) ppm: 47.78, 66.60, 114.56, 117.55, 118.51, 122.04, 124.74, 126.42, 127.54, 128.00, 128.45, 133.26, 133.37, 133.64, 134.09, 134.32, 149.59, 153.19, 156.91, 182.52, 185.21; MALDI-TOF: mass calcd. for \(\hbox {C}_{25}\hbox {H}_{19}\hbox {N}_{3}\hbox {O}_{3}\) [\(\hbox {M}^{+}\)]: 409.14; found: 410.24; anal. Calcd. for \(\hbox {C}_{25}\hbox {H}_{19}\hbox {N}_{3}\hbox {O}\_{3}\): C, 73.34; H, 4.68; N, 10.26% Found: C, 73.62; H, 4.24; N, 9.95%.
3 Results and Discussion
3.1 Synthesis and characterization
The synthesis of the target compounds 1–5 based on 2-phenyl-anthra[1,2-d]imidazole-6,11-dione, is illustrated in Scheme 1.
The starting bromo derivative 2-(4-bromo)phenyl-anthra[1,2\(-d\)]imidazole-6,11-dione (6) required for the present study was synthesized by the reported procedure.[17]\(^\mathrm{d}\)4-bromobenzaldehyde was oxidatively coupled with 1,2-diaminoanthraquinone to obtain the precursor compound 6. The compound 6 was conveniently converted to triarylamine derivatives 1–5 by Buchwald-Hartwig coupling reaction in the presence of \(\hbox {Pd}_{2}\hbox {(dba)}_{3}\) (5–8 mol%) as the source of the palladium catalyst, SPhos (2-dicyclohexylphoshpino-2\(^\prime \),\(6^\prime \)-dimethylbiphenyl) as co-ligand and sodium t-butoxide as a base. Characterization methods which include \(^{1}\)H NMR, \(^{13}\)C NMR, MALDI-TOF mass, FTIR, etc. Further, it was observed that the yield of the reaction was significantly affected by the substituents attached to the diarylamine; when electron-donating –\(\hbox {OCH}_{3 }\) group was present on diarylamine resulted in better yield as compared to electron-withdrawing \(\hbox {-NO}_{2}\) group. The five target compounds were obtained in quantitative yield as yellow to red solids. Compounds 1–5 showed good solubility in common organic solvents.The identity and purity of all new compounds were confirmed by various spectroscopic techniques. \(^{1}\)H NMR (600 MHz) and \(^{13}\)C NMR(75 MHz) of 2-(4-bis(4- methoxy phenyl) amino)phenyl)\(-1H\)-anthra[1,2-d]imidazole-6,11-dione (3) recorded in \(\hbox {CDCl}_{3 }\) (see Supporting Information, Figure S3). A characteristic singlet peak due to imidazole proton was observed at 11.14 ppm. Two doublets due to two protons of phenyl ring (b,\(\hbox {b}^\prime )\) appeared at 8.34 and 8.27 ppm confirming the phenyl substitution on the imidazole ring. Two doublets due to two protons (c and d) of the anthraquinone moiety appear at 8.21 and 8.05 ppm while a doublet of 2 protons (e,\(\hbox {e}^\prime )\) was observed at 7.93 ppm and a multiplet due to two protons (f,\(\hbox {f}^\prime )\) at 7.80 ppm showing the presence of the anthraquinone moiety in the molecule. The presence of singlet at 3.83 ppm indicates the presence of –\(\hbox {OCH}_{3}\) in the molecule. Characteristic signal of two carbonyl groups was observed at 185.27 and 182.57 ppm and methoxy carbon appeared at 55.51 ppm in \(^{13}\)C–NMR. All other peaks observed in \(^{13}\)C–NMR spectra can be assigned to aromatic carbons.
3.2 Photophysical properties
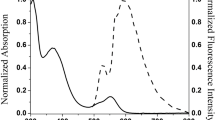
UV–Vis absorption and fluorescence emission spectra of dyes 1–5 were carried out in solvents of varying polarity and in neat solid film. Absorption and emission spectra in dichloromethane solution of dyes 1–5 along with parent compound for comparison are presented in Figure 2(a) and 3(a) while the pertinent data are summarized in Table 1. The absorption spectra of core molecule 2-phenyl-anthra[1,2-d]imidazole-6,11-dione (Std) possesses two absorption maxima at 273 nm which arises due to overlap of n-\(\uppi \)*and \(\uppi \)-\(\uppi \)* transitions within the entire molecule, while a broad and low-intensity peak at 405 nm arises due to intramolecular charge transfer transition (ICT).[19] The absorption spectra of 1–5 are dominated by multiple overlapping bands owing to the presence of different chromophoric segments. Dyes 1–5 display a high intensity band in the range of 261–293 nm and a new distinguished band in the range of 312–339 nm emerges in 1–5 compared to Std and these are assigned to n-\(\uppi \)* and \(\uppi \)-\(\uppi \)*transitions from entire molecule due to extension in conjugation by the introduction of triarylamines into imidazoanthraquinone core.

(a) Normalized absorption spectra of compounds 1–5 in dichloromethane (\(2\,\times \,10^{-6}\,\hbox {M}\)) and (b) neat solid film (thickness of the film was not measured). Molar extinction coefficients (\(\upvarepsilon )\) of 1–5 are given in Table 1. Std molecule is 2-phenyl-anthra[1,2\(-d\)]imidazole-6,11-dione (Figure 1).
The lowest absorption maximum for all the dyes appear in the range of 436–502 nm which is assigned to intramolecular charge transfer (ICT) transitions from electron–donor triarylamine subunit to electron–acceptor imidazoanthraquinone core. ICT transitions in 1–5 were found to be red-shifted with an increase in the absorption intensity as compared to Std. This increase in absorbance indicates that the transition probability for charge transfer increased upon substitution of amines on the imidazoanthraquinone segment while the red-shift is justified considering the auxochromic effect exerted by the diarylamine unit.
A bathochromic and hyperchromic shift in ICT transition was observed in the absorption spectra of 3 and 5 compared with the other derivatives indicating the influence of electron donating –\(\hbox {OCH}_{3}\) substituent on diarylamine and cyclic aliphatic morpholine amine, respectively. While a blue and more hyperchromic shift for ICT was found in 4 compared to that in 3 and 5, which is due to the strong electron acceptor \(\hbox {-NO}_{2}\) substituent on diphenylamine, thus showing the effect of electron donating/withdrawing substituent present on the triarylamine moieties in absorption spectra. Further slight blue shift were also observed in 1 and 2 as compared to 3 and 5, which again confirms the effect of substituent on absorption profile of triarylamines. Bathochromic shift of the ICT absorption maxima was observed in the more polar solvent, which indicates that the excited state is more polar than the ground state, suggesting an ICT nature in 1–5.[8] In the absorption spectra of 1–5 as neat solid film form (Figure 2(b)), the ICT absorption band is slightly broadened and red shifted by 14, 61, 24, 8 and 21 nm for the dyes 1, 2, 3, 4 and 5, respectively, in comparison to that observed in dichloromethane, which could be due to the intermolecular aggregation. The absorption onset extended up to 700 nm for dyes 3, 4 and 5 containing substitution on triarylamine ring indicating advantageous absorption window proliferation.

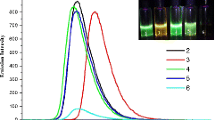
(a) Normalized emission spectra of Std and 1–5 in dichloromethane (2 x 10\(^{-6}\) M) excited at \(\uplambda _{\mathrm{ex}}=405~\hbox {nm}\). (b) Tapping mode AFM topographical 3D images of compounds 1 and 4 as neat solid film coated (1000 rpm) from chloroform solution on quartz coverslips.
The emission peak for 1–5 in dichloromethane solutions appear in the green region in the range of 512–560 nm (Figure 3a) upon excitation (\(\uplambda _{\mathrm{ex}})\) at 405 nm. The emission characteristics of the dyes were found to be affected by the auxochromic effect of diarylamines incorporated in Std. The red shift by 2–48 nm in the emission profiles of dyes was observed compared to Std. The emission colour of 1–5 are greenish in nature while the shift in emission depends on the nature of triarylamine unit. Dyes 1–5 do not show emission in neat solid film form, this can be attributed to aggregation based quenching of emission in the solid state by fast interchain electron transfer from donor to acceptor subunit via close spatial contact.[20]
No specific solvent-dependent absorption and emission behaviour was observed for the dyes but showed 5–30 nm shift in the absorption ICT peak on varying the nature of solvents irrespective of their polarity (see Supporting Information). Considerable Stokes shift of about 467–5078 \(\hbox {cm}^{-1 }\) was observed for 1–5 indicating the change in the ground and excited state geometry, and also suggesting the possibility of formation of a charge–transfer state. The Stokes shifts recorded for the dyes 1–5 were lower than that of Std and observed to increase on substituting the respective diarylamine unit in Std. The quantum yields of 1–5 were calculated using fluorescein (\(\Phi _{\mathrm{F}}=0.79\) in 0.1 M NaOH)[21] as a reference. The relatively low quantum yield observed for 1–5 suggest a prominent nonradiative deactivation pathway for the excited state and were found to be dependent upon the electron donating/withdrawing nature of substituents on triarylamines and on the polarity of solvent (see Supporting Information). The morphological characteristics of the neat solid film of compounds 1–5 were studied by atomic force microscopy (AFM) in the tapping mode. Figure 3(b) shows the AFM results of compound 1 and 4 (for other compounds, see Supporting Information). The AFM results show a smooth film with a root mean square (rms) roughness value of 7.69–33.2 nm, while micrometre-sized phase separation was not observed.
3.3 Electrochemical properties
In order to estimate the charge transport capability and feasibility of electron injection and transport of 1–5, cyclic voltammetry (CV) measurements were performed. The corresponding redox potentials of 1–5 are listed in Table 2 and selected CVs are displayed in Figure 4. The CV wave did not undergo substantial modification after the repeated scans. Cyclic voltammetry of Std showed two irreversible oxidation peaks at 1.96 and 2.19 V, which are expected due to the oxidation of the imidazole moiety. While on cathodic sweep, one quasi-reversible and one irreversible reduction peaks were observed at −1.58 and −2.31 V, respectively.

Cyclic Voltammograms in acetonitrile. (a) (anodic sweep) of Std and 1–5, and (b) full scan of compound 1.
These reduction peaks are attributed to the reduction of carbonyl groups of anthraquinone moiety.[22] Anodic sweep of compounds 1–5, except compound 4, show similar oxidation pattern as that of 1 but the first oxidation peak of 1–5 appear at a slightly lower potential than Std and found in the range of 0.94–1.89 V, while the second peak is found to be shifted more positive potential than Std with potentials in the range 2.24–2.37 V. This shift in the oxidation potential is attributed to introduction of diarylamine moiety in the Std. An additional oxidation peak at lower potential appears for compound 3 and 5 at 1.28 V and 0.94 V, respectively. This is due to the oxidation of electron-rich –\(\hbox {OCH}_{3}\) substituent in 3 and morpholine in 5. Compound 4 shows only one oxidation peak at 1.89 V which is probably due to the effect of \(\hbox {-NO}_{2}\) substituent on diphenylamine ring. The oxidation potential of 1–5 increases in the following order 5<3<1<2<4. This trend is attributed to the electron donating nature of triarylamines in 1–5. On a cathodic sweep, 1–5 showed similar two-wave reduction pattern with a slight change in potentials as compared to Std, one quasi-reversible and second an irreversible reduction peak due to the reduction of carbonyl groups of the anthraquinone moiety.

Schematic representation of theoretically calculated frontier molecular orbitals of Std and 1–5.
The HOMO and LUMO energy levels of the materials are crucial parameters for most of the electro-optical devices, which guides the interfacial charge transport kinetics. The energies of highest occupied and lowest unoccupied (HOMO and LUMO) energy levels of 1–5 (Table 2) were calculated from first oxidation and reduction potential and were found in the range of \(-\,5.67\) to \(-\,6.62\) eV and \(-\,3.18\) to \(-\,3.42\) eV, respectively, by setting \(\hbox {Fc/Fc}^{+}\) at \(-\,5.1\) eV vs. vacuum.[23] It is clearly evident that the nature of the triarylamine unit significantly alters the HOMO/LUMO energies. The low-lying LUMO energy level in the range \(-\,3.0\) to \(-\,4.0\)- eV is essential for the efficiency and stability of n-type materials.[24] The LUMO energy levels of the dyes are found to be lower than that of the most widely used electron-transport (small n-type) materials; such as metal chelates like Alq3 (tris(8-hydroxyquinoline)aluminium) (LUMO=-3.10 eV),[25] organic materials such as 2,4,7,9-tetraphenylpyrido[2,3-g]quinoline (LUMO = –3.10 eV)[26] and 2,5-di([1,10-biphenyl]-4-yl)-1,1-dimethyl-3,4-diphenyl-1H-silole (LUMO = –3.30 eV)[27] and some polymers such as polyquinoxalines (LUMO = –3.0 to –3.24 eV)[28] polyphenylquinoxalines and thiophene linked polyphenylquinoxalines (LUMO=-3.0 to –3.30 eV)[29] and thus making these molecules as potential candidates for n-type materials. The energy band gap calculated from the cyclic voltammetry measurements were in the range of 2.49–3.40 eV. A decrease in band gap was observed in compound 3 (2.81 eV) and 5 (2.49 eV), compared with the other derivatives indicating the influence of electron donating –\(\hbox {OCH}_{3}\) substituent on diarylamine and cyclic aliphatic morpholine amine moiety, respectively (Table 2). Thus, the strength of electron-donating nature of the amine in 1–5 results in a decrease in the HOMO and LUMO energy levels and the energy band gap.
3.4 Theoretical studies
To gain more insight into the electronic structure, dyes 1–5 along with Std were modelled using density functional theoretical (DFT) calculations using Gaussian 03 software package.[30] The geometry of the dyes was optimized by using B3LYP as exchange-correlation functional and 6-311 G (d,p) basis set. The electronic distribution in the frontier molecular orbitals HOMO and LUMO of Std and 1–5 are shown in Figure 5. The optimized structure, frontier molecular orbitals (FMOs) along with the Cartesian coordinates for all the molecules are shown in Supporting Information. The energies of HOMO and LUMO levels, HOMO-LUMO gap, first ionization potential, electron affinity and ground-state dipole moment computed for Std and 1–5 are collected in Table 3. In the parent compound (Std), HOMO is delocalized over the 2-phenyl-benzimidazole ring while the LUMO is localized on the entire anthraquinone moiety and extended to the imidazole ring.

Thermogram (a) and DSC plot (b) of Std and 1–5.
In case of 1–5, the introduction of triarylamine redistributes the HOMO/LUMO energy level. HOMO in 1–5 is mainly constituted by the triarylamine segment while LUMO is localized on the anthraquinone and spread over the imidazole ring as well. The visual representation of HOMO and LUMO of the dyes clearly support the formation of the push-pull system in the molecule that has been commonly observed in the molecules featuring donor-acceptor architecture[31] and is considered an indication of the prominent HOMO to LUMO charge transfer absorption band in the compounds 1–5. The calculated HOMO and LUMO energy of the compounds are found in the range \(-\,5.18\) to \(-\,5.99\) and \(-\,3.17\) to \(-\,3.46\) eV, respectively, and the HOMO-LUMO gap of the dyes are ranged in 2.00–2.52 eV. The calculated LUMO energies were very close to those estimated from CV. The first vertical ionization potential (VIP) and the electron affinity (EA) have been computed as the difference in the total energies of neutral and cationic/anionic species respectively. Thus, the computed LUMO energy values suggest that these compounds may be effective as electron-transport layer and can be used as n-type materials for electroactive devices. The ionization potential and electron affinity computed for the dyes are also in agreement with the above observation.
The dipole moment calculated for the derivatives in gaseous phase is small but found to be high for 4 (9.02) as compared to other derivatives due to the presence of polar \(\hbox {-NO}_{2 }\) group attached to diphenylamine.
3.5 Thermal properties
Thermal properties of 1–5 were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) as these phenomena play a crucial role in the stability and lifetime of photo-electronic devices. Materials possessing good thermal stability and high decomposition temperature found to improve the device performance. The optoelectronic devices should withstand temperature expeditions as high as \(80\,~ {^{\circ }}\hbox {C}\). [32] Observed thermal data of 1–5 is listed in Table 2 and thermograms are displayed in Figure 6. Thermogravimetric analysis revealed that compounds 1–5 are thermally stable materials, and the onset of the decomposition occurred above \(250\,~{^{\circ }}\hbox {C}\) with no weight loss at low temperature.
Melting points of 1–5 were determined by DSC and are found in the range of \(198{-}316\,~{^{\circ }}\hbox {C}\). No glass phase transition temperature was observed in 1–5. It is found that these compounds are thermally stable and could be used in optoelectronic devices.
4 Conclusions
We have synthesized a new series of imidazoanthraquinone based triarylamines, donor-acceptor derivatives via palladium-catalysed Buchwald–Hartwig C–N bond formation amination reaction. These compounds were thoroughly analyzed by routine spectral methods and subjected to photophysical, electrochemical, thermal and theoretical studies. The photophysical and electrochemical properties of the dyes are significantly influenced by the nature of triarylamine segment attached to the imidazoanthraquinone core displaying a broad absorption ICT band attributed to charge transfer transitions from the electron donating triarylamine to electron acceptor imidazoanthraquinone core. These molecules were found to emit in the green region. Electrochemical studies reveal the stability of these compounds under redox conditions and low-lying LUMO energy levels in the range of –3.18 to –3.42 eV which are very similar to well-known n-type materials. The donor-acceptor architecture and HOMO-LUMO energies were further rationalized using DFT calculations. Optoelectronic properties and thermal stability of these compounds along with theoretical calculations suggest that these molecules have potential to be used as n-type materials in optoelectronic devices. Further, we are studying the ion sensing ability of these molecules as chemosensors.
References
(a) Shirota Y 2000 Organic materials for electronic and optoelectronic devices J. Mater. Chem. 10 1; (b) Shirota Y 2005 Photo and electroactive amorphous molecular materials—molecular design, syntheses, reactions, properties, and applications J. Mater. Chem. 15 75; (c) Koene B E, Loy D E and Thompson M E 1998 Asymmetric Triaryldiamines as Thermally Stable Hole Transporting Layers for Organic Light-Emitting Devices Chem. Mater. 10 2235; (d) Wu C, Djurovich P I and Thompson M E 2009 Study of Energy Transfer and Triplet Exciton Diffusion in Hole-Transporting Host Materials Adv. Func. Mater. 19 3157
(a) Song Y, Di C, Yang X, Li S, Xu W, Liu Y, Yang L, Shuai Z, Zhang D and Zhu D 2006 A Cyclic Triphenylamine Dimer for Organic Field-Effect Transistors with High Performance J. Am. Chem. Soc. 128 15940; (b) Cravino A, Roquet S, Aleveque O, Leriche P, Frere P and Roncali J 2006 Triphenylamine—Oligothiophene Conjugated Systems as Organic Semiconductors for Opto-Electronics Chem. Mater. 18 2584; (c) Saragi T P I, Lieker T F and Salbeck J 2006 Comparison of Charge-Carrier Transport in Thin Films of Spiro-Linked Compounds and Their Corresponding Parent Compounds Adv. Funct. Mater. 16 966
(a) Mishra A, Fischer M K R and Baüerle P 2009 Metal-free organic dyes for dye-sensitized solar cells: from structure: property relationships to design rules Angew. Chem. Int. Ed. 48 2474; (b) Roncali J, Leriche P and Cravino A 2007 From One-to Three-Dimensional Organic Semiconductors: In Search of the Organic Silicon Adv. Mater. 19 2045
(a) Bordeau G, Lartia R, Metge G, Fiorini-Debuisschert C, Charra F and Teulade-Fichou M.–P 2008 Trinaphthylamines as Robust Organic Materials for Two-Photon-Induced Fluorescence J. Am. Chem. Soc. 130 16836; (b) Bhaskar A, Ramakrishna G, Lu Z, Twieg R, Hales J M, Hagan D J, Van Stryland E and Goodson T 2006 Investigation of Two-Photon Absorption Properties in Branched Alkene and Alkyne Chromophores J. Am. Chem. Soc. 128 11840
(a) Chi L, Wu Y, Zhang X, Ji S, Shao J, Guo H, Wang X and Zhao J 2010 Ethynylated Triphenylamine Monoboronic acid Chemosensors: Experimental and Theoretical Studies J. Fluoresc. 20 1255; (b) Sreenath K T G T and Gopidas K R 2011 Cu(II) Mediated Generation and Spectroscopic Study of the Tris(4-anisyl)amine Radical Cation and Dication. Unusually Shielded Chemical Shifts in the Dication Org. Lett. 13 1134; (c) Ghosh K, MasantaG, Fröhlich R, Petsalakis I D and Theodorakopoulos G 2009 Triphenylamine-Based Receptors in Selective Recognition of Dicarboxylic Acids J. Phys. Chem. B 113 7800
Thelakkat M 2002 Star-Shaped, Dendrimeric and Polymeric Triarylamines as Photoconductors and Hole Transport Materials for Electro-Optical Applications Mater. Eng. 287 442
(a) Shen J-Y, Yang X-L, Huang T-H, Lin J T, Ke T-H, Chen L-Y, Wu C-C and Yeh M-C P 2007 Ambipolar Conductive 2,7-Carbazole Derivatives for Electroluminescent Devices Adv. Funct. Mater. 17 983; (b) Thomas K R J, Velusamy M, Lin J T, Tao Y-T and Chuen C-H 2004 Cyanocarbazole Derivatives for High-Performance Electroluminescent Devices Adv. Funct. Mater. 14 387; (c) Thomas K R J, Lin J T, Tao Y-T and Ko C-W 2001 Light-Emitting Carbazole Derivatives: Potential Electroluminescent Materials J. Am. Chem. Soc. 123 9404; (d) Xia Z Y, Su J H, Fan H H, Cheah K W, Tian H and Cheah C H J 2010 Multifunctional Diarylamine-Substituted Benzo[\(k\)]fluoranthene Derivatives as Green Electroluminescent Emitters and Nonlinear Optical Materials J. Phys. Chem. C 114 11602; (e) Shaikh A M, Sharma B K, Chacko S and Kamble R M 2017 Novel electroluminescent donor–acceptors based on dibenzo[\(a,c\)]phenazine as hole-transporting materials for organic electronics New. J. Chem. 41 628
(a) Meng H, Sun F P, Goldfinger M B, Jaycox G D, Li Z G, Marshall W J and Blackman G S 2005 High-Performance, Stable Organic Thin-Film Field-Effect Transistors Based on Bis-5‘-alkylthiophen-2‘-yl-2,6-anthracene Semiconductors J. Am. Chem. Soc. 127 2406; (b) Chung D S, Park J W, Park, J–H, Moon D, Kim G H, Lee H-S, Lee D H, Shim H-K, Kwon S-K and Park C E 2010 High mobility organic single crystal transistors based on soluble triisopropylsilylethynyl anthracene derivatives J. Mater. Chem. 20 524; (c) Teng C, Yang X, Yang C, Li S, Cheng M, Hagfeldt A and Sun L 2010 Molecular Design of Anthracene-Bridged Metal-Free Organic Dyes for Efficient Dye-Sensitized Solar Cells J. Phys. Chem. C 114 9101; (d) Sharma B K, Shaikh A M and Kamble R M 2015 Synthesis, photophysical, electrochemical and thermal investigation of Triarylamines based on 9\(H\)-Xanthen-9-one: Yellow–green fluorescent materials J. Chem. Sci. 127 2063; (e) Shaikh A M, Sharma B K and Kamble R M 2015 Photophysical, Electrochemical and Thermal Studies of 5-methyl-5\(H\)-Benz[\(g\)]indolo[2,3-\(b\)] quinoxaline Derivatives: Green and Yellow Fluorescent Materials Can. Chem. Trans. 3 158; (f) Shaikh A M, Sharma B K and Kamble R M 2015 Synthesis, Photophysical, Electrochemical and Thermal Studies of Triarylamines based on benzo[\(g\)]quinoxalines J. Chem. Sci. 127 1571; (g) Shaikh A M, Chacko S and Kamble R M 2017 Synthesis, Optoelectronic and Theoretical Investigation of Anthraquinone Amine-Based Donor-Acceptor Derivatives ChemistrySelect. 2 7620
Doi H, Kinoshita M, Okumoto K and Shirota Y 2003 A Novel Class of Emitting Amorphous Molecular Materials with Bipolar Character for Electroluminescence Chem. Mater. 15 1080
Chen C-T, Wei Y, Lin J-S, Moturu M V R K, Chao W-S, Tao Y-T and Chien C-H 2006 Doubly Ortho-Linked Quinoxaline/Diphenylfluorene Hybrids as Bipolar, Fluorescent Chameleons for Optoelectronic Applications J. Am. Chem. Soc. 128 10992
Tao Y, Wang Q, AoL, Zhong C, Yang C, Qin J and Ma D 2010 Highly Efficient Phosphorescent Organic Light-Emitting Diodes Hosted by 1,2,4-Triazole-Cored Triphenylamine Derivatives: Relationship between Structure and Optoelectronic Properties J. Phys. Chem. C 114 601
Ge Z, Hayakawa T, Ando S, Ueda M, Akiike T, Miyamoto H, Kajita T and Kakimoyo M 2008 Novel Bipolar Bathophenanthroline Containing Hosts for Highly Efficient Phosphorescent OLEDs Org. Lett. 10 421
Sharma B K, Shaikh A M, Agarwal N and Kamble R M 2016 Synthesis, photophysical and electrochemical studies of acridone-amine based donor–acceptors for hole transport materials RSC Adv. 6 17129
Shaikh A M, Sharma B K, Chacko S and Kamble RM 2016 Synthesis and opto-electrochemical properties of tribenzo[a,c,i]phenazine derivatives for hole transport materials RSC Adv. 6 94218
Thomas K R J, Lin J T, Tao Y-T and Chuen C-H 2004 New Carbazole—Oxadiazole Dyads for Electroluminescent Devices: Influence of Acceptor Substituents on Luminescent and Thermal Properties Chem. Mater. 16 5437
(a) da Silva Jr E N, Cavalcanti B C, Guimarães T T, Carmo M D, Pinto F R, Cabral I O, Pessoa C, Costa-Lotufo L V, de Moraes M O, de Andrade C K Z, dos Santos M R, de Simone C A, Goulart M O F and Pinto A V 2011 Synthesis and evaluation of quinonoid compounds against tumor cell lines Eur. J. Med. Chem. 46 399; (b) Huang H-S, Chen T-C, Chen R-H, Huang K-F, Huang F-C, Jhan J-R, Chen C-L, Lee C-C, Lo Y and Lin J-J 2009 Synthesis, cytotoxicity and human telomerase inhibition activities of a series of 1,2-heteroannelated anthraquinones and anthra[1,2-\(d\)]imidazole-6,11-dione homologues Bio. Org. Med. Chem. 17 7418
(a) Peng X, Wu Y, Fan J, Tian M and Han K 2005 Colorimetric and Ratiometric Fluorescence Sensing of Fluoride: Tuning Selectivity in Proton Transfer J. Org. Chem. 70 10524; (b) Hernández C M, Figueroa L E S, Moragues M E, Raposo M M M, Batista R M F, Costa S P G, Pardo T, Máñez R M and Sancenón F 2014 Imidazoanthraquinone Derivatives for the Chromofluorogenic Sensing of Basic Anions and Trivalent Metal Cations J. Org. Chem. 79 10752; (c) Saha S, Ghosh A, Mahato P, Mishra S, Mishra S K, Suresh E, Das S and Das A 2010 Specific Recognition and Sensing of \(\text{CN}^{-}\) in Sodium Cyanide Solution Org. Lett. 12 3406; (d) Kumari N, Jha S and Bhattacharya S 2011 Colorimetric Probes Based on Anthraimidazolediones for Selective Sensing of Fluoride and Cyanide Ion via Intramolecular Charge Transfer J. Org. Chem. 76 8215
Sharma B K, Dixit S, Chacko S, Kamble R M and Agarwal N 2017 Synthesis and Studies of Imidazoanthraquinone Derivatives for Applications in Organic Electronics Eur. J. Org. Chem. 30 4389
Li G Y, Zhao G J, Liu Y H, Han K-L and He G–Z 2010 TD-DFT study on the sensing mechanism of a fluorescent chemosensor for fluoride: Excited-state proton transfer J. Comput. Chem. 311 759
Sun S–S and Sariciftci N S 2005 Organic Photovoltaics: Mechanism, Materials and Devices (United States: Taylor and Francis) p.199
(a) Dawson W R and Windsor M W 1968 Fluorescence yields of aromatic compounds. J. Phys. Chem. 72 3251 (b) Hamai S and Hirayama F1983 Actinometric determination of absolute fluorescence quantum yields J. Phys. Chem. 87 83
Ajloo D, Yoonesi B and Soleymanpour A 2010 Solvent Effect on the Reduction Potential of Anthraquinones Derivatives. The Experimental and Computational Studies Int. J. Electrochem. Sci. 5459; (b) Shaikh A M, Sharma B K and Kamble R M 2016 Electron-deficient molecules: photophysical, electrochemical and thermal investigations of naphtho[2,3-\(f\)]quinoxaline-7,12-dione derivatives Chem. Heterocycl. Comp. 52 110; (c) Shaikh A M, Sharma B K, Chacko S and Kamble R M 2016 Synthesis and optoelectronic investigations of triarylamines based on naphtho[2,3-\(f\)]quinoxaline-7,12-dione core as donor–acceptors for n-type materials RSC Adv. 6 60084
(a) Schulz G L, Kar P, Weidelener M, Vogt A, Urdanpilleta M, Lind’en M, Osteritz E M and Bäuerle A M P 2016 The influence of alkyl side chains on molecular packing and solar cell performance of dithienopyrrole-based oligothiophenes J. Mater. Chem. A 4 10514; (b) Cekli S, Winkel R W, Alarousu E, Mohammed O F and Schanze K S 2016 Triplet excited state properties in variable gap \(\pi \)-conjugated donor–acceptor–donor chromophores Chem. Sci. 7 3621
(a) Usta H, Facchetti A and Marks T J 2011 n—Channel Semiconductor Materials Design for Organic Complementary Circuits Acc. Chem. Res. 44 501; (b) Gao X and Hu Y 2014 Development of n—type organic semiconductors for thin film transistors: a viewpoint of molecular design J. Mater. Chem. C 2 3099; (c) Wang Z, Kim C, Facchetti A and Marks T J 2007 Anthracenedicarboximides as Air-Stable N-Channel Semiconductors for Thin-Film Transistors with Remarkable Current On—Off Ratios J. Am. Chem. Soc. 129 13362; (d) Jones B A, Facchetti A and Wasielewski M R 2007 Tuning Orbital Energetics in Arylene Diimide Semiconductors. Materials Design for Ambient Stability of n-Type Charge Transport J. Am. Chem. Soc. 129 15259
Tang C W and VanSlyke S A 1987 Organic Electroluminescent devices Appl. Phys. Lett. 51 913
Tonzola C J, Alam M M, Kaminsky W and Jenekhe S A 2003 New n-Type Organic Semiconductors:-Synthesis, Single Crystal Structures, Cyclic Voltammetry, Photophysics, Electron Transport, and Electroluminescence of a Series of Diphenylanthrazolines J. Am. Chem. Soc. 125 13548
Tabatake S, Naka S, Okada H, Onnagawa H, Uchida M, Nakano T and Furukawa K 2002 Low operational voltage of electroluminescent devices with a high bipolar conducting silole derivative Jpn. J. Appl. Phys. 41 6582
Fukuda T, Kanbara T, Yamamoto T, Ishikawa K, Takezoe H and Fukuda A 1996 Polyquinoxaline as an excellent electron injecting material for electroluminescent device Appl. Phys. Lett. 68 2346
(a) Brien D O, Weaver M S, Lidzey D G and Bradley D D C 1996 Use of poly(phenyl quinoxaline) as an electron transport material in polymer light-emitting diodes Appl. Phys. Lett. 69 881; (b) Cui Y, Zhang X and Jenekhe S A 1999 Thiophene-Linked Polyphenylquinoxaline. A New Electron Transport Conjugated Polymer for Electroluminescent Devices Macromolecules 32 3824
Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Montgomery J A, T Jr, Vreven, Kudin K N, Burant J C, Millam J M, Iyengar S S, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson G A, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox J E, Hratchian H P, Cross J B, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann R E, Yazyev O, Austin A J, Cammi R, Pomelli C, Ochterski J W, AyalaP Y, Morokuma K, Voth G A, Salvador P, Dannenberg J J, Zakrzewski V G, Dapprich S, Daniels A D, Strain M C, Farkas O, Malick D K, Rabuck A D, Raghavachari K, Foresman J B, Ortiz J V, Cui Q, Baboul A G, Clifford S, Cioslowski J, Stefanov B B, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin R L, Fox D J, Keith T, Al-Laham M A, Peng C Y, Nanayakkara A, Challacombe M, Gill P M W, Johnson B, Chen W, Wong M W, Gonzalez C and Pople J A 2004 Gaussian 03 Revision D.02 (Gaussian, Inc.: Wallingford CT,)
Zhu Y, Kulkarni A P, Wu P-T and Jenekhe S A 2008 New Ambipolar Organic Semiconductors: Synthesis, Single-Crystal Structures, Redox Properties, and Photophysics of Phenoxazine-Based Donor—Acceptor Molecules Chem. Mater. 20 4200
Yu M X, Duan J P, Lin C H, Cheng C H and Tao Y T 2002 Diaminoanthracene Derivatives as High-Performance Green Host Electroluminescent Materials Chem. Mater. 14 3958
Acknowledgements
The authors thank the Science and Engineering Research Board (SERB), New Delhi, India for financial support [SERB Scheme No. SB/EMEQ–507/2014]. BKS and AS thank the University Grant Commission, India for research fellowships. We thank the Micro-Analytical Laboratory, Department of Chemistry, University of Mumbai, Mumbai for providing instrumentation facility. We also thank the Tata Institute of Fundamental Research, Mumbai for MALDI-TOF and \(^{1}\)H–NMR facilities.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sharma, B.K., Shaikh, A.M., Chacko, S. et al. Synthesis and optoelectronic investigation of triarylamines based on imidazoanthraquinone as donor–acceptors for n-type materials. J Chem Sci 130, 49 (2018). https://doi.org/10.1007/s12039-018-1443-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-018-1443-2