
Abstract
An extensive theoretical study on the molecular structure and vibrational analysis of 3-amino-4-methoxy benzamide (3A4MBA) was undertaken using density functional theoretical (DFT) method. The possibility of formation of intramolecular hydrogen bonding was identified from structural parameter analysis and confirmed with the natural bond orbital (NBO), molecular electrostatic potential (MEP) and natural charge analysis. The harmonic oscillator model of aromaticity (HOMA) index elucidated the impact of hydrogen bonding in the ring. Intramolecular hydrogen bonding energy has been calculated from topological study. The low wavenumber vibrational modes obtained from experimental FT-Raman spectrum also supported the presence of hydrogen bonding. Bands are assigned for vibrational frequencies using DFT/B3LYP/6-311 ++G** level of theory.

An extensive theoretical study on the molecular structure and vibrational analysis of 3-amino-4-methoxy benzamide was undertaken using density functional theoretical (DFT) method. The formation of intramolecular hydrogen bonding was identified from structural parameter analysis and confirmed with the natural bond orbital (NBO), molecular electrostatic potential (MEP) and natural charge analysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Amides contain the −CONH 2 group in which a carbon atom is attached to oxygen by a covalent double bond.[1] Benzamide is the simplest aromatic carboxylic amide, which is used in the synthesis of various organic compounds. Benzamide is slightly soluble in water but easily soluble in many organic solvents.[2]
Benzamide derivatives have therapeutic utility.[3] Benzamide derivatives are known to cure antipsychotic and antiemetic ailments. Owing to its neurotropic characteristics, this class of compounds finds use in imaging melanoma and melanoma meta states.[4] N,N-diethyl benzamide and N,N-diethyl 3-methyl benzamide are used in mosquito repellents. A benzamide derivative is used as an antipsychotic drug for many depressive disorders.[5] Owing to various applications of benzamide and its derivatives,[5] identifying the structure and vibrations of benzamide molecules by quantum chemical calculations are necessary for understanding many of its related properties.[6‐12] The present work deals with 3-amino-4-methoxy benzamide (3A4MBA). This is an intermediate product of dyes and pigments. Structural studies on this molecule are scanty. Hence, an extended theoretical analysis on the structural and vibrational properties of the 3A4MBA molecule is undertaken using density functional theory (DFT) calculations. An effort has been made to calculate the N–H- - -O intramolecular hydrogen bonding, its nature, bond distances and related frequencies by calculating the topological parameters at the bond critical points using atoms-in-molecule theory.
2 Experimental and Computational
The fine polycrystalline sample of 3A4MBA was procured from Alfa Aesar Company with a stated purity of 98% and used as such. Infrared spectrum of the compound by KBr pellet method was recorded with JASCO FT-IR 4100 spectrometer in the region 400–4000 cm−1. The FT-Raman spectrum of 3A4MBA was recorded on a BRUKER IFS-66V model interferometer equipped with FRA–106 FT-Raman equipment. The spectrum was recorded in the range 4000–0 cm−1. For Stokes region, 1064 nm line of a Nd:YAG laser was used for the excitation with an operating power of 200 mW. The reported transitions are accurate in the order ±1 cm−1.
The molecular geometries of 3A4MBA were fully optimized with DFT methods using the 6-311 ++G** basis set and further calculations were carried out with the same basis set. Theoretical calculations were performed with Gaussian-03[13] program package, invoking gradient geometry optimization on Intel core i5/2.50 GHz processor. DFT with the three-parameter hybrid function (B3) for the exchange part and the Lee–Yang–Parr (LYP) correlation function[14‐16] were utilised for three-dimensional potential energy scan (3DPES), molecular structure optimization, vibrational frequencies, natural bond orbital (NBO), molecular electrostatic potential (MEP) and UV-Vis spectral analysis.
3 Results and Discussion
3.1 Conformational study
Estimation of hydrogen bonding in the present compound is due to the presence of adjacent amino group and methoxy group in the aromatic ring. To find out the impact of hydrogen bonding in defining conformational flexibility of the molecule along with its type of bonding, HF/6-31G** calculations were carried out by applying rotations in the amino and methoxy groups. Minimum energy corresponding to a particular geometry was obtained using a three dimensional potential energy scan. The structure of 3A4MBA molecule is shown in Figure 1. The dihedral angle of H18–C17–O16–C4 of methoxy group and H15–N13–C3–C4 of amino group with the aromatic ring was varied by every 20∘. Two 3DPES were performed, one for the amide group C2–C1–C7–O8 with the dihedral angle of 180∘ (Figure 2) and other one for C2–C1–C7–O8 with the dihedral angle of 0∘ (Figure 3). The total energies obtained for these possible conformers are listed in Table 1. From the two 3DPES figures, we observed more number of saddle points, and 11 optimized minimum energy values for each scan and are been tabulated. Minimum energy of −567.1727 Hartrees was obtained for the conformer number 332 when C2–C1–C7–O8 has the dihedral angle of 180∘ and the methoxy group H18–C17–O16–C4 at 180∘ and the amino group H15–N13–C3–C4 at 360∘. There is no imaginary frequencies in the optimized structure (N im=0) and positive harmonic vibrations are only obtained for the minimum energy conformer number 332, indicating that conformer number 332 is the most stable structure.

Structure of 3A4MBA.

Three dimensional potential energy scan for C2–C1–C7–O8 dihedral angle 180∘. Here, SC1 is the dihedral angle of C4-O16-C17-H18, SC2 is the dihedral angle of C4-C3-N13-H15.

Three dimensional potential energy scan for C2–C1–C7–O8 dihedral angle 0∘. Here, SC1 is the dihedral angle of C4-O16-C17-H18, SC2 is the dihedral angle of C4-C3-N13-H15.
3.2 Structural properties
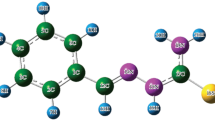
The structure and numbering scheme for atoms of 3A4MBA are shown in Figure 4. The optimized structural parameters for the geometry of 3A4MBA determined at B3LYP/6-311 ++G** level are presented in Table S1 (in Supplementary Information) for the molecule shown in Figure 4. Adjacent methoxy and amino groups indicate the possibility of formation of intramolecular hydrogen bonding.

Optimized molecular structure of 3-amino-4-methoxy benzamide.
From the structural data, it was observed that the calculated C–C bond distances of the ring carbon atoms and the C–H bond lengths were consistent with the literature values except for C3–C4 bond.[17] The substitution of methoxy and amino groups were responsible for the elongation of C3–C4 bond. C4–O16 bond length also got elongated (1.37 Å compared to the normal bond length of 1.347 Å)[17] which might be due to the presence of intramolecular hydrogen bond with the adjacent amino group. The distortion in the symmetry of the ring is caused by intra molecular hydrogen bond. Some of the endocyclic angles of the ring C3–C4–C5, C4–C5–C6 and C5–C6–C1 were 120∘ whereas the angles of C1–C2–C3, C2–C3–C4 and C6–C1–C2 are 121.5∘, 118.3∘ and 119.3∘, respectively. In addition, the bond length of H15- - -O16 was found to be 2.431 Å which would validate the formation of intramolecular hydrogen bonding. Deviation from computation occurred only at the exocyclic angle C2–C3–C4, as the molecule in the present study possessed intramolecular hydrogen bonding.
3.3 Natural bond orbital analysis
NBO analysis was carried out to explain the charge transfer and delocalization of charges due to inter- and intramolecular interaction among bonds. Delocalization of electron occurred between the occupied Lewis-type and non-Lewis-type NBO orbitals.[17,18] The delocalization would explain the details of conjugation in the whole system. Transitions for which stabilization energy is high are selected for the molecule and are given in Table 2.
Interestingly, charge transfer was identified through the predicted intramolecular hydrogen bonding O16- - - H15–N13 in the geometrical structure of the molecule. The lone pair transition LP (O16) →σ* (N13–H15) had very small hyper conjugative interaction energy/ stabilization energy of 1.25 kcal/mol with the donor occupancy of 1.96108 and acceptor occupancy of 0.01090 which would confirm the weak nature of intramolecular interaction.
3.4 Molecular electrostatic potential
The MEP diagram with the contour of the molecule is shown in Figure 5. It clearly depicts the reactive sites of oxygen and hydrogen atoms which emphasize the electrophilic and nucleophilic attacks in the molecule. These sites have been identified with different colours for better clarity.[19,20] The localization of the negative region over the oxygen atom (O8) of the carbonyl moiety and the maximum positive region on the hydrogen atom (H10 and H11) of the amide group clearly indicate a possible site for a nucleophilic attack. The oxygen atom (O16) in the methoxy group lost more of its red colour grading than O8 atom due to the weak intramolecular hydrogen bonding between O16 and H15 in the amino group.

Molecular electrostatic potential and contour of 3-amino-4-methoxy benzamide.
3.5 Natural charges
Natural atomic charges of the molecule were calculated by DFT method and are presented in S2 (in Supplementary Information). The observed charge value of O8 was −0.62462 and that of O16 was −0.53873. The decreased value of charge in oxygen atom (O16) may be due to the intramolecular hydrogen bond formation. In addition, the carbon atomic charges were found to be positive and negative, while hydrogen atomic charges were only positive. At the same time, the recorded negative charge values of O and N atoms indicate a charge transfer from carbon to these atoms.
3.6 Aromaticity
Aromaticity is a measure of the π-electron delocalization in cyclic systems. Of the many quantitative measures of aromaticity in the ring, harmonic oscillator model of aromaticity (HOMA) can be used. The HOMA index can be analytically separated into two independent terms, namely GEO and EN, describing quantitatively whether the decrease of π-electron delocalization is due to an increase of bond alternation (GEO) or the elongation of the mean bond length (EN). The HOMA index[21] is defined as:
Where,
R av, the average bond length was found to be 1.393 Å for the ring C–C while R i the running bond length was 1.41 Å. The normalisation constant α for C–C bonds was 257.7 and n is the number of bonds which is 6. The calculated GEO was 0.01241 and EN was 0.00644 which gave rise to a HOMA index of 0.98115 for the present system. This only indicated that there was no large deviation from aromaticity in the benzene ring. Hence, the geometry of the molecule was not altered much by intramolecular hydrogen bonding, giving an indication of its weakness in the present system.
3.7 Electronic structure and UV spectrum
Using the fully ground state optimized structure, TD-DFT calculation was used at B3LYP/6-311 ++G** level in CPCM model for solvent ethanol to determine the low lying excited state of the molecule. The simulated and experimental UV-Vis spectra, absorbance vs wavelength, are displayed in Figure 6. The possible computed transitions are listed in Table 3. The π→π* transition in the aromatic ring was observed experimentally at 235 nm and by TD-DFT method, at 264 nm with an oscillator strength f = 0.0411. The absorbance at 318 nm with an oscillator strength f = 0.1330 belongs the π→π* transition caused by C =O in the amide group which is experimentally found at 305 nm.

Experimental and calculated UV-Vis spectra of 3-amino-4-methoxy benzamide. (A) Experimental (Ethanol conc. 1×10−5 M; pathlength, 1 cm); (B) Calculated.
3.8 Vibrational analysis
MOLVIB version V7.0-G77 was utilized for scaling of the observed frequencies for total energy distribution. The calculated Raman activities (S i) were suitably adjusted by the scaling procedure with MOLVIB and subsequently, the relative Raman intensities (I i) were converted.[22‐24] The full set of 84 standard internal coordinates defined for 3A4MBA are given in Table S3 (in SI), from which constructions of a non-redundant set of 84 local symmetry coordinates were undertaken.[25,26] These coordinates are summarized in Table S4 (in SI). The title molecule has 22 atoms with 60 fundamental modes of vibrations which belong to C1 point group symmetry. The FT-IR and FT-Raman spectra are presented in Figures 7 and 8, respectively, which give a visual comparison between the observed and simulated spectra. The experimental assignments of FT-IR and FT-Raman for different vibrational frequencies of 3A4MBA are shown in Table S5 (in SI). The root mean square (RMS) values of frequencies were found[23] and the error (unscaled /B3LYP/6-311 ++G**) obtained for 3A4MBA molecule was found to be 76.7 cm−1. The observed wavenumbers were reproduced using scaling factors and optimized through least square refinement algorithm. The average RMS deviation obtained is 6.4 cm−1.

FT-Raman spectra of 3-amino-4-methoxy benzamide. (a) Calculated; (b) Observed.

FT-IR spectra of 3-amino-4-methoxy benzamide. (a) Calculated; (b) Observed.
3.8.1 Ring vibrations
There are six equivalent C–C bonds in benzene and, relevantly, six C–C stretching vibrations, in addition to, in-plane and out-of-plane bending C–C–C based bond vibrations in the ring carbons. The skeletal C–C stretching bands of benzene derivatives usually occur in the region 1400–1650 cm−1.[27] The bands observed at 1631, 1614, 1606, 1561, 1515, 1486 cm−1 were assigned to C–C stretching vibration and were in good agreement with the theoretically computed values.
3.8.2 Amine and Amide group vibrations
In aromatic primary amines, asymmetric and symmetric stretching of vibrations of N–H are in the regions 3520–3420 cm−1 and 3420–3340 cm−1, respectively.[28] Owing to the result of intramolecular hydrogen bonding, the asymmetric stretching of N–H vibration got slightly lowered from the higher value to 3366 cm−1 in this molecule. The theoretically scaled down frequency at 3366 cm−1 by B3LYP/6-311 ++G** method implied a good agreement with the experimentally observed value. In primary amides, two sharp bands observed in the region 3375–3320 cm−1 and 3205–3155 cm−1 were assigned to N–H asymmetric and symmetric stretching vibrations, respectively. The peaks observed at 3433 cm−1 and 3175 cm−1 were assigned to amide group N–H asymmetric and symmetric stretching vibrations.
The NH 2 deformation vibration in the FT-IR spectrum presented a very strong peak at 1614 cm−1 and was assigned to NH 2 scissoring, and the theoretically scaled down value being 1613 cm−1 which was well within the region of 1650–1590 cm−1 as reported in literature.[29] The scaled down frequency at 1069 cm−1 may be assigned to NH 2 rocking vibration and in the recorded FT-IR spectrum, it was observed at 1080 cm−1. The peak at 579 cm−1 in FT-Raman could be attributed to NH 2 wagging mode and it was correlated with the computed value of 577 cm−1.
The carbonyl stretching vibration of C =O group is expected to appear at the region of 1680–1630 cm−1 for amides. The very strong C =O band observed at 1735 cm−1 corresponds to the stretching vibration of C =O group of 3A4MBA. In-plane deformation of C =O possibly occurs in the region of 750–695 cm−1 and it is observed as a medium intensity band at 726 cm−1 in FT-Raman. The calculated vibrations for these bands are in excellent consistency with the experimentally obtained results.
Band mixing leads to difficulty in identifying the C–N vibrations. However, with the help of theoretical calculations and relative percentage of weight stretching and deformation, vibrations of C–N are identified and assigned in this study. The vibrational assignments for amides appeared in the region 1420–1450 cm−1.[28] In 3A4MBA, intense C–N stretching bands are found at 1457, 1287 cm−1 in FT-IR spectrum. DFT at B3LYP/6-311 ++G** level computed frequencies showed an excellent agreement with the corresponding experimental ones. Amino group C–N stretching vibration got slightly lowered due to the formation of intramolecular hydrogen bonding.
3.8.3 Methoxy group vibrations
Basically, nine fundamentals are assigned to the CH 3 group, namely, symmetric stretch; in-plane stretch (i.e., in-plane hydrogen deformation modes), symmetric bending, in-plane rocking, out-of-plane rocking, twisting hydrogen bending modes and out-of-plane hydrogen bending modes inclusive of CH 3 out-of-plane stretch. CH 3 out-of-plane bending modes of the CH 3 group are also found. CH 3 stretching modes appear at 2959, 2919, 2841 cm−1 in the FT-Raman spectrum of 3A4MBA. Symmetric bending vibration was observed at 1343 cm−1 in FT-Raman, in-plane and out-of-plane bending vibrations are at 1448, 1380 cm−1, respectively. Torsion vibration occurred at 249 cm−1 in the FT-Raman spectrum. In-plane bending vibration of methoxy group appeared at 472 and 384 cm−1 in the FT-Raman spectrum and these appear to have good agreement with the calculated spectrum.
3.9 Low frequency hydrogen bonding vibrations
The interaction between the hydrogen donor and acceptor groups leads to the formation of new vibrational degrees of freedom which appear at lower frequencies in the range between 50 and 300 cm−1.[30]These modes arise due to the lattice vibrations and are referred to as external modes. Rotational and translational vibrations along with the vibrations of hydrogen modes are usually involved in external modes. The modes of hydrogen bond are found to be weak in the FT-Raman spectrum with overlapping of liberation modes. The peak in the FT-Raman at 72 cm−1 is assigned to the translational motion of the hydrogen bonded molecules while the intense band at 107 cm−1 is assigned to the rotational motion. In general, these lattice modes are found to be stronger in intensity than other higher frequency bands.
3.10 Topological study (AIM)
‘Atoms in molecules’ (AIM) is an attractive theory for the illustration of the molecular topology as it determines the Bond Critical Points (BCP) between neighboring atoms.[31,32] Localizing the bond critical points, several properties can be calculated at their position in space. Amongst these, the most important one is ρ BCP or the charge density at the bond critical point which has a minimal value along the line of the bond. The next useful quantity to characterize a bond is the Hessian (3×3) matrix of second order partial derivatives with respect to coordinates of the charge density. The coordinate invariant (ordered) eigenvalues λ1<λ2<λ3 arise from the diagonalization of this matrix. Using the Eigenvalues, the Laplacian of the charge density (∇2 ρ BCP) at the bond critical point can be calculated. According to Koch and Popelier criteria,[33] for closed-shell interactions found in ionic bonds, hydrogen bonds, and van der Waals molecules, the ∇2ρ BCP should be positive (in the range 0.015–0.15 a.u.) and low value of ρ (0.002–0.040 a.u.). For electron density analysis of atoms, the positions of (3, −3) critical points are nearly identical to nuclear positions, hence, (3, −3) is called nuclear critical point (NCP) which usually occurs in heavy atoms whereas (3, −1) is called the bond critical point (BCP). (3, + 1) and (3, + 3) are ring critical point (RCP) and cage critical point (CCP), respectively. The molecular graph of the 3A4MBA using Multiwfn program[34] shown in Figure 9 gives BCPs, RCPs and NCPs (topological parameters) along with the bond paths.

Molecular graph of 3A4MBA. Bond critical points (small orange spheres), ring critical points (small yellow spheres), nuclear critical point (small magenta spheres) and bond paths (brown lines) using Multiwfn program.
The energy of the N-H- - -O hydrogen bond has been calculated by the relationship E HB= V(r BCP)/2 described by Espinosa et al.[35] Intramolecular hydrogen bonding energy of 3A4MBA has been calculated to be −20.053 kcal/mol. Hydrogen bond length is 2.431 Å which is less than van der Waals radius (2.7 Å). Figure 9 shows one more BCP between H11 and H12 which in turn gives one more ring critical point also. This H–H interaction arises because of the inter atomic distance (2.02A) is less than the sum of their van der Waals radii (2.4 Å). This results in an increase in the repulsive contributions of their energies and this change is dominated by an increase in the magnitude of the attractive interaction of the protons with the electron density distribution.[36]
Johnson and co-workers[37] developed an approach to investigate the weak interactions in real space based on the electron density and its derivatives. The reduced density gradient (RDG) is a fundamental dimensionless quantity coming from the density and its first derivative:
The weak interactions can be isolated as regions with low electron density and low RDG value. The density values of the low-gradient spikes (the plot of RDG versus ρ) appear to be an indicator of the interaction strength. The sign of λ2 is utilized to distinguish the bonded (λ2 < 0) from nonbonded (λ2 > 0) interactions.
RDG is a way to distinguish weak interaction region from other regions. RDG provides a rapid and rich representation of van der Waals interactions, hydrogen bonds and steric clashes. The three-dimensional molecular structure defines covalent bonds, however, the noncovalent interactions are hidden within the voids in the bonding network. RDG is one of the tools to view and analyze the covalent and electrostatic interactions.[37] The plot of the RDG versus the electron density, ρ multiplied by the sign of λ2 can allow analysis and visualization of a wide range of interaction types. One or more spikes are found in the low-density, low-gradient region which is indicative of weak noncovalent interactions. In the present case, as seen in Figure 10, such a weak noncovalent interaction between O16- - -H15-N13 is observed. Liberation modes higher negative values of (λ2)ρ are indicative of stronger attractive interactions, while large and positive values indicate repulsive interaction. Spikes found in the large positive region show H11-H12 type of non-bonded repulsive interaction while values near zero indicate very weak, van der Waals interactions.

Plots of the RDG vs (λ2)ρ, the electron density q multiplied by k2 for 3A4MBA.
4 Conclusions
Intramolecular hydrogen bonding with a distance of 2.431 Å between the O16 and H15 atoms was confirmed in the title molecule. The stabilization energy and the values of donor and acceptor occupancies resulting from the lone pair transition LP (O16) →σ* (N13–H15) confirmed very well the weak intramolecular hydrogen bonding from NBO analysis. Further, the effective π-electron delocalization was confirmed through the transitions in the ring. Evidence was found for the existence of weak intramolecular hydrogen bonding from MEP and natural charge analysis. The lesser impact of hydrogen bonding in the ring system was indicated by HOMA index. Topological analysis was used to calculate the intramolecular hydrogen bonding energy of 3A4MBA which was found to be −20.053 kcal/mol. The theoretically predicted vibrational modes are in good agreement with the experimental ones. The incidence of low frequency bands at 72 and 107 cm−1 in the experimental FT-Raman spectrum further confirmed the intramolecular hydrogen bonding.
References
Clark J 2004 http://www.chemguide.co.uk/ organicprops/amides/background.html (accessed on 28 February 2016)
http://en.wikipedia.org/wiki/Benzamide (accessed on 10 October 2016)
Powers J P, Yosup Rew, Xuelei Yan 2010 Benzamide derivatives and uses related thereto US Patent 7659287 B2
Gawronski J, Kołbon H and Kwit M 2002 J. Stereo Chem. 7 85
Dewis M L, Merrit D, Miller K, Chen Z and Reiber L 2009 Benzamide Compounds Useful as High Potency Sweet Taste Enhancers US Patent 20090047379
Kawski P, Kochel A, Perevozkina M G and Filarowski A 2006 The intramolecular hydrogen bond in 2-hydroxy-benzamides J. Mol. Struct. 790 65
Arslan H, Florke U, Kulcu N and Binzet G 2007 The molecular structure and vibrational spectra of 2-chloro-N-(diethylcarbamothioyl)benzamide by Hartree-Fock and density functional methods Spectrochim. Acta A 68 1347
Arjunan V and Mythili C V 2011 Experimental and theoretical investigations of benzamide oxime Spectrochim. Acta A 79 245
Muthu S, Rajamani T, Karabacak M and Asiri A M 2014 Vibrational and UV spectra, first order hyperpolarizability, NBO and HOMO-LUMO analysis of 4-chloro-N-(2-methyl-2,3-dihydroindol-1-yl)-3-sulfamoyl-benzamide Spectrochim. Acta A 122 1
Jezierska A, Panek J J and Mazzarello R 2009 Structural and electronic structure differences due to the O–H ⋯O and O–H ⋯S bond formation in selected benzamide derivatives: A first-principles molecular dynamics study Theor. Chem. Acc. 124 319
Ushakumari L, Varghese H T, Panicker C Y, Ertan T and Yildiz I 2008 Vibrational spectroscopic studies and DFT calculations of 4-fluoro-N-(2-hydroxy-4-nitrophenyl)benzamide J. Raman Spectrosc. 39 1832
Arjunan V, Kalaivani M, Ravindran P and Mohan S 2011 Structural, vibrational and quantum chemical investigations on 5-chloro-2-hydroxybenzamide and 5-chloro-2-hydroxybenzoic acid Spectrochim. Acta A 79 1886
Frisch M J et al. 2004 Gaussian, Inc., Wallingford CT
Becke A D 1993 Density-functional thermochemistry. III. The role of exact exchange J. Chem. Phys. 98 5648
Becke A D 1998 Density-functional exchange-energy approximation with correct asymptotic behaviour Phys. Rev. A 38 3098
Pulay P, Fogarasi G, Pang F and Boggs J E 1979 Systematic ab initio gradient calculation of molecular geometries, force constants, and dipole moment derivatives J. Am. Chem. Soc. 101 2550
Arjunan V, Suja Ravi Isaac A, Rani T, Mythili C V and Mohan S 2011 Density functional theory studies on vibrational and electronic spectra of 2-chloro-6-methoxypyridine Spectrochim. Acta A 78 1625
James C, Amal Raj A, Reghunathan R, Hubert Joe I and Jayakumar V S 2006 Structural conformation and vibrational spectroscopic studies of 2,6-bis(p- N,N-dimethyl benzylidene)cyclohexanone using density functional theory J. Raman Spectrosc. 37 1381
Scrocco E and Tomasi J 1978 Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Euristic Interpretation by Means of Electrostatic Molecular Potentials Adv. Quantum Chem. 11 115
Murray J S and Sen K 1996 In Molecular Electrostatic, Potentials Concepts and Applications (Amsterdam: Elsevier)
Sobczyk L, Grabowski S J and Krygowski T M 2005 Interrelation between H-bond and Pi-electron delocalization Chem. Rev. 105 3513
Sundius T 2002 Scaling of ab initio force fields by MOLVIB Vib. Spectrosc. 29 89
Sundius T 2002 MOLVIB (v.7.0); Calculation of Harmonic Force Fields and Vibrational Modes of Molecules, QCPE, Program No. 807 (2002)
Sundius T 1990 MOLVIB: A program for harmonic force field calculations, CPE Program No. 604 J. Mol. Struct. 218 321
Fogarasi G, Pulay P and Durig J R 1985 In Vibrational Spectra and Structure 4 (Amsterdam: Elsevier) Ch. 3 p. 125
Fogarasi G, Zhov X, Taylor P W and Pulay P 1992 The calculation of ab initio molecular geometries: Efficient optimization by natural internal coordinates and empirical correction by offset forces J. Am. Chem. Soc. 114 8191
Varsanyi G 1969 In Vibrational spectra of Benzene Derivatives (New York: Academic press)
Socrates G 2001 In Infrared and Raman Characteristic Group Frequencies–Tables and Charts 3rd ed. (Chichester: John Wiley)
Tellez C A, Hollauer E, Felcman J, Lopes D C N and Cattapan R A 2002 Fourier-transform infrared and Raman spectra, and ab initio calculations for cadmium-n-di-iso-propylphosphorylguanidine-di-chloride (CdDPGCl2) complex Spectochim. Acta A 58 1853
Padmaja L, Vijayakumar T, Hubert Joe I, Reghunadhan Nair C P and Jayakumar V S 2006 Vibrational spectral studies and the non-linear optical properties of a novel NLO material L-prolinium tartrate J. Raman Spectrosc. 37 1427
Bader R F W 1990 In Atoms in Molecules: A Quantum Theory (Oxford: Oxford University Press)
Bader R F W 1991 A quantum theory of molecular structure and its applications Chem. Rev. 91 893
Koch U and Popelier P L A 1995 Characterization of C–H–O Hydrogen Bonds on the Basis of the Charge Density J. Phys. Chem. 99 9747
Lu T and Chen F 2012 Multiwfn: A multifunctional wavefunction analyzer J. Comput. Chem. 33 580
Espinosa E, Molins E and Lecomte C 1998 Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities Chem. Phys. Lett. 285 170
Matta C F, Hernandez-Trujillo J, Tang T -H and Bader R F W 2003 Hydrogen- Hydrogen Bonding: A Stabilizing Interaction in Molecules and Crystals Chem. Eur. J. 9 1940
Johnson E R, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen A J and Yang W 2010 Revealing noncovalent interactions J. Am. Chem. Soc. 132 6498
Acknowledgements
The authors are thankful to Sophisticated Analytical Instrumentation Facility (SAIF), IIT Madras for FT-Raman studies. The authors are indebted to the management of Sri Paramakalyani College for the facilities provided through the research centre.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information (SI)
All additional information pertaining to geometrical parameters of 3-amino-4-methoxy benzamide are given in Table S1 and Natural charge in Table S2. Information regarding vibrational studies are given in Tables S3, S4 and S5. Supplementary Information is available at www.ias.ac.in/chemsci.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
SUBHAPRIYA, G., KALYANARAMAN, S., GANDHIMATHI, S. et al. Structural, intramolecular hydrogen bonding and vibrational studies on 3-amino-4-methoxy benzamide using density functional theory. J Chem Sci 129, 259–269 (2017). https://doi.org/10.1007/s12039-017-1227-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12039-017-1227-0