
Abstract
Two new inorganic-organic hybrid materials based on heteropolyoxometalates, (C4H10N)6(P2 Mo18O62).4H2O I, and (C4H10N)6(As2Mo18O62).4H2O II, where C4H10N is protonated pyrrolidine have been synthesized and structurally characterized by physic-chemical methods. Single-crystal X-ray diffraction method, infrared, ultraviolet spectroscopy, Thermogravimetricanalysis andcyclic voltammetry measurements of the title hybrid materials indicate that there are hydrogen bond interaction between O atoms of the heteropolyoxometalates and water molecules as well as the N and O atoms of the organic compound. The molecular structures of synthesized hybrid materials contain discrete entities of pyrrolidinumion and water molecules surround every [X2Mo18O62]6− anion over the extended crystalline network that the [X2Mo18O62]6− anion retains its “Dawson structure”. Crystal data: I monoclinic, space group P21/a, a = 13,453(1) Å, b = 24,046 (1) Å, c = 24,119(1) β = 97,99(1)°, V = 7726,30(5) Å3 and Z = 4; II monoclinic, space group P21/a, a = 13.4900(1) Å, 24.0900(1) Å, 24.2740(1) Å, β = 98.320(1)°, V = 7805.40(7) Å3 and Z = 4.

Two new inorganic-organic hybrid materials based on the inorganic cluster, [X2Mo18O62]6- (X = P, As), crystallize in monoclinic space group P21/a.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The polyoxometalates (POMs) form a significant class of inorganic metal–oxygen clusters with intriguing architectures and diverse physicochemical properties.[1–4]These materials have attracted increasing attention worldwide because of their nanosized structures and their potential applications in many fields such as catalysis, pharmacology, medicine, nanotechnology, and molecular electronics.[5–9]Recently, the interest is extended to the modification of POM structure by introduction of organic derivatives[10,11]or metal–organic complexes.[12] Thus, various organic–inorganic hybrid materials were synthesized based on hydrogen, electrostatic or coordination bonding between the organic and inorganic moieties. The organic units can also be attached to the polyanion by covalent bond and several functionalized POM were synthesized and characterized.[13] Such POM-based hybrids demonstrate fascinating structural diversity and provide an important type of functional materials with original physical properties.[14–16]
To date, a number of hybrid materials based on polyoxometalate clusters, such as the Lindquist, Keggin and Dawson[17–19]type and six isomers of octamolybdates have been studied and exhibited potential applications in catalysis, electrical conductivity and biological chemistry. Generally, there are two routes to prepare such POM-based hybrid materials: (a) organic ligands graft onto POMs directly; (b) organic ligand and a second metal ion form complex subunits which link POMs through M-O bonds.
This contribution describes for the first time the syntheses and characterization of two inorganic hybrid based on heteropolyoxometalates: (C4H10N)6 (P2Mo18O62).4H2O (I) and (C4H10N)6(As2Mo18O62). 4H2O (II).
2 Experimental
2.1 Materials and measurements
Sodium molybdatedihydrate Na2MoO4.2H2O (99%), H3PO4(85%), H3AsO4 (85%) and pyrrolidine (98%) were purchased from Sigma-Aldrich andused without further purification. The elemental compositionof the synthesized compound was identified by scanning electron microscopy analysis; a JEOL JSM 6700F scanning electron microscope equipped with a field-effectgun and operated with a 0.5–30 kV scale acceleration voltage was used. The infrared spectra were recorded as KBr pellets, in the 4000–400 cm−1 range on a Nicolet 470FT-IR spectrophotometer. The UV–visible absorption spectrum was recorded on a Perkin-Elmer Lambda 19 spectrophotometer. The thermogravimetric and differential thermal analyses (TGA–DTA) were carried out on a Setaram TG-DTA 92–16.18 thermal analyzer, under nitrogen at a heating rate of 10°C min−1.
2.2 Synthesis
2.2.1 Synthesis of Compound I
The compound, I was synthesized employing solution methods. Na2 MoO4.2H2O (1 g, 4 mmol) was dissolved in water (25 mL) with stirring. H3PO4 (0.5 mL, 0.2 mmol) and pyrrolidine (0.2 mmol) were added to the mixture (if a light turbidity occurred, it cleared with time and then the next drop was added) and the pH was adjusted to about 2.5–2.6 by adding 6 M HCl. The mixture was refluxed at 353 K for 3 h and then cooled to room temperature. Slow evaporation of the solvent at room temperature led to green prismatic crystals of I suitable for X-ray diffraction. Qualitative analysis of these crystals by electron microscope revealed the presence of the Mo, P, O, N and C atoms.
2.2.2 Synthesis of Compound II
Compound II was prepared following the procedure described for compound I, but H3AsO4 was used instead of H3PO4. Qualitative analysis of these crystals by electron microscope probe revealed the presence of the all atoms.
3 Results and discussion
3.1 X-ray data collection
Both compounds are stable under ambient conditions and two single crystals was carefully selected and glued on the end of a glass capillary. The intensities of the diffraction data were measured using an Enraf-Nonius CAD-4 diffractometer with monochromated graphite Mo K α radiation (λ = 0.71073 Å)[20]at 293 K.
For compound I, the number of collected and independent reflections were respectively, 18516 and 16819. Unit cell dimensions were obtained by least-square refinement of the angular settings in the 2.22°<𝜃<26.97°. The reflections were corrected for Lorentz and polarization effects; an empirical absorption correction was also applied using Ψ-scan data. The structure was successfully developed in the centrosymmetric space group P21/a, solved by Patterson method using SHELXS-97[21] and refined with anisotropic temperature factorsfor non-hydrogen atoms, by full matrix least-squares based on F2 using SHELXL-97.[21] The positions of the hydrogen atoms were determined from a difference Fourier map and were refined isotropically. The final full-matrix least-squares refinement on F2 converged with R = 0.086 and wR(F2)=0.186 for 12822 unique observed reflections [I > 2 σ (I)].
For the compound II the number of collected and independent reflections were respectively, 21432 and 16577. Unit cell dimensions were obtained by least-square refinement of the angular settings in the 2.04°<𝜃<24.95°. Following analysis similar to compound I, the final full-matrix least-squares refinement on F2 converged with R = 0.071 and wR(F2)=0.175 for 8984 unique observed reflections [I > 2 σ (I)]. A summary of the crystal data and structure refinement for compounds I and II are provided in table 1.
3.2 Structure description
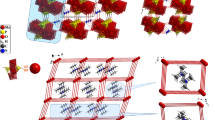
Singlecrystal X-ray analysis reveals that this material consists of one α-Dawsonpolyoxomolybdates[ α-X2Mo18O62]6− (abbreviated as {X2Mo18}, X = P and As) anion, six pyrrolidinium ions (abbreviated as Pyr+) and four water molecules. As shown in figure 1, each [X2Mo18O62]6− anion is encapsulated by six pyrrolinium cations and connected interactively with eight adjacent clusters [X2Mo18O62]6−. In the two structures, the [X2Mo18O62]6− clusters are linked by pyrrolinium cations and water molecules to form a 3D supramolecular structure (figure 2).

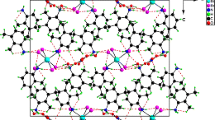
View showing the strong hydrogen-bonding interactions betweenpyrrolidinium and terminal and central oxygen atoms of polyoxoanions.

View of three-dimensional network in the compound.
The {X2Mo18} anion is formed by close packing of oxygen atoms with X and Mo atoms in the distorted octahedral voids. The centrosymmetric polyoxoanion is a classical α-Dawson isomer [ α-X2Mo18O62]6−, consisting of two central XO4 tetrahedra which are surrounded by six vertex-sharing Mo3O15 trimers, which may be described as two [ α-XMo9O31]3− units, generated from the well known [ α-XMo12O40]3− by removal of a set of three corner-sharing MoO6 octahedra and fused into a cluster of virtual D 3h symmetry. In the Dawson type POM, there are two structurally distinct types of Mo atoms: six ‘cap’ atoms on vertical mirrorplanes, grouped in two sets of three, and twelve equatorial Mo atoms are grouped in two sets of six atoms, but do not lie on mirror-planes.[22]
Four kinds of oxygen atoms exist in the cluster according to the manner of oxygen coordination: (i) There are 18 terminal O atoms which are only bonded to one Mo atom and the Mo–O distances are in the range 1.660(6)–1.701(2) Å for I and 1.655(8) - 1.711(8) Å for II; (ii) There are 36 O atoms which are shared by two Mo atoms and the Mo–O distances vary from 1.800(7) to 2.072(4) Å for I and from 1.812(5) to 2.086(7) Å for II; (iii) There are six O atoms which are combined with one X atom and two Mo atoms. The Mo–O distances vary from Å for 2.292 to 2.358 Å I and from 2.257(1) to 2.311(3) Å for II; (iv) There are two O atoms which are coordinated to one X atom and three Mo atoms. The Mo–O distances are between 2.356(2) and 2.389(1) Å for I and between 2.330 (4) and 2.360(4) Å for II. The O–Mo–O angle deviations, from 90° for cis-standing oxygen atoms and from 180° for trans-standing oxygen atoms, also reflect the extent of distortion within the MoO6 octahedra. Indeed, the angle ranges cover 71.80(1)–105.13(1)° (I) and 72.07(1)–107.01(1)° (II) for cis-standing oxygen atoms and 154.97–173.74(1)° (I) and 154.16(1)–171.25(1)° (II) for trans-standing oxygen atoms.
The Mo …Mo distances between corner-sharing Mo O6 octahedra vary in the range of 3.668(1)–3.866(4) Å and 3.682(1)–3.882(4) Å for I and II, respectively. They are obviously greater than those between edge-sharing MoO6 octahedra [d(Mo …Mo) = 3.318(4)–3.387(4) Å and 3.319(4)–3.379(4) Å]. The bond distances within the polyoxomolybdates unit of the two hybrid compounds are in accord with the elsewhere reported values for Dawson type compounds.[23,28]
The X–Mo distances are in the 3.312(2)–3.327(7) Å range. The X-O bond lengths and O-X-O bond angles differ only slightly in the compounds and are in good agreement with those bond lengths and angles found in other compounds containing the (X2Mo18O62)6− anion.[29]
BVS calculations using the Brown and Altermatt method[30] revealed that all the molybdenum atoms have valence sums ranging from 5.979 to 6.106, with an average value of 6.028, close to the ideal value of 6 for MoVI. The bond valences of arsenic and phosphorus atoms are, respectively, 4.983, 5.043 and the bond valences of oxygen atoms in the polyanion are in the range of 1.683–1.869 valence units for terminal oxygen atoms and 1.649–2.024 valence units for bridging oxygen atoms.
Owing to the presence of organic molecules and the molecular nature of the compounds, a large number of hydrogen bond interactions have been observed. The majority of the interactions are between the terminal oxygen atoms of the clusters and the hydrogen atoms of the amine molecule. Thus, N …O distances are in the range 2.79–3.46 Å with the majority of D–H …O bond angles >150°. Similar hydrogen bond interactions have also been observed for II. The important hydrogen bond interactions observed in I and II are listed in table 2a and 2b.
3.3 IR spectroscopy
There are four characteristic asymmetric vibrations resulting from heteropolyanions with Dawson structure, namely, υ as(Mo =Ot) (Ot is the terminal oxygen atom), υ as(Mo-Ob) (Ob refers to the oxygen atom located in the shared corners between two Mo3O13 units), υ as (Mo-Oc) (Oc refers to the oxygen atom connecting edge-sharing MoO6 octahedra in the Mo3O13 units), and υ as(X-Oa) (Oa refers to the oxygen atom connecting the X and Mo atoms).
As shown in figure 3, the spectra of compounds I and II can be divided into the following typical regions: 3500-2700 cm−1 (C-H and N-H stretchings), 1650-1380 cm−1 (O-H and N-H bendings), and 1250 to 1000 cm−1 (C–H in-plane bending). The strong band at 1090 cm−1 could be ascribed to υ (P–O).

IR Spectrum (a) of (I) and (b) of (II).
The low-wave number peaks are attributed to the Dawson anion [X2Mo18O62]6−: l030-800 cm−1 (Mo = Ot vibrations), 750-550 cm−1 (fundamentally Mo-Ob modes) and <450 cm−1 (Mo-Oc, and some other modes). Between 550 and 500 cm−1, it is possible to observe some bands attributable to water libration modes.
3.4 UV–visible spectra
The UV–vis absorption of II was analyzed in the 190–800 nm range using aqueous solution (5×10−5 M) of pH 5. The spectrum (figure 4) reveals two absorption bands centered at 200 nm and 280 nm, which are characteristic of the Dawson-type polyoxotungstates.[31] The strong higher energy band can be assigned to the ligand-to-metal charge transfer (LMCT) from terminal oxygen to the molybdenum center, whereas the broad lower energy band is attributed to the bridging oxygen-to-molybdenum LMCT. The absorption onset of II was about 305 nm, indicating an optical energy gap (Eg) of 4.06 eV, which is commonly observed in Dawson-type polyoxotungstates.

UV spectrum of (C4H10N)6(As2Mo18O62).4H2O (5×10−4 M, pH 5, path length 10 mm).
3.5 Thermal analysis
The two curves corresponding to DTA and TGA analysis of compound II are given in figure 5. The DTA curve shows that this compound shows a succession of several endothermic and exothermic phenomena. The first peak at 120°C is accompanied by a weight loss obviously observed in the TGA curve. This weight loss corresponds to the removal of all water molecules (water: experimental 2.33%; calculated 2.12%). The second loss of 9.1% occurred between 250 and 360°C which is consistent with the calculated value attributable to the degradation of the four organic moieties. The DTA curve also exhibits exothermic peak at 470°C accompanied by a weight loss 11.2% in the TGA curve which is ascribed to the loss of most of the As2O3 and combustion of two organic ligands.

DTA-TGA thermograms of (C4H10N)6(As2Mo18O62).4H2O.
3.6 Electrochemical behavior
Cyclic voltammetry of the compound I was carried out in H2 SO 4 aqueous solution in the potential range from −200 to 800 mV at scan rate 50 mV s−1 and was recorded in anodic scan. The voltammetric pattern showed three redox waves, (I–I’), (II–II’) and (III–III’), corresponding to Molybdenum redox process[32] (figure 6). Because of the poor reversibility of the waves, we could determine the mean peak potentials (Ef = (Epa+ Epc)/2) approximately as 483 mV (I–I’), 354 mV (II–II’) and 197 mV (III–III’) (vs. Ag/AgCl) and the difference of the peak potentials between each pair of peaks (ΔE p) is about 30 mV. The number of the electrons involved in each reduction step is 2 as calculated from ΔE p(V) = 0.06/n. So, we conclude that the reduction is a process of three reversible, two-electron steps.

Cyclic voltammogram of compound II.
4 Conclusions
In summary, two new compounds have been successfully synthesized by slow evaporation at room temperature and characterized by X-ray diffraction method, UV, IR spectra, thermal analyses (TG-DTA) and cyclic voltammetry. This compound crystallizes in the monoclinic system, space group of P21/a. The main geometrical feature of this structure is the existence of [X2Mo18O62]6− (X = P and As) anionic clusters and these groups are linked via hydrogen bonds to form a 3D framework.
References
Müller A, Peters F, Pope M T and Gatteschi D 1998 Chem. Rev. 98 239
Pope M T 1983 In Heteropoly and Isopoly Oxometalates (Springer: Berlin)
Gouzerh P and Proust A 1998 Chem. Rev. 98 77
Yamase T 1998 Chem. Rev. 98 307
Müller A, Shah S Q N, Bogge H and Schmidtmann M 1999 Nature 397 48
Rhule J T, Hill C L, Judd D A and Schinazi R F 1998 Chem. Rev. 98 327
Proust A, Thouvenot R and Gouzerh P 2008 Chem. Commun. 16 1837
Katsoulis D E 1998 Chem. Rev. 98 359
Kozhenikov I V 1998 Chem. Rev. 98 171
Razak I A, Raj S, Chantrapromma H-K, Fun Y-S and Zhou X-Z You 2001 J. Chem. Crystallogr. 31 255
Legagneux N, Jeanneau E, Basset J M and Lefebvre F 2009 J. Mol. Struct. 21 300
Ma H, Gong L, Wang F and Wang X 2008 Struct. Chem. 19 435
Dablemont C, Hamaker C G, Thouvenot R, Sojka Z, Che M, Maatta E A and Proust A 2006 Chem. Eur. J. 12 9150
Galan-Mascaros J R, Gimenez-Saiz C, Triki S, Gomez-Garcia C J, Coronado E and Ouahab L 1995 Angew. Chem. Int. Ed. 34 1460
An H Y, Lan Y, Li Y G, Wang E B, Hao N, Xiao D R, Duan L Y and Xu L 2004 Inorg. Chem. Commun. 7 356
Li L C, Liao D Z, Jiang Z H and Yan S P 2002 Inorg. Chem. 41 1019
(a) Reinoso S, Vitoria P, Lezama L, Luque A and Gutiérrez- Zorrilla J M 2003 Inorg. Chem. 42 3709; (b) Lu Y, Xu Y, Wang E B, Lu J, Hu C W and Xu L 2005 Cryst. Growth Des. 5 257; (c) Bonhomme F, Larentzos J P, Alam T M, Maginn E J and Nyman M 2005 Inorg. Chem. 44 1774; (d) Vasylyeva M, Popovitz-Birob R, Shimonc L J W and Neumanna R 2003 J. Mol. Struct. 656 27
(a) Niu J Y, Wei M L, Wang J P and Dang D B 2004 Eur. J. Inorg. Chem. 1 160; (b) Wang J P, Zhao J W and Niu J Y 2004 J. Mol. Struct. 697 191
Hagrman P J, Hagrman D and Zubieta J 1999 Angew. Chem. Int. Ed. 38 3165
CAD-4 Express Software, Enraf-Nonius, Delft (1994) The Netherlands
Sheldrick G M, SHELXS97 and SHELXL97 (1997) Program for Crystal Structure Solution and Refinement (University of Goettingen: Goettingen, Germany)
Wang J P, Zhao J W and Niu J Y 2004 J. Mol. Struct. 697 191
Yu H, Zhang X, Kong L and Xu J 2009 Acta Cryst. E65 m1698
Soumahoro T, Burkholder E, Ouellette W and Zubieta J 2005 Inorg. Chim. Acta 358 606
Jinghua L, Jun P, Enbo W, Lihua B and Shurong G 2000 J. Mol. Struct. 525 71
Pope M T 1976 Inorg. Chem. 15 2068
D’Amour H and Allmann R 1974 Naturwiss. 61 34
Strandberg R 1975 Acta Chem. Scand. A 29 350
Yang Y, Xu L, Jia L, Gao G, Li F, Qu X and Qiu Y 2007 Cryst. Res. Technol. 42 10
Brown I and Altermatt D 1985 Acta Crystallogr. Sect. B 4 1244
Pope M T and Papaconstantinou E 1967 Inorg. Chem. 6 1147
Liu D, Tan H Q, Chen W L, Li Y G and Wang E B 2010 Cryst. Eng. Comm. 12 2044
Acknowledgments
The crystal data collection of I and II was done in the Laboratoire de MateriauxetCristallochimie, Faculté des Sciences, El Manar, 2092, Tunis, Tunisia. We are grateful to Ahmed Driss who supervised this experiment.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information
The cif and checkCIF files for X-ray crystallography of the both compounds are available in supplementary information at www.ias.ac.in/chemsci. Crystallographic data for the structures of I and II have been deposited in the Cambridge Crystallographic Data center as publication number CCDC 1031359 and 1031358, respectively. Copy of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB21EZ, UK (fax: (+ 44) 1223-336-033; e-mail: deposit@ccdc.com.ac.uk).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
HMIDA, F., AYED, M., AYED, B. et al. Two new inorganic-organic hybrid materials based on inorganic cluster, [X2Mo18O62]6− (X=P, As). J Chem Sci 127, 1645–1651 (2015). https://doi.org/10.1007/s12039-015-0931-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12039-015-0931-x