
Abstract
Duchenne muscular dystrophy (DMD) is an X-linked genetic disease primarily affecting boys causing loss of the dystrophin protein, ultimately leading to muscle wastage and death by cardiac or respiratory failure. The genetic mutation involved can be overcome with antisense oligonucleotides which bind to a pre-mRNA and results in reading frame restoration by exon skipping. Phosphorodiamidate morpholino oligonucleotides (PMOs) are a class of antisense agents with a neutral backbone derived from RNA which can induce effective exon skipping. In this review, the evolution of PMOs in exon skipping therapy for the last two decades has been detailed with the gradual structural and functional advancements. Even though the success rate of PMO-based therapy has been high with four FDA approved drugs, several key challenges are yet to overcome, one being the dystrophin restoration in cardiac muscle. The current scenario in further improvement of PMOs has been discussed along with the future perspectives that have the potential to revolutionize the therapeutic benefits in DMD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Duchenne muscular dystrophy (DMD) is an X-linked recessive muscular dystrophy affecting roughly 1 in 3500 boys, which is caused by mutations in the dystrophin gene (Zubrzycka-Gaarn et al. 1988). The dystrophin gene is the largest gene identified with 79 exons (Roberts et al. 1993) (figure 1). DMD patients have different forms of mutations in varying positions of the protein, resulting in the production of functionally compromised dystrophin protein. Some mutations in the gene cause disruption of the open reading frame (ORF) or introduction of a premature stop codon leads to a complete absence of a functional dystrophin protein.

Full-length dystrophin protein and gene. (A) Schematic of key functional domains of dystrophin protein from the N-terminus to the C-terminus: actin-binding domain containing two calponin homology domains (CH): Exons 2–8; central rod domain containing 24 rod domains (numbered 1–24) and four hinge regions (H): Exons 9–61; WW domain: Exons 62–63; cysteine-rich domain (CYS): Exons 64–70 (dystroglycan binding site); C-terminal domain (CT): Exons 71–79 (syntrophin and dystrobrevin binding sites). (B) Representation of the 79 spliced exons of full-length dystrophin gene colour coded parallel to the functional domains of protein represented in (A). The shape of exons indicates which exons can be spliced together to maintain the ORF of the protein. When the shapes fit together like jigsaw puzzle, the ORF is maintained.
This disease causes loss of ambulation at the age of around 10–12 years, followed by respiratory difficulties and progressive DMD associated dilated cardiomyopathy (DCM) leading to heart failure and early death. Conversely, a milder form of dystrophinopathy known as Becker muscular dystrophy (BMD) also exists, where genetic mutations do not disrupt the reading frame, leading to the expression of a truncated yet functional dystrophin protein (Monaco et al. 1988).
The life expectancy of DMD patients has improved with the development of treatment over time for cardiopulmonary dysfunction and essential guidelines for patient care and management (Ryder et al. 2017). Nevertheless, the life-limiting and devastating nature of this disease has fuelled the development of many potential treatment approaches (Wagner et al. 2007). Among the spectrum of approaches for DMD treatment, a significant clinical potential has been observed in ‘exon skipping therapy’ (Heemskerk et al. 2009; Nakamura and Takeda 2009). Briefly, this therapeutic approach employs antisense oligonucleotides (AONs) to specifically manipulate the splicing event of dystrophin pre-mRNA such that the mutated exon or the surrounding exons in the hotspot region is skipped, thus restoring the correct reading frame and expression of shortened functional dystrophin protein.
This review particularly focusses on the regime of exon skipping therapy using phosphorodiamidate morpholino oligonucleotides (PMOs). Before venturing into the therapeutic aspects, it is of paramount importance to understand the underlying structural mutations and associated pathophysiology of the disease.
2 DMD: An overview
2.1 Dystrophin structure and function
The DMD gene encoded on Xp21.1 has several internal promoters enabling it to produce multiple truncated isoforms of dystrophin. The full-length dystrophin protein (Dp427m) is expressed on striated muscle tissue and translated from a 2.2 Mb mRNA bearing 79 exons and has four functional domains: (1) an actin binding domain1 (ABD1) in the N-terminus which interacts with F-actin via its two calponin homology (CH) domains; (2) a central rod domain consisting of 24 spectrin-like repeats which can extend from 21 nm to 84 nm adding an extensible feature to the protein and four distributed hinge domains (H) which provide flexibility to the protein; (3) a cysteine-rich domain (CR) which binds to ankyrin-B and helps in costameric localization of dystrophin (figure 1). It also contains a WW domain which links the interior of the cell to the extracellular matrix by binding to β-dystroglycan, and (4) the C-terminal domain (CT) which further stabilizes the sarcolemal localization of dystrophin by binding α-dystrobrevin primarily and also α1, β1 syntrophin. The whole assembly of components that interacts with dystrophin in the sub-sarcolemmal region is known as the dystrophin glycoprotein complex (DGC), which is necessary for the maintenance of membrane stability (Wilson et al. 2022).
Among the different domains across the 79 exons, the actin-binding domains at the N-terminus and the dystroglycan-binding domain at the C-terminus are practically indispensable; mutations in these regions are associated with a more severe DMD. The most commonly mutated regions are exons 45–55 and 2–19, which are the cluster of mutation hotspots that identify about 98% of all deletions. The central and distal rod domains are practically dispensable; in-frame deletions in exons 38–44, 48–51, or 48–53 lead to near normal dystrophin concentrations (Muntoni et al. 2003).
2.2 Mutations in dystrophin
Mutations in muscular dystrophy can be broadly divided into two subtypes depending on the reading frame. BMD is typically associated with in-frame mutations that do not disrupt functional protein synthesis. In DMD, deletions and duplications usually disrupt the reading frame, which abruptly terminate functional protein synthesis. Nonsense point mutations lead to termination of translation because of the stop codon incorporation (figure 2), whereas missense mutations found in the N- or C-terminus connecting domains vital for protein function may lead to the DMD phenotype (Singh et al. 2010). Either way, the cysteine-rich domain and the crucial C-terminal domains are missing in the truncated protein. Statistically, exon 51 is the most mutated exon with 14% of the total mutations and 21% of exon deletions, followed by exon 53 with 10% of total mutations and 14% of exon deletions. Point mutations represent around 26% of DMD cases (Happi Mbakam et al. 2022).

Mutations in BMD and DMD. Schematic representation of different mutations in DMD gene taking an example of a mutational hotspot region, their effect on the functionality and production of dystrophin protein, and their clinical outcome.
An example of point mutation is the mdx mice model (Sicinski et al. 1989), in which there is a single base substitution; a cytosine is replaced by a thymine at nucleotide position 3185 resulting in a termination codon (TAA) instead of a glutamine codon (CAA). This results in premature termination of translation at 27% of the full-length protein.
Several pathophysiological features are associated with DMD, such as necrotic or degenerating muscle fibres which are surrounded by macrophages and CD4+ lymphocytes. At the advanced stages, the muscle fibres are gradually replaced by adipose and connective tissue (Deconinck and Dan 2007). The collapse of the dystrophin-associated protein complex leads to membrane destabilization and failure to regenerate causing wasting, fibrosis, and fat deposition. Since muscles that have been wasted do not recover, an early therapeutic intervention is required for optimum functions. Onset of dystrophy is seen earlier in skeletal muscles, and cardiomyocytes are affected only at a later stage (Dongsheng et al. 2021).
For therapeutic benefit in DMD, correction of mRNA is required so that the reading frame is restored. Apart from genome editing methods, such as CRISPR technology, mRNA correction is possible in the pre-mRNA stage in the nucleus before splicing, where the out-of-frame exons can be ‘skipped’ to give a shorter but functional dystrophin with the important domains intact as in BMD. This skipping could be induced by short complementary oligonucleotides which could bind to the pre-mRNA and impact splicing.
After the first report on mRNA splicing in thalassemia induced by 2′-OMe-phosphorothioate antisense oligonucleotides (PS-AONs) in vitro (Sierakowska et al. 1996), similar studies utilizing the same class of AONs in case of DMD were explored in mdx mice models (Mann et al. 2001) and in patient samples (van Deutekom et al. 2001). The positive outcomes in this paved the way for further research in the therapeutic applications of exon skipping, especially in DMD. Although PS-AONs were pursued initially as the therapeutic for DMD, drisapersen was rejected in clinical trials eventually. For the purpose of this review, we will focus only on the evolution of morpholinos in DMD treatment.
3 PMOs in DMD treatment: Insights from the past
3.1 PMOs: Structure and mechanism of action in exon skipping therapy
PMOs are chemically modified RNA derivatives where the ribose sugar unit is replaced by a morpholine ring and the phosphodiester linkages are replaced by phosphorodiamidate linkages (figure 3). These two modifications primarily impart the stability and neutral characters of the oligonucleotide, the former being an important prerequisite for translation to therapy. Developed by Summerton, PMOs are typically 18-30-mer in length and have shown enhanced enzymatic stability over an extended period of time. Mechanistically, PMOs work by steric blocking by binding to the complementary mRNA or pre-mRNA sequences to hinder the translation or splicing machinery (Summerton and Weller 1997), respectively. Target knockdown by PMOs is traditionally used to reveal the role of different genes involved in the developmental stages of vertebrate (e.g., zebrafish) (Nasevicius and Ekker 2000; Corey and Abrams 2001).

Structural comparison of RNA and PMO. The phosphodiester linkage in RNA is replaced by phosphorodiamidate linkage and the 5-membered sugar ring is replaced by a 6-membered morpholine ring. B represents the nucleobases A, G, C and T.
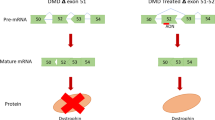
Since PMOs do not induce RNase H-mediated cleavage of their target, they have been particularly utilized for pre-mRNA splicing modulation or exon skipping therapy. Correction of aberrant splicing in DMD patients by PMOs is a mutation-specific approach. This can be achieved by designing a short stretch of antisense oligonucleotides (AONs) that can bind to the target exon on the splice sites or cis-regulatory sequences so that the sequences get masked from interacting with the trans-splicing regulatory factors and the remaining splicing machinery. As a consequence, it induces a switch to an alternative splice site and the targeted exon gets skipped, resulting in an mRNA isoform with a restored reading frame for protein synthesis (Siva et al. 2014). The exon to be skipped depends on the size and location of the mutation, which is shown with eteplirsen as an example (figure 4). Hence, application of PMOs in DMD treatment was explored further.

Mechanism of action of PMO in exon skipping with eteplirsen as an example. Schematic depiction of dystrophin pre-mRNA, mature mRNA, and protein fate in normal individuals, Δexon50 DMD patients, and eteplirsen-treated Δexon50 patients along with the mechanism of action of PMO in exon skipping.
3.2 PMOs as splice switching agents: Earliest research
An inherent drawback of the otherwise therapeutically safe PMOs is their neutral nature, which prevents its interaction with negatively charged cell membranes and impairs delivery by endocytosis. For optimum intracellular concentration of efficient splicing activity, the delivery has to be enhanced by attachment of a moiety capable of interacting with cell membranes or incorporation of similar modifications in the PMO itself. The former approach has been the subject of most of the research in this field, which will be discussed chronologically in the following sections (figure 5).

Timeline of PMO development in exon skipping therapy.
Wilton and group (Gebski et al. 2003) showed for the first time that PMOs could induce exon 23 skipping in the mdx mouse model at low concentrations after annealing to a sense oligonucleotide, termed as ‘leash’, which showed intracellular delivery after lipoplex formation. Although these leashes showed superior activity in vitro, later in vivo experiments showed that the single-stranded PMO fared better when administered intramuscularly or intravenously. Addition of PMO at 100 mg/kg dose with various dosing regimens showed greater exon 23-skipped RNAs in quadriceps, abdominals, intercostals, and gastrocnemii, and to a lesser extent in the diaphragm, biceps, triceps, and tibialis anterior, but there was no effect in the heart (Fletcher et al. 2006).
3.3 Unassisted PMO treatment
It was subsequently reported that naked PMOs showed better histopathological results as compared to those with leashes, where the leashes induced tissue damage without significant increase in dystrophin fibres. Dystrophin levels < 20% of the normal were restored in tibialis anterior (TA) muscles, and there were no histological signs of damage in lung, liver and kidney after repeated intravascular injections (Alter et al. 2006).
Further studies included the injection of PMOs in cardiac muscles, which could not reproduce the exon skipping efficiency in TA muscles, and this was attributed to a lower intracellular availability in cardiomyocytes (Vitiello et al. 2008). It was also found that the multiple intravascular injections of naked PMO at low doses (50 mg/kg, four times a week) showed more dystrophin-positive fibres than a single dose of the same amount (200 mg/kg) (Malerba et al. 2009).
Long-term treatments with PMO in mdx mice were also reported almost simultaneously by two groups with 5 and 50 mg/kg (Malerba et al. 2011), and 15 and 60 mg/kg doses (Wu et al. 2011), by intravenous injection. Wu et al. also showed that 60 mg/kg biweekly administration induced <2% dystrophin expression in the heart but improved cardiac functions as demonstrated by hemodynamics analysis. However, it was also found that enhanced locomotor functions due to long-term systemic administration of PMOs led to a more severe cardiac pathology compared with untreated mice. This postulated that dystrophin restoration in cardiac muscles is required simultaneously in conjunction with the skeletal muscles for improving longevity.
Even though cardiac improvement was minimal with PMO treatment, the improved skeletal functions with low toxicity even on systemic administration led to initiation of clinical trials. It is worth mentioning that the four PMO-based drugs: eteplirsen (exon 51), casimersen (exon 45), golodirsen (exon 53), and viltolarsen (exon 53), that have been approved by FDA since 2016 are all unconjugated PMOs (figure 6). All four have successfully completed phase III trials, and long-term benefit of longitudinal phase III is underway (Dongsheng et al. 2021).

FDA-approved PMO drugs. General structure of PMO-based drugs: their respective sequence and length (n) along with their 5′ modification (R).
3.4 Conjugated PMO treatment
Even though naked PMOs showed exon skipping activity, there were two major drawbacks, i.e., requirement of high doses and inactivity in cardiac muscles. To facilitate delivery, cell penetrating peptides (CPPs), which were emerging promisingly at the time, were explored for PMOs as well as PS-AONs. In this review, we will be focussing only on the evolution of CPP–PMO conjugates.
The first study regarding the pharmacokinetics, biodistribution, stability, and toxicity of a CPP–PMO conjugate was reported by Iversen and group (Amantana et al. 2007), where it was shown that the conjugate did not show any apparent toxicity until 15 mg/kg of dose, and adverse events such as lethargy and weight loss were observed only at 150 mg/kg dose, which were primarily attributed to the CPP moiety. This study provided a cautionary approach to the development of subsequent CPP–PMO conjugates for therapeutic application.
The first ever evaluation of a CPP–PMO conjugate in mdx mice was done by Fletcher et al. (2007), where the peptide (RXR)4XB (where R is arginine, X is 6-aminohexanoic acid, and B is β-alanine) was used for conjugation with exon 23 PMO. The incorporation of X or B in the parent octa-arginine peptide (R8) decreased transfection but increased splice correction activity and reduced cytotoxicity (Wu et al. 2007). The conjugate showed activity after intraperitoneal injection at markedly lower dose (weekly once 125 µg/adult mouse) than those previously used for unconjugated PMO (weekly thrice 625 µg/adult mouse), and there were no adverse effects at the doses used in the study. The same peptide was also injected intravenously at 25 mg/kg single dose, after which dystrophin was restored at 25–100% of wild type levels in skeletal muscles and 10–20% of the wild type levels in cardiac muscles (Yin et al. 2008).
The validity of the same CPP–PMO was further confirmed in double knockout mice (dKO) lacking both utrophin and dystrophin, which represents a more severe and progressive model of DMD with lifespan reduction from 18 months to 8.2 weeks. Treatment with 25 mg/kg/week for six weeks induced almost complete exon 23 skipping in all muscles except the heart, and this could be due to the intraperitoneal route of administration, which was the only mode possible in neonatal dKO mice (Goyenvalle et al. 2010).
The above peptide was also modified to (RXRRBR)2XB, called the B-peptide, which was compared with the parent peptide after intravenous administration. The B-peptide was less efficient at exon skipping at the same dose compared with the parent (Yin et al. 2008). In another study around the same time by Jearawiriyapaisarn et al. (2008), the CPPs were first screened in vivo by an enhanced green fluorescent protein (EGFP) reporter system where splicing enhanced the GFP signal, signifying the efficacy of the conjugate. The B-peptide was found to be highly potent in the heart, diaphragm, and quadriceps as per the reporter assay. It was subsequently conjugated to exon 23 PMO and showed persistent exon skipping with lower serum creatinine kinase levels after systemic delivery with 12 mg/kg/day for four days. Similarly, the same peptide conjugated PMO (PPMO) was reported to restore the dystrophin protein to near normal levels in the TA muscle two weeks after a single 30 mg/kg injection intravenously via the retro-orbital route (Wu et al. 2008). Truncated dystrophin levels analysed by RT-PCR showed >80% skipping in all skeletal muscles and about 50% in cardiac muscles. Six injections at the same dose at biweekly intervals for three months led to higher amounts of exon-skipped mRNAs and protein. The study also showed improvement in heart function by a hemodynamic model, where dobutamine, a stimulator of β1-adrenoceptors of the heart, was used to induce cardiac stress. All PPMO-treated mice survived the challenge as opposed to 33% survivability in untreated mdx mice.
The long-term benefit of the same PPMO was further elucidated by another study (Jearawiriyapaisarn et al. 2010) which showed that 12 mg/kg dose injected intravenous (IV) in two cycles of four once-daily injection with 2-week intervals in 16-week-old mdx mice (the age just before onset of cardiomyopathy) resulted in significantly reduced cardiac creatine kinase-myocardial band (CK-MB) levels with dystrophin expression throughout the heart. It was established that PPMO treatment initiated before the onset of cardiomyopathy that may effectively prevent or slow down cardiac hypertrophy and diastolic dysfunction with significant long-term impact.
To prevent the major accumulation of PPMOs in the liver and kidneys, the concept of a muscle homing peptide was developed. By phage display screening of a peptide library, ASSLNIAX was identified as a muscle-specific peptide (MSP), which was conjugated with B-PMO to give B-MSP-PMO (Yin et al. 2009). Body-wide restoration of dystrophin protein with improvement in muscle pathology was found with weekly three low doses of 6 mg/kg administered intravenously in mdx mice. Up to 25% of the dystrophin protein level in skeletal muscles was obtained with B-MSP-PMO as opposed to about 10% of the levels with B-PMO. It was also found that reversing the peptide attachment sequence to MSP-B-PMO led to complete loss of activity.
Further studies with the B-MSP-PMO conjugate used two doses, 3 and 6 mg/kg, where the lower dose could not reach the critical dose threshold required for high-level correction in mdx mice even after multi-injections biweekly (Yin et al. 2010). The higher dose of 6 mg/kg administered biweekly for six weeks resulted in enhanced correction of the dystrophin defect at the RNA level, with decreased serum CK levels, improved grip functions, restored DAPC localization, and other physiological responses, without any detectable immune response or toxicity. However, dystrophin correction in the heart was again poor, and this was attributed to the high affinity of MSP to skeletal muscles and the lower doses used.
A novel 12-mer muscle homing peptide, M12 (RRQPPRSISSHP), was reported by Gao et al. (2014), which was designed after in vitro bio-panning in myoblasts and showed preferential binding to skeletal muscles than the liver. When only M12 was conjugated to PMO against exon 23 and injected intravenously at 25 mg/kg, body-wide skeletal muscles showed dystrophin restoration levels up to 25% after three weekly injections but the effect was absent in the heart. This was the first report where a muscle homing peptide alone led to dystrophin restoration without any overt toxicity until 75 mg/kg.
Since the natural peptidic backbone of the CPPs led to degradation in vivo, other non-peptidic transporters were explored for PMO delivery. Vivo morpholino (vPMO) was designed with a triazine core which held a dendritic structure whose arms culminated in eight guanidinium groups which are responsible for cellular entry (Li and Morcos 2008). Intravenous administration of the vPMO for an EGFP reporter assay in transgenic mice showed GFP expression in a broad range of tissues and organs. Its application in PMO delivery in mdx mice was also explored, where a single, IV injections of 6 mg/kg vPMO exon 23 generated dystrophin expression in skeletal muscles at levels higher than 300 mg/kg of the unmodified exon 23 (Wu et al. 2009). Repeated injections at biweekly intervals led to protein restoration of about 50% and 10% in skeletal and cardiac muscles, respectively, without eliciting any immune response. The effect of vPMO was also illustrated in the canine model of DMD by simultaneously targeting exons 6 and 8 (Yokota et al. 2012). vPMOs of 120 µg–1.2 mg were used for intramuscular injections in this study. However, it was found that vPMOs could be highly toxic and could induce blood clotting and cardiac arrest in mice (Ferguson et al. 2014).
Another series of cell-penetrating peptides called peptide nucleic acid internalization peptides (also known as Pip peptides) were discovered by Michael Gait (Ivanova et al. 2008) (figure 7). Initially inspired from Penetratin, a known CPP, the Pip series of peptides were designed to have three domains: an N-terminal oligoarginine terminal, a central more hydrophobic region, and a C-terminal more basic region. Certain modifications for stability led to Pip-2b, which showed exon skipping and dystrophin production after conjugation with a PNA against exon 23 on intramuscular injection in mdx mice. The Pip-2b peptide was further modified and shortened to give the Pip-5 series of peptides (Yin et al. 2011), of which the Pip5e was shown to be most active after a single IV injection at 25 mg/kg dose. The maximum increase in dystrophin production was observed in the cardiac muscle, with >50% levels of normal dystrophin protein following a single dose. This remarkable effect in the heart was attributed to the core sequence, ILFQY, which was presumed to enhance the nuclear delivery in cardiomyocytes. Further modifications to the Pip-5 series in the hydrophobic core led to Pip-6 series of peptides (Betts et al. 2012), where it was proved that partial deletions of the hydrophobic core abolished the dystrophin production in the heart. It was found that the Pip6 series fared better than the previous one at half the dose (12.5 mg/kg).

Different approaches to PMO-conjugates. Cellular transporters are conjugated at the 3′-N-terminal of PMO.
Since the peptide sequence is crucial for the desired functions of the conjugate, methods for library synthesis of such peptides followed by easy conjugation methods and purification were also explored (O’Donovan et al. 2015). The activity of the screened CPP–PMO conjugates was verified in the mdx mice model by exon 23 skipping and was called SELection of PEPtide CONjugates (SELPEPCON).
Since PMOs are neutral, nanoparticle-mediated delivery has not been explored as actively as in charged oligonucleotides. However, it has been reported that lipid-containing CPPs can form nanoparticles with PMOs and show exon skipping in vitro as well as in patient-derived primary cell lines of spinal muscular atrophy and DMD (Järver et al. 2015). Lipids were conjugated based on their interaction with the non-polar PMO backbone, and the particle formation was measured by nanoparticle tracking analysis.
Polyethylenimine-conjugated pluronic copolymers were also evaluated for exon 23 PMO delivery in mdx mice (Wang et al. 2013) with molecular weights ranging from 2–6 kDa, with a range of hydrophilic/lipophilic balanced polymers tested in vivo. It was found that more hydrophobic copolymers induced better exon skipping but an amphiphilic balance was also essential. Dystrophin expression up to 15% in skeletal muscles and 5% in cardiac muscles was observed.
Single exon skipping is more of a mutation-specific approach applicable to only a small subset of patients (8–13%), but multiple exon skipping in mutation hotspot regions, such as exons 45–55, could treat a higher percentage (~40%) of patients and lead to a milder BMD phenotype. This was first verified with PMO cocktails followed by DG9-peptide conjugation to target the exon 45–55 region in humanized mice by skipping five exons (Lim et al. 2022).
Sinha and co-workers (Das et al. 2023b) have reported a novel self-penetrating PMO where the first or last four phosphorodiamidate linkages have been replaced by a guanidine linkage, which is responsible for membrane interaction and internalization, to form the GMO chimera, where GMO stands for guanidinium-linked morpholino. The GMO–PMO showed efficient knockdown of stemness factor Nanog, both in vitro and in vivo. Since this strategy rules out the requirement of any external conjugation and permits at-a-stretch synthesis, it could be a cost-effective and easier alternative to all the PMO-based therapeutics discussed. Sinha and co-workers have also shown that conjugation of an internal oligo-guanidinium transporter (IGT) to a PMO also increases the uptake and shows efficient Nanog downregulation in cancer cells (Kundu et al. 2020). Thus, IGT could also be explored as a CPP alternative in exon skipping of the mdx mice model. They have also reported an unnatural amino acid, azaproline-based flexible transporter (Gupta et al. 2022) which has successfully delivered PMO intracellularly (figure 7).
4 Present challenges in PMO-based therapeutics
Even though four drugs have been given accelerated approval for DMD, long-term trials are still underway and the efficacy and complications after prolonged administration, if any, will only be known after a few years. Unconjugated PMOs have impressive safety profiles with respect to dose escalation studies, but reduction in dosage amount with the right distribution and efficacy are the challenges that are yet to be overcome. Furthermore, the current FDA-approved PMO drugs are exorbitantly expensive, to the tune of 750,000 USD (INR 6 crore) per patient per year, making this therapy unaffordable to most patients in India and globally as well.
4.1 Skeletal muscle delivery
Exon skipping in skeletal muscles has been fairly successful with unconjugated PMOs as well as conjugated ones, and almost completely skipped mRNA has been restored in many muscle groups in most cases (Table 1). However, the corresponding protein-level expressions have been consistently lower as compared with mRNA, and this is attributed to the nonsense-mediated decay in the nucleus. It is known that >15% dystrophin levels in patients are associated with milder BMD, and DMD characterization corresponds to <3% of the normal levels. Most of the data in mdx mice show that low-level improvements in protein expression can substantially change the pathophysiology; 5–15% restoration protects the muscle cells from contraction-induced damage, whereas about 40% restoration is required for improvement in force. Apart from the overall dystrophin levels, their distribution also has to be kept in mind, whether the restorative effect is marginal for a whole range of fibres or substantial for some. In the patient samples of early clinical trials of PS-AON and PMOs, it was found that the former resulted in a more widespread dystrophin restoration compared with PMOs, which showed a patchy distribution (Aartsma-Rus et al. 2019). It is yet to be seen how this change in distribution will impact the therapy. Therapeutic benefits have been impressive even with partial protein restoration, but the longevity of the restored protein has to be cautiously maintained with the correct dose and intervals for improving the quality of life in the long run.
4.2 Cardiac muscle delivery
Unconjugated PMOs have failed miserably in dystrophin restoration in the heart of the mdx mice model, which has led to accelerated cardiomyopathy due to enhanced locomotor functioning after skeletal muscle recovery. Specific CPP–PMO conjugates have successfully overcome this drawback in in vivo models as discussed earlier. The most recent example on this is the proprietary peptide conjugate of eteplirsen, SRP-5051, which is currently in phase II trials, and has shown a 6.5% increase in dystrophin levels as compared with the 0.4% increase in the case of only eteplirsen (Aartsma-Rus 2022). The same peptide has also shown about 41% cardiac dystrophin levels in mdx mice after a single-dose intravenous injection at 80 mg/kg dose (Gan et al. 2022). However, phase II trials were put on hold for a few months after hypomagnesemia was observed, but the trials have been restarted recently. The hypomagnesemia was attributed to proximal tubular toxicity, and so, the renal clearance of these conjugates has to be observed carefully (Aartsma-Rus 2022).
The exploration of the Pip series of PMOs, specifically the Pip6 series, has led to a more critical understanding of why cardiac muscle delivery is not as straightforward as skeletal muscle delivery (Betts et al. 2012). The hydrophobic–cationic balance, as well as their separation in the chimera, is important for in vivo delivery systems, and the results mostly do not correlate with in vitro data. The placement of cationic or hydrophobic groups could also change the secondary structure of the conjugate, which could alter its serum interaction and circulatory half-life. These parameters are hard to predict without the experimental data and exhaustive structure–activity analysis is crucial to identifying the perfect peptide candidate.
4.3 Toxicity of PMO conjugates
Unconjugated PMOs have not shown much toxicity except in the kidney where tubular basophilia and vacuolation have been observed in non-human primates (Sazani et al. 2011; Carver et al. 2016). However, the effects were considered non-adverse and unlikely to develop into nephrotoxicity until the highest dose of 320 mg/kg. Immune response to PMOs has not been observed (Miyagoe-Suzuki and Takeda 2010).
As discussed in case of SRP-5051, the toxicity concerns in the case of peptide–PMO conjugates are real and serious. Even though CPPs have been known since the early 2000s and their application in PMO delivery in mdx mice was explored within 5–6 years, the progress from research to clinical trials has not been that fruitful (Järver et al. 2010). Since CPP–PMO conjugates show internalization by endocytosis, endosomal sequestration reduces the overall availability to a large extent, limiting their efficiency. Other ways to improve transfection with small molecules such as saponins (Wang et al. 2018), fructose (Cao et al. 2016), and aminoglycosides (Wang et al. 2019) have been tried mostly with only PMOs. One such report is available on PPMOs (Han et al. 2019), where B-MSP-PMO was used at 20 mg/kg in a formulation with 1:1 glucose:fructose and induced a two-fold higher dystrophin production in the cardiac muscle in adult mdx mice but not in aged mice. It has to be determined whether this type of formulation will improve pharmacological aspects of PPMOs further. Another issue with PPMOs is the adverse immune response in vivo, which is a serious point of contention in therapeutics (Järver et al. 2010).
4.4 Serum interaction of PMOs or their conjugates for DMD treatment
The reason for PMOs having an impressive safety profile in vivo is that they are not metabolized. No enzyme is known that recognizes and cleaves the unnatural backbone (Hudziak et al. 1996), and they are not metabolized in the body (Amantana et al. 2007). However, intravenous delivery also leads to high renal clearance as their molecular weight (~6–8 KDa) lies much below the glomerular filtration cut-off (>30 kDa) (Cirak et al. 2011). This requires more frequent and higher dosing, elevating treatment costs. One significant difference between PMOs and PS-AONs is the serum interaction of the latter by virtue of the phosphorothioate moieties, which leads to greater circulation half-life (Crooke et al. 2020). If PMOs could also interact with the serum, thereby binding to albumins or other high-molecular weight proteins, then the renal clearance would be lower and much slower. In this direction, Caruthers and co-workers (Le et al. 2022) have reported the thiophosphoramidate morpholinos (TMOs) and evaluated their efficacy in inducing exon skipping in DMD in vitro, which was found to be better than that of PMOs. CPP–PMOs usually have a serum interaction due to the cationic/hydrophobic peptide portion, but they are also degraded because of the same. Lipid conjugation with other oligonucleotides such as siRNA is well known to increase serum binding and in vivo efficacy (Sarett et al. 2017), but a similar application in PMOs is not known.
4.5 Immune response due to dystrophin restoration
PMO exon-skipping-mediated truncated dystrophin restoration has also been shown to elicit a dystrophin-specific immune response in the mdx mice model, with serum-circulating antibodies that recognized both full-length and truncated dystrophin in mouse skeletal muscles (Vila et al. 2019). However, a recent report in patients has not established this fully, and it has been suggested that baseline T-cell response to dystrophin should be considered first before evaluating the same in the case of restored dystrophin (Anthony et al. 2023). These observations have extended the purview of DMD treatment beyond the restorative effects, and further research with larger sample size is necessary to establish any unfavourable immune responses.
5 Future perspectives: How do we make PMOs better?
Considering the overall scenario, the evolution of PMOs as modified oligonucleotides from basic research to therapy has been impressive and systematic when compared with other similar members such as siRNA, where a plethora of modifications (sugar and backbone) continue to this date. With the background of past research and an insight into the current challenges, several avenues for improvement can be ventured into for better therapeutic applications.
5.1 Modified PPMOs
It is imperative to think that conjugation of CPPs to PMOs, from the initial B-peptide to the latest Pip-6 derivatives has been the most beneficial aspect considering the extent of the restored dystrophin protein in skeletal as well as cardiac muscles. Many groups are currently working to find the best peptide sequences for optimum uptake, endosomal escape as well as activity in cardiac muscle, but the peptide backbone has mostly been natural and hence susceptible to degradation in vivo (Youngblood et al. 2007). Alternative strategies such as vivo-PMO suffer from toxicity associated with too many positive charges, and hence, a non-peptidic non-dendrimeric structure (Kundu et al. 2020) could be explored for similar activity without these drawbacks. Another way of doing this would be the introduction of unnatural amino acids within the peptide, such as D-amino acids (Schissel et al. 2022) and azaproline-based CPP (Gupta et al. 2022), for bypassing the peptidase-mediated degradation. This would also avoid the immunogenicity of the peptide, and if un-degraded, the conjugate would be much less toxic like PMOs.
Modifications like the GMO–PMO chimera have shown promising results without the need of an external conjugation and are expected to follow a PMO-like profile with respect to stability and toxicity with as minimum as four guanidinium groups. So, an approach beyond CPP could also be useful in the search for next-generation PMO therapy.
5.2 Improving serum interaction: Utilizing the immune system
As discussed earlier, improved serum interaction of uncharged PMOs could reduce the dose as well as dosing frequency, and this can also be enhanced with the help of immune system in DMD. Since muscle cells are inflamed, macrophage presence at the site of inflammation has been shown to act as a ‘reservoir’ of PMOs with slow release, for several days after injection (Novak et al. 2017). If PMOs have a longer circulatory half-life in the blood then they are more prone to phagocytic uptake and will stay intact in the macrophages until they reach the site of inflammation of the muscles. Hence, utilization of the phagocytic system could be a novel approach to PMO-based DMD therapeutics.
5.3 Effective PMO design
Several factors should be kept in mind before arriving at the correct target pre-mRNA sequence for exon skipping in case of a specific mutation. First, the secondary structure of the pre-mRNA has to be studied since PMO binding is more plausible at more accessible sites; stem-loop structures should be avoided. Closely related to this factor is the length of the PMO: longer PMOs provide greater binding to the pre-mRNA. Moreover, if the free energy of binding is sufficiently high then the pre-mRNA secondary structures can also change after binding to the PMO (Popplewell et al. 2009). It has also been observed that many exons contain exonic splicing enhancer (ESE) sites that are the binding sites of SR (serine-arginine) proteins, facilitate the incorporation of other splicing factors to promote splicing. Hence, ESE sites are favourable targets for AONs to induce exon skipping. An ESE finder has also been developed as a Web source to find the potential binding sites of SR proteins, and targeting ESE motives closer to the 5ʹ-end of exons which has shown to be more successful (Aartsma-Rus et al. 2005). An in silico screening tool has also been developed which could predict effective PMO sequences with 89% success rate (Echigoya et al. 2015). A shorter 25-mer PMO sequence was designed to target the same region of exon 51 and found to be equally effective as the 30-mer PMO sequence of eteplirsen (Akpulat et al. 2018).
Since binding energies of the mRNA–PMO duplex is important for effective skipping, a suitably designed modification in the PMO backbone which enhances the hybridization has immense therapeutic benefits. In this context, 5-substituted pyrimidine morpholino unit-incorporated PMOs have been reported to increase the duplex stability by >10°C (Das et al. 2023a). This would lead to shorter PMOs which provide the same effect as longer PMOs, and will be easier to synthesize and more cost-effective (figure 8).

Schematic depiction of increasing mRNA binding by nucleobase modifications. (A) Increase of melting temperature (Tm) upon incorporation of functionalized nucleobase in a PMO oligomer. (B) Uracil and cytosine as examples to show the site that can be functionalized.
5.4 Small-molecule-mediated enhancement
As reported previously, if small molecules which are usually well tolerable, such as hexose/fructose, can increase the exon skipping even by two-fold, then it could be a very promising option for in vivo application (Cao et al. 2016; Han et al. 2019). The effect should be observed with PPMOs since cardiac skipping is an important criterion. Other adjuvants such as saponins or aminoglycosides are much less favourable (Wang et al 2018, 2019) but if they can enhance the activities of PPMOs at lower doses then they could also have a beneficial effect.
6 PMO-based drugs: Laboratory research for patient benefit
It goes without saying that academic research on PMO-based therapies in DMD has shown several avenues for improvement of the immediate benefit of patients within very limited time. For aggressive transition from research to clinic, several strategies have to be employed by the government with the wholesome participation of scientists, clinicians, and the pharmaceutical industry.
First, an accurately maintained database of patients with specific mutations should be available to the scientist for proper sequence design. After sequence validation and synthesis, it is the clinician’s job to test the drug on patient samples for further validation of efficacy. Once this is established, technology should be transferred to the industrial setting for scaling up of clinical trials. In India, scaling up is still challenging because of the insufficient technological advancement in PMO synthesis and indigenous production is hampered by limited industry–academia collaboration, leading to a high cost of treatment. Lack of funds has also been an issue but there is a ray of hope in the form of the National Policy for Rare Diseases drafted in 2021, which also covers DMD. Perhaps, in the next few years, the production of DMD drugs will be started in India and patients will have a chance to improve their longevity and quality of life.
7 Conclusion
Many different approaches for DMD therapy have emerged over the years but morpholino-based drugs dominate the category of FDA-approved therapeutics, the only other drug being elevidys, which was approved in June 2023 for ambulatory pediatric patients. Elevidys has been given accelerated approval and involves the adeno-associated virus-mediated delivery of micro-dystrophin but there have been concerns about the immunogenicity of vectors previously (Agarwal et al. 2020). CRISPR-Cas9-mediated gene editing also holds a great promise in DMD for all types of mutation but requires AAV-mediated delivery for the Cas9 protein (Choi and Koo 2021). The small-molecule drug ataluren, which promotes a read through of the nonsense mutation in DMD, has been approved by the European Medicines Agency but not by the FDA (Dongsheng et al. 2021). However, long-term off-target effects, if any, are yet to be seen since the molecule can practically act everywhere in the body system. A multi-faceted approach combining efficient delivery and endosomal escape (Li et al. 2023) as well as enhanced mRNA binding of PMOs has to be pursued simultaneously. Considering the overall picture and status of on-going clinical trials, it can be postulated that modified PMOs with better stability and cell permeability have a better chance when it comes to the next-generation PMO-based therapeutics for DMD, and it is high time that leading research groups in the area collaborate to fast-track this development.
As research continues, the future of DMD treatment seems increasingly optimistic, with the potential to transform the lives of individuals affected by this devastating condition and improve their quality of life. Innovative research on combination therapies, including advancements in gene editing and regenerative medicine, more effective exon-skipping reagents, stem cell therapies, and muscle regeneration strategies continue to progress providing newer treatment avenues. While it is crucial to conduct rigorous clinical trials and ensure the safety and efficacy of these novel treatments, collaborative efforts between researchers, clinicians, and pharmaceutical companies can pave the way for transformative treatments in the future.
References
Aartsma-Rus A 2022 Good news for the mdx mouse community: Improved dystrophin restoration after skipping mouse dystrophin exon 23. Mol. Ther. Nucleic Acids 30 355–356
Aartsma-Rus A, De Winter CL, Janson AA, et al. 2005 Functional analysis of 114 exon-internal AONs for targeted DMD exon skipping: indication for steric hindrance of SR protein binding sites. Oligonucleotides 15 284–197
Aartsma-Rus A, Morgan J, Lonkar P, et al. 2019 Report of a TREAT-NMD/world Duchenne organisation meeting on dystrophin quantification methodology. J. Neuromuscul. Dis. 6 147–159
Agarwal S 2020 High-dose AAV gene therapy deaths. Nat. Biotechnol. 38 910
Akpulat U, Wang H, Becker K, et al. 2018 Shorter phosphorodiamidate morpholino splice-switching oligonucleotides may increase exon-skipping efficacy in DMD. Mol. Ther. Nucleic Acids 13 534–542
Alter J, Lou F, Rabinowitz A, et al. 2006 Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 12 175–177
Amantana A, Moulton HM, Cate ML, et al. 2007 Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide−morpholino oligomer conjugate. Bioconjugate Chem. 18 1325–1331
Anthony K, Ala P, Catapano F, et al. 2023 T cell responses to dystrophin in a natural history study of Duchenne muscular dystrophy. Hum. Gene Ther. 34 439–448
Betts C, Saleh AF, Arzumanov AA, et al. 2012 Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol. Ther. Nucleic Acids 1 e38
Cao L, Han G, Lin C, et al. 2016 Fructose promotes uptake and activity of oligonucleotides with different chemistries in a context-dependent manner in mdx mice. Mol. Ther. Nucleic Acids 5 e329
Carver MP, Charleston JS, Shanks C, et al. 2016 Toxicological characterization of exon skipping phosphorodiamidate morpholino oligomers (PMOs) in non-human primates. J. Neuromuscul. Dis. 3 381–393
Choi E and Koo T 2021 CRISPR technologies for the treatment of Duchenne muscular dystrophy. Mol. Ther. 29 3179–3191
Cirak S, Arechavala-Gomeza V, Guglieri M, et al. 2011 Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378 595–605
Corey DR and Abrams JM 2001 Morpholino antisense oligonucleotides: tools for investigating vertebrate development. Genome Biol. 2 1–3
Crooke ST, Vickers TA and Liang XH 2020 Phosphorothioate modified oligonucleotide–protein interactions. Nucleic Acids Res. 48 5235–5253
Das A, Ghosh A and Sinha S 2023a C5-pyrimidine-functionalized morpholino oligonucleotides exhibit differential binding affinity, target specificity and lipophilicity. Org. Biomol. Chem. 21 1242–1253
Das U, Kundu J, Shaw P, et al. 2023b Self-transfecting GMO-PMO chimera targeting Nanog enable gene silencing in vitro and suppresses tumor growth in 4T1 allografts in mouse. Mol. Ther. Nucleic Acids 32 203–228
Deconinck N and Dan B 2007 Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatr. Neurol. 36 1–7
Dongsheng D, Nathalie G, Eugenio M, et al. 2021 Duchenne muscular dystrophy (Primer). Nat. Rev. Dis. Primers 7 1–19
Echigoya Y, Mouly V, Garcia L, et al. 2015 In silico screening based on predictive algorithms as a design tool for exon skipping oligonucleotides in Duchenne muscular dystrophy. PLoS One 10 e0120058
Ferguson DP, Dangott LJ and Lightfoot JT 2014 Lessons learned from vivo-morpholinos: How to avoid vivo-morpholino toxicity. Biotechniques 56 251–256
Fletcher S, Honeyman K, Fall AM, et al. 2006 Dystrophin expression in the mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J. Gene Med. 8 207–216
Fletcher S, Honeyman K, Fall AM, et al. 2007 Morpholino oligomer–mediated exon skipping averts the onset of dystrophic pathology in the mdx mouse. Mol. Ther. 15 1587–1592
Gan L, Wu LC, Wood JA, et al. 2022 A cell-penetrating peptide enhances delivery and efficacy of phosphorodiamidate morpholino oligomers in mdx mice. Mol. Ther. Nucleic Acids 30 17–27
Gao X, Zhao J, Han G, et al. 2014 Effective dystrophin restoration by a novel muscle-homing peptide–morpholino conjugate in dystrophin-deficient mdx mice. Mol. Ther. 22 1333–1341
Gebski BL, Mann CJ, Fletcher S, et al. 2003 Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum. Mol. Genet. 12 1801–1811
Goyenvalle A, Babbs A, Powell D, et al. 2010 Prevention of dystrophic pathology in severely affected dystrophin/utrophin-deficient mice by morpholino-oligomer-mediated exon-skipping. Mol. Ther. 18 198–205
Gupta A, Gupta S, Das U, et al. 2022 Guanidinium-functionalized flexible azaproline transporter for efficient intracellular delivery of proapoptotic peptide and PDL1 antisense morpholino oligo in human carcinoma cells in vitro. Bioconjugate Chem. 33 907–917
Han G, Gu B, Lin C, et al. 2019 Hexose potentiates peptide-conjugated morpholino oligomer efficacy in cardiac muscles of dystrophic mice in an age-dependent manner. Mol. Ther. Nucleic Acids 18 341–350
Happi Mbakam C, Lamothe G and Tremblay JP 2022 Therapeutic strategies for dystrophin replacement in Duchenne muscular dystrophy. Front. Med. 9 774
Heemskerk H, De Winter CL, Van Ommen GJB, et al. 2009 Development of antisense mediated exon skipping as a treatment for Duchenne muscular dystrophy. Ann. N. Y. Acad. Sci. 1175 71–79
Hudziak RM, Barofsky E, Barofsky DF, et al. 1996 Resistance of morpholino phosphorodiamidate oligomers to enzymatic degradation. Antisense Nucleic Acid Drug Dev. 6 267–272
Ivanova GD, Arzumanov A, Abes R, et al. 2008 Improved cell-penetrating peptide–PNA conjugates for splicing redirection in HeLa cells and exon skipping in mdx mouse muscle. Nucleic Acids Res. 36 6418–6428
Järver P, Mäger I and Langel Ü 2010 In vivo biodistribution and efficacy of peptide mediated delivery. Trends Pharmacol. Sci. 31 528–535
Järver P, Zaghloul EM, Arzumanov AA, et al. 2015 Peptide nanoparticle delivery of charge-neutral splice-switching morpholino oligonucleotides. Nucleic Acid Ther. 25 65–77
Jearawiriyapaisarn N, Moulton HM, Buckley B, et al. 2008 Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice. Mol. Ther. 16 1624–1629
Jearawiriyapaisarn N, Moulton HM, Sazani P, et al. 2010 Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc. Res. 85 444–453
Kundu J, Banerjee P, Bose C, et al. 2020 Internal oligoguanidinium transporter: mercury-free scalable synthesis, improvement of cellular localization, endosomal escape, mitochondrial localization, and conjugation with antisense morpholino for NANOG inhibition to induce chemosensitization of taxol in MCF-7 cells. Bioconjugate Chem. 31 2367–2382
Le BT, Paul S, Jastrzebska K, et al. 2022 Thiomorpholino oligonucleotides as a robust class of next generation platforms for alternate mRNA splicing. Proc. Natl. Acad. Sci. USA 119 e2207956119
Li X, Kheirabadi M, Dougherty PG, et al. 2023 The endosomal escape vehicle platform enhances delivery of oligonucleotides in preclinical models of neuromuscular disorders. Mol. Ther. Nucleic Acids 33 273–285
Li YF and Morcos PA 2008 Design and synthesis of dendritic molecular transporter that achieves efficient in vivo delivery of morpholino antisense oligo. Bioconjugate Chem. 19 1464–1470
Lim KRQ, Woo S, Melo D, et al. 2022 Development of DG9 peptide-conjugated single-and multi-exon skipping therapies for the treatment of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 119 e2112546119
Malerba A, Boldrin L and Dickson G 2011 Long-term systemic administration of unconjugated morpholino oligomers for therapeutic expression of dystrophin by exon skipping in skeletal muscle: implications for cardiac muscle integrity. Nucleic Acid Ther. 21 293–298
Malerba A, Thorogood FC, Dickson G, et al. 2009 Dosing regimen has a significant impact on the efficiency of morpholino oligomer-induced exon skipping in mdx mice. Hum. Gene Ther. 20 955–965
Mann CJ, Honeyman K, Cheng AJ, et al. 2001 Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl. Acad. Sci. USA 98 42–47
Miyagoe-Suzuki Y and Takeda SI 2010 Gene therapy for muscle disease. Exp. Cell Res. 316(18) 3087–3092
Monaco AP, Bertelson CJ, Liechti-Gallati S, et al. 1988 An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2 90–95
Muntoni F, Torelli S and Ferlini A 2003 Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2 731–740
Nakamura A and Takeda SI 2009 Exon skipping therapy for Duchenne muscular dystrophy. Neuropathology 29 494–501
Nasevicius A and Ekker SC 2000 Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 26 216–220
Novak JS, Hogarth MW, Boehler JF, et al. 2017 Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nat. Commun. 8 941
O’Donovan L, Okamoto I, Arzumanov AA, et al. 2015 Parallel synthesis of cell-penetrating peptide conjugates of PMO toward exon skipping enhancement in Duchenne muscular dystrophy. Nucleic Acid Ther. 25 1–10
Popplewell LJ, Trollet C, Dickson G, et al. 2009 Design of phosphorodiamidate morpholino oligomers (PMOs) for the induction of exon skipping of the human DMD gene. Mol. Ther. 17 554–561
Roberts RG, Coffey AJ, Bobrow M, et al. 1993 Exon structure of the human dystrophin gene. Genomics 16 536–538
Ryder S, Leadley RM, Armstrong N, et al. 2017 The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J. Rare Dis. 12 1–21
Sarett SM, Werfel TA, Lee L, et al. 2017 Lipophilic siRNA targets albumin in situ and promotes bioavailability, tumor penetration, and carrier-free gene silencing. Proc. Natl. Acad. Sci. USA 114 E6490–E6497
Sazani P, Ness KPV, Weller DL, et al. 2011 Repeat-dose toxicology evaluation in cynomolgus monkeys of AVI-4658, a phosphorodiamidate morpholino oligomer (PMO) drug for the treatment of duchenne muscular dystrophy. Int. J. Toxicol. 30 313–321
Schissel CK, Farquhar CE, Malmberg AB, et al. 2022 Cell-penetrating d-peptides retain antisense morpholino oligomer delivery activity. ACS Biol. Med. Chem. Au. 2 150–160
Sicinski P, Geng Y, Ryder-Cook AS, et al. 1989 The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science 244 1578–1580
Sierakowska H, Sambade MJ, Agrawal S, et al. 1996 Repair of thalassemic human β-globin mRNA in mammalian cells by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 93 12840–12844
Singh SM, Kongari N, Cabello-Villegas J, et al. 2010 Missense mutations in dystrophin that trigger muscular dystrophy decrease protein stability and lead to cross-β aggregates. Proc. Natl. Acad. Sci. USA 107 15069–15074
Siva K, Covello G and Denti MA 2014 Exon-skipping antisense oligonucleotides to correct missplicing in neurogenetic diseases. Nucleic Acid Ther. 24 69–86
Summerton J and Weller D 1997 Morpholino antisense oligomers: design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 7 187–195
Van Deutekom JC, Bremmer-Bout M, Janson AA, et al. 2001 Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum. Mol. Genet. 10 1547–1554
Vila MC, Novak JS, Benny Klimek M, et al. 2019 Morpholino induced exon skipping stimulates cell-mediated and humoral responses to dystrophin in mdx mice. J. Pathol. 248 339–351
Vitiello L, Bassi N, Campagnolo P, et al. 2008 In vivo delivery of naked antisense oligos in aged mdx mice: analysis of dystrophin restoration in skeletal and cardiac muscle. Neuromuscul. Disord. 18 597–605
Wagner KR, Lechtzin N and Judge DP 2007 Current treatment of adult Duchenne muscular dystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 1772 229–237
Wang M, Wu B, Lu P, et al. 2013 Polyethylenimine-modified pluronics (PCMs) improve morpholino oligomer delivery in cell culture and dystrophic mdx mice. Mol. Ther. 21 210–216
Wang M, Wu B, Shah SN, et al. 2018 Saponins as natural adjuvant for antisense morpholino oligonucleotides delivery in vitro and in mdx mice. Mol. Ther. Nucleic Acids 11 192–202
Wang M, Wu B, Shah SN, et al. 2019 Aminoglycoside enhances the delivery of antisense morpholino oligonucleotides in vitro and in mdx mice. Mol. Ther. Nucleic Acids 16 663–674
Wilson DGS, Tinker A and Iskratsch T 2022 The role of the dystrophin glycoprotein complex in muscle cell mechanotransduction. Commun. Biol. 5 1022
Wu B, Li Y, Morcos PA, et al. 2009 Octa-guanidine morpholino restores dystrophin expression in cardiac and skeletal muscles and ameliorates pathology in dystrophic mdx mice. Mol. Ther. 17 864–871
Wu B, Moulton HM, Iversen PL, et al. 2008 Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. USA 105 14814–14819
Wu B, Xiao B, Cloer C, et al. 2011 One-year treatment of morpholino antisense oligomer improves skeletal and cardiac muscle functions in dystrophic mdx mice. Mol. Ther. 19 576–583
Wu RP, Youngblood DS, Hassinger JN, et al. 2007 Cell-penetrating peptides as transporters for morpholino oligomers: effects of amino acid composition on intracellular delivery and cytotoxicity. Nucleic Acids Res. 35 5182–5191
Yin H, Moulton HM, Betts C, et al. 2010 Functional rescue of dystrophin-deficient mdx mice by a chimeric peptide-PMO. Mol. Ther. 18 1822–1829
Yin H, Moulton HM, Betts C, et al. 2009 A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 18 4405–4414
Yin H, Moulton HM, Seow Y, et al. 2008 Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum. Mol. Genet. 17 3909–3918
Yin H, Saleh AF, Betts C, et al. 2011 Pip5 transduction peptides direct high efficiency oligonucleotide-mediated dystrophin exon skipping in heart and phenotypic correction in mdx mice. Mol. Ther. 19 1295–1303
Yokota T, Nakamura A, Nagata T, et al. 2012 Extensive and prolonged restoration of dystrophin expression with vivo-morpholino-mediated multiple exon skipping in dystrophic dogs. Nucleic Acid Ther. 22 306–315
Youngblood DS, Hatlevig SA, Hassinger JN, et al. 2007 Stability of cell-penetrating peptide− morpholino oligomer conjugates in human serum and in cells. Bioconjugate Chem. 18 50–60
Zubrzycka-Gaarn EE, Bulman DE, Karpati G, et al. 1988 The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature 333 466–469
Acknowledgements
SS thanks SERB, New Delhi, Government of India (Grant No. TTR/2021/000044) for grant support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: Alok Bhattacharya
This article is part of the Topical Collection: The Rare Genetic Disease Research Landscape in India.
Rights and permissions
About this article

Cite this article
Gupta, S., Sharma, S.N., Kundu, J. et al. Morpholino oligonucleotide-mediated exon skipping for DMD treatment: Past insights, present challenges and future perspectives. J Biosci 48, 38 (2023). https://doi.org/10.1007/s12038-023-00365-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-023-00365-z