
Abstract
The extremely high mortality of both lung cancer and Idiopathic pulmonary fibrosis (IPF) is a global threat. Early detection and diagnosis can reduce their mortality. Since fibrosis is a necessary process of cancer, identifying the common potential prognostic genes involved in these two diseases will significantly contribute to disease prevention and targeted therapy. Microarray datasets of IPF and lung cancer were extracted from the GEO database. GEO2R was exploited to retrieve the differentially expressed genes (DEGs). The intersecting DEGs were obtained by the Venn tool. DAVID tools were used to perform GO and KEGG pathway enrichment analysis of DEGs. Then, the Kaplan–Meier plotter was employed to determine the prognostic value and verify the expression, pathological stage, and phosphorylation level of the hub gene in the TCGA and GTEx database. Finally, the extent of immune cell infiltration in lung cancer was estimated by the TIMER2 tool. The Venn diagram revealed 1 upregulated gene and 15 downregulated genes from GSE32863, GSE43458, GSE118370, and GSE75037 of lung cancer, as well as GSE2052 and GSE53845 of IPF. CytoHubba identified the top three genes [TEK receptor tyrosine kinase (TEK), caveolin 1 (CAV1), and endomucin (EMCN)] as hub genes following the connectivity degree. Survival analysis claimed the association of only TEK and CAV1 expression to both overall survival (OS) and first progression (FP). Pathological stage analyses revealed the relationship of only CAV1 expression to the pathological stage and the significant correlation of only CAV1 phosphorylation expression level for lung cancer. Furthermore, a statistically positive correlation was observed between the immune infiltration of cancer-associated fibroblasts, endothelial, and neutrophils with the CAV1 expression in lung cancer, whereas the contradictory result was noted for the immune infiltration of T cell follicular helper. Early detection and diagnostic potential of lung cancer are ameliorated by the combined selection of key genes among IPF and lung cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Lung cancer is highly malignant, and its morbidity and mortality are increasing at an alarming rate with the passing years (Qi et al. 2014). Early detection, diagnosis, and treatment can effectively minimize the mortality rate of lung cancer (Bade and Dela Cruz 2020). Since the discovery of cancer biomarkers plays a key role in the early diagnosis, treatment guidance, and prognostic monitoring of cancer, it is of prime importance to elucidate effective markers for lung cancer (Hoy et al. 2019). Idiopathic pulmonary fibrosis (IPF) is mainly fibrotic lesions in lung tissue resulting from alveolar epithelial cell damage and abnormal repair, affecting about 3 million people worldwide (Martinez et al. 2017; Glass et al. 2020).
A large body of epidemiological evidence substantiates the positive correlation of IPF to lung cancer (Tzouvelekis et al. 2019). One study reported the preferential development of lung tumors in the immediate vicinity of the fibrotic area of IPF patients, compared with the non-IPF-related lung tumors (Kinoshita and Goto 2019). Moreover, Jang et al. documented that the risk of malignancy in IPF patients is approximately eight times that of the general population and determined the impact of the severity of IPF on the clinical outcome of lung cancer patients (Jang et al. 2021).
Both IPF and lung cancer are serious lung diseases. Many studies have reflected several pathogenic similarities, including genetics, epigenetic markers, and biological pathways between these two diseases (Karampitsakos et al. 2017; Kim et al. 2021; Ballester et al. 2019). Therefore, screening out the target genes shared by these two diseases and understanding their related pathophysiological mechanisms and molecular pathways are more conducive to the diagnosis and targeted therapy of diseases.
GEO (Gene Expression Omnibus) plays a pivotal role in bioinformatics analysis (Barrett et al. 2013). The GEO database was employed in this study to elucidate the shared hub gene of IPF and lung cancer with thorough bioinformatics analysis.
2 Materials and methods
2.1 Data source
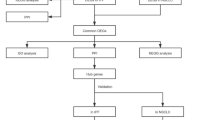
To identify differentially expressed genes and enriched pathways in lung cancer, we first entered “lung cancer” and selected “There are 46365 results for “lung cancer” in the GEO DataSets Database”. Next, we selected “Series (2040)”, and further selected “Homo sapiens (1704)”. Finally, GSE118370, GSE75037, GSE32683, and GSE43458 were opted. Thereafter, for screening and isolating the key candidate genes and pathways of pulmonary fibrosis, we logged into the GEO database website entering “idiopathic pulmonary fibrosis”, and GSE53845 and GSE2052 were selected.
2.2 Data processing of DEGs
The GEO2R online analysis tool helped to retrieve the DEGs (P < 0.05, |logFC| ≥ 1.0) between lung cancer and normal tissue samples and between IPF and control samples. Based on adjusted P < 0.05, upregulated and downregulated genes were initially picked out and visualized with a volcano map. The Venn tool was then adopted to identify the intersecting part.
2.3 Enrichment analysis of DEGs
DAVID tools examined the results of GO and KEGG pathway enrichment analysis of DEGs, which were finally visualized with the chart function of the excel tool. FDR < 0.05 and gene counts ≥ 3 in GO annotation analysis as well as P < 0.05 and gene counts ≥ 40 in KEGG pathway enrichment analysis were considered as statistically significant.
2.4 PPI network and hub gene analysis
The potential PPI (protein-protein interaction) relationship of the DEGs identified from combined lung cancer vs. normal tissue samples, and IPF vs. control samples was analyzed. We selected “low confidence (0.15)” in the “minimum required interaction score” module in the STRING database and established the PPI network by Cytoscape. The hub genes were recognized by CytoHubba (a plugin in Cytoscape).
2.5 Survival analysis of hub genes
In Kaplan–Meier plotter website, the hub genes (TEK (TEK receptor tyrosine kinase), CAV1 (caveolin 1), and EMCN (endomucin)) were fed into the “use multiple genes” module. As to “Smoking history”, we selected “only those never smoked”.
2.6 Hub gene expression analysis
TIMER2 and GEPIA2 tools were employed to ascertain the difference in expression of CAV1 or TEK between lung cancer and adjacent normal tissues. For differential pathological stages, violin plots of the CAV1 or TEK expression were also obtained through GEPIA2. Moreover, the expression level of the total protein or phosphoprotein of CAV1 and TEK was explored by the lung cancer datasets of the UALCAN portal.
2.7 Immune infiltration analysis
The morality between CAV1 expression and immune infiltrates was investigated by TIMER2. The immune cells of cancer-associated fibroblasts, endothelial, neutrophil, and T cell follicular helper were selected. Finally, a heatmap and a scatter plot were used to visualize the data.
3 Results
3.1 Identification of DEGs
Differentially expressed genes and enriched pathways in lung cancer were elucidated by screening four gene expression profiles (GSE32863, GSE43458, GSE118370, and GSE75037). GSE32863 contained 58 lung cancer tissue specimens (T) and 58 normal tissue specimens (N), GSE43458 included 40 T and 30 N, GSE75037 comprised 83 T and 83 N, and GSE118370 encompassed 6 T and 6 N (figure 1A). Furthermore, GSE2052 and GSE53845 were selected to screen and identify the key candidate genes and pathways of IPF. There were 40 IPF samples and eight control samples in GSE2052, and GSE43458 included 13 IPF specimens and 11 control specimens (figure 1B). First, upregulated and downregulated genes were picked out based on adjusted P < 0.05 and visualized via a volcano map (figure 1A, B). Subsequently, based on the criteria of P < 0.05 and |logFC| ≥ 1.0, we identified and got the intersection of the DEG profiles by Venn analysis (figure 1C, D, E).

Identification of DEGs and Venn diagram of DEGs common to all six GEO datasets. (A) DEGs among lung cancer vs. normal tissues. (B) DEGs among IPF vs. control. (C) Upregulated genes and downregulated genes among lung cancer vs. normal tissues. (D) Upregulated genes and downregulated genes among IPF vs. control. (E) Upregulated genes and downregulated genes among the intersection gene of lung cancer and IPF. DEGs, differentially expressed genes; GEO, Gene Expression Omnibus; IPF, idiopathic pulmonary fibrosis.
3.2 Enrichment analysis of DEGs
GO function enrichment analysis (table 1 and figure 2A) highlighted dominant enrichment of DEGs in BPs, including angiogenesis (GO:0001525) and negative regulation of endothelial cell proliferation (GO:0001937), and also dominant enrichment in CCs, including membrane raft (GO:0045121) and plasma membrane (GO:0005886). Furthermore, KEGG pathways enrichment analysis (table 2 and figure 2B) revealed that DEGs were dominantly enriched in the following pathways: rheumatoid arthritis (hsa05323), cardiac muscle contraction (hsa04260), bacterial invasion of epithelial cells (hsa05100), adherens junction (hsa04520), viral myocarditis (hsa05416), and ferroptosis (hsa04216).

GO and KEGG enrichment analysis of the overlapping DEGs. (A) The significantly enriched GO terms of down-and upregulated DEGs. The x-axis signifies the adjusted P value, bubble size represents the number of DEGs involved in GO terms, and the y-axis indicates the terms of the significant enriched GO function. (B) KEGG pathway enrichment analysis of down-and upregulated DEGs. The x-axis represents the P value, and the y-axis denotes the terms of the significant KEGG pathway, and the y-axis from bottom to top indicates a gradual increase in the enrichment number of DEGs involved in KEGG terms. GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
3.3 PPI network and hub gene analysis
The protein interactions among the DEGs were explored by STRING tools and visualized by Cytoscape software. The results documented a total of 16 nodes and 31 edges present in the PPI network (figure 3A). According to the degree of connectivity, the top three most prominent genes identified in the PPI network included TKE (50), CAV1 (40), and EMCN (31) (figure 3B).

Protein-protein interaction (PPI) network construction and hub gene identification. (A) PPI network constructed with the DEGs. Note: green nodes represent downregulated genes. (B) Top three hub genes (TEK, CAV1, and EMCN) with a higher degree of connectivity. Note: green nodes denote downregulated genes. The nodes colors of TEK, CAV1, and EMCN symbolize the degree of connectivity.
3.4 Survival analysis of hub genes
Eliminating the smoking factor, we tried to explore the impact of the hub genes on the survival and prognosis of lung cancer patients. Therefore, the three potential hub genes were considered to assess the prognostic values of lung cancer with never smoking. A total of 141 lung cancer patients with never smoking were retrieved for the analysis of first progression (FP). As illustrated in figure 4A, the low expression of TEK, CAV1, and EMCN was associated with unfavorable FP of lung cancer patients who have never smoked.

The Kaplan–Meier plotter was employed to perform a series of survival analyses, including (A) FP and (B) OS, via the expression level of CAV1, EMCN, and TEK genes in lung cancer cases.
Furthermore, we also retrieved 141 lung cancer patients who have never smoked and were subjected to the overall survival (OS) analysis. After analysis, we reported the correlation of only the low expression of TEK and CAV1 among the selected three hub genes to the unfavorable OS in lung cancer patients who had never smoked (figure 4B).
3.5 CAV1 and TEK genes expression analysis
The expression status of CAV1 and TEK in LUAD (Lung adenocarcinoma) and LUSC (Lung squamous cell carcinoma) of TCGA was estimated by the TIMER2 tool. As substantiated by figure 5A, the expression levels of CAV1 and TEK in LUAD and LUSC (P < 0.001) were found to be lower than the corresponding control tissues. Similar results (P < 0.05) were obtained for match TCGA normal and GTEx data, as evident in figure 5B. Moreover, a mutual correlation was reflected only between CAV1 expression and the pathological stages of LUSC (figure 5C, D). Figure 5E claimed significant differences in all the CAV1 phosphorylation sites (S9, S37, Y25, and T46) and total protein levels. However, TEK exhibited no phosphorylation sites, but the total protein level highlighted the significant differences.

Expression levels of CAV1 and TEK genes in LUAD and LUSC and pathological stages, and phosphorylation analysis. (A) The expression status of CAV1 and TEK genes in LUAD and LUSC were assessed through TIMER2. ***P < 0.001. (B) Match TCGA normal and GTEx data; the box plot data were supplied. *P < 0.05. Log2 (TPM + 1) was applied for log-scale. (C, D) Based on the TCGA data, the expression levels of CAV1 and TEK genes were evaluated by the fundamental pathological stages (stage I, stage II, stage III, and stage IV) of LUAD and LUSC. Log2 (TPM + 1) was applied for log-scale. (E) The phosphoprotein sites with positive results and the total proteins levels of CAV1 and TEK are illustrated in the schematic diagram of CAV1 protein.
3.6 Analysis of CAV1 gene immune infiltration
Tumor-infiltrating immune cells, integral components of the tumor microenvironment, significantly contribute to tumor diagnosis, survival outcome, and clinical therapeutic sensitivity. The present study examined the relationship between CAV1 expression and the infiltration level of cancer-associated fibroblasts, endothelial, neutrophils, and T cell follicular helper across LUAD and LUSC in TCGA. Heat maps based on all or most algorithms, illustrated in figure 6, confirmed that the immune infiltration of cancer-associated fibroblasts, endothelial, and neutrophils manifested a statistically positive correlation to the CAV1 expression in LUAD and LUSC, whereas the opposite result was observed for the immune infiltration of T cell follicular helper. Figure 6 also demonstrated the scatterplot data of LUAD and LUSC generated with one algorithm based on the most correlation.

Different algorithms were exploited to investigate the potential correlation between the expression level of the CAV1 gene and the infiltration level of cancer-associated fibroblasts, endothelial, neutrophil, and T cell follicular helper across LUAD and LUSC in TCGA.
4 Discussion
Lung cancer is a malignant tumor, seriously endangering human health (Qi et al. 2014). IPF is an interstitial disease with insidious onset and gradually deteriorating condition (Tang et al. 2019). The progressive lung scar formation of IPF aggravates the risk for lung cancer (Kinoshita et al. 2019). The significant similarities between IPF and lung cancer in terms of pathophysiological pathways and potential pathogenic factors (Karampitsakos et al. 2017; Kim et al. 2021; Ballester et al. 2019) motivated the scientists in the present study to identify the hub genes shared by the above two diseases with the goal to determine new targeted therapies for lung cancer.
This study used the GEO database to screen out the DEGs between IPF or lung cancer and healthy lung tissue. Overall, we identified 16 DEGs that were shared among IPF and lung cancer. Enrichment analysis based on the above 16 DEGs revealed that these DEGs were concerned with GO BP and CC terms (plasma membrane GO: 0005886 and membrane raft GO: 0045121). Predominant enrichment of these DEGs was witnessed in rheumatoid arthritis, cardiac muscle contraction, viral myocarditis, and Ferroptosis, as per KEGG analysis. PPI network analysis determined the relationship between DEGs, and three hub genes, including TEK (connectivity degree = 50), CAV1 (connectivity degree = 40), and EMCN (connectivity degree = 31), were elucidated according to the degree of connectivity. Subsequently, the survival analysis claimed that the low expression of all the hub genes was involved in the first progression of lung cancer patients with the Kaplan-Meier plotter online tool. Nonetheless, only the low expression of TEK and CAV1 were unfavorable factors for the overall survival of lung cancer patients. Immediately afterward, the low expression of TEK and CAV1 in lung cancer tissues in the TCGA and GTEx databases were verified, and association of the only CAV1 with the pathological stage of LUSC (P = 0.0285) was documented. Additionally, the UALCAN portal confirmed that only CAV1 showed significant alterations in phosphorylation sites between primary tumor and normal tissues. CAV1 was thus selected for further analysis. Finally, tumor immune cell infiltration analysis indicated a statistically positive correlation of the immune infiltration of cancer-associated fibroblasts, endothelial, and neutrophils to the CAV1 expression in LUAD and LUSC, while the opposite result was obtained for the immune infiltration of T cell follicular helper.
The main component of caveolin is Caveolin 1 (CAV1), which is involved in the regulation of cell signal transduction and endocytosis and is a potential target for the treatment of fibrotic diseases. Research has validated the down-regulation of inflammasomes activity and prevention of bleomycin-mediated pulmonary fibrosis resulting from gene transfer of CAV1 (Lin et al. 2019). Fibrosis is generally regarded to be a precursor to lung cancer. It is worth noting that, as far as human risk assessment is concerned, fibrosis is an intermediate step in microparticle-induced lung cancer (Gvaramia et al. 2013). Furthermore, CAV1 with two isoforms of α and β is a complete oncogenic membrane protein related to extracellular matrix tissue, cell migration, and signal transduction. It is engaged in a variety of cellular functions, such as vesicle transport and cell adhesion, and the regulation of signal transduction during growth and survival (Quest et al. 2013). Previous studies have highlighted the alteration of CAV1 in several cancer types, and it also affects the occurrence or onset of tumors by controlling metabolism (Nwosu et al. 2016). On the contrary, some reports have documented the activity of CAV1 as a tumor suppressor (Díaz et al. 2020; Sanhueza et al. 2020; Zhou et al. 2018), such as CAV1 inhibits tumor formation by impeding unfolded protein responses (Díaz et al. 2020), and its mediated tumor suppression is associated with HIF1αS-nitrosylation and decreased transcriptional activity under hypoxia (Sanhueza et al. 2020). Usually, the presence of CAV1 on the plasma membrane is essential for its tumor suppressor activity. The tumor suppressor activity of this protein is also believed to mediate through the interaction of CAV1 with a variety of proteins (Díaz et al. 2020). Henceforth, more and more evidence has established the role of CAV1 as a clinical biomarker of cancer progression. However, there are limited studies on CAV1 as a prognostic factor of lung cancer. Air dust particles are an important factor followed by smoking that induces pulmonary fibrosis. In our study, compared with normal lung tissue, CAV1 expression was found to be low in lung cancer, and it was related to the overall survival of lung cancer patients who exclude smoking. This justifies that CAV1 could be a more important target factor.
Immune cell infiltration is found to be closely associated with the clinical outcome, and thus, it is mostly targeted by drugs to improve the survival rate of patients. Tumor tissues are not pure tumor cells but also immune-related cells. For example, cancer-associated fibroblasts (CAF), the main matrix component of cancer, take part in the regulation of the infiltration function of various tumor immune cells (Wang et al. 2021; Takahashi et al. 2017). In addition, the microenvironment outside the tumor and its behavior are regulated by various factors secreted by numerous tumor immune cells, such as macrophages, T cells, NK cells, endothelial, neutrophils, T cell follicular helper, etc. (Gu-Trantien et al. 2013; Ino et al. 2013; Domingues et al. 2016). In this study, based on all or most algorithms, we found a statistically positive correlation of the immune infiltration of cancer-associated fibroblasts, endothelial, and neutrophils with the CAV1 expression in LUAD and LUSC, whereas the opposite result was witnessed for the immune infiltration of T cell, follicular helper. This is beneficial to study the mechanism of CAV1 in lung cancer.
The present study exploited a variety of bioinformatics analyses to identify the most noteworthy hub gene CAV1, which may be nurtured as an effective therapeutic target for the early diagnosis and prevention of IPF and lung cancer. However, biological experiments are required to corroborate these findings, and further research is essential to decipher the underlying molecular etiology.
References
Bade BC and Dela Cruz CS 2020 Lung Cancer 2020: Epidemiology, etiology, and prevention. Clin. Chest Med. 41 1–24
Ballester B, Milara J and Cortijo J 2019 Idiopathic pulmonary fibrosis and lung cancer: mechanisms and molecular targets. Int. J. Mol. Sci. 20 593
Barrett T, Wilhite SE, Ledoux P, et al. 2013 NCBI GEO: archive for functional genomics data sets – update. Nucleic Acids Res. 41 D991–D995
Díaz MI, Díaz P, Bennett JC, Urra H, Ortiz R, Orellana PC, Hetz C and Quest AFG 2020 Caveolin-1 suppresses tumor formation through the inhibition of the unfolded protein response. Cell Death Dis. 11 648
Domingues P, González-Tablas M, Otero Á, et al. 2016 Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav. Immun. 53 1–15
Glass DS, Grossfeld D, Renna HA, et al. 2020 Idiopathic pulmonary fibrosis: Molecular mechanisms and potential treatment approaches. Respir. Investig. 58 320–335
Gu-Trantien C, Loi S, Garaud S, et al. 2013 CD4+ follicular helper T cell infiltration predicts breast cancer survival. J. Clin. Invest. 123 2873–2892
Gvaramia D, Blaauboer ME, Hanemaaijer R and Everts V 2013 Role of caveolin-1 in fibrotic diseases. Matrix Biol. 32 307–315
Hoy H, Lynch T and Beck M 2019 Surgical Treatment of lung cancer. Crit. Care Nurs. Clin. North Am. 31 303–313
Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y and Hiraoka N 2013 Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 108 914–923
Jang HJ, Park MS, Kim YS, Chang J, Lee JH, Lee CT, Lee SH and Yoon HI 2021 The relationship between the severity of pulmonary fibrosis and the lung cancer stage. J. Cancer. 12 2807–2814
Karampitsakos T, Tzilas V, Tringidou R, et al. 2017 Lung cancer in patients with idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 45 1–10
Kim HC, Lee S and Song JW 2021 Impact of idiopathic pulmonary fibrosis on clinical outcomes of lung cancer patients. Sci. Rep. 11 8312
Kinoshita T and Goto T 2019 Molecular mechanisms of pulmonary fibrogenesis and its progression to lung cancer: A review. Int. J. Mol. Sci. 20 1461
Lin X, Barravecchia M, Matthew Kottmann R, Sime P and Dean DA 2019 Caveolin-1 gene therapy inhibits inflammasome activation to protect from bleomycin-induced pulmonary fibrosis. Sci. Rep. 9 19643
Martinez FJ, Collard HR, Pardo A, et al. 2017 Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 3 17074
Nwosu ZC, Ebert MP, Dooley S and Meyer C 2016 Caveolin-1 in the regulation of cell metabolism: a cancer perspective. Mol. Cancer. 15 71
Qi W, Li X and Kang J 2014 Advances in the study of serum tumor markers of lung cancer. J Cancer Res. Ther. 10 C95–C101
Quest AF, Lobos-González L, Nuñez S, et al. 2013 The caveolin-1 connection to cell death and survival. Curr. Mol. Med. 13 266–281
Sanhueza C, Bennett JC, Valenzuela-Valderrama M, et al. 2020 Caveolin-1-mediated tumor suppression is linked to reduced HIF1α S-nitrosylation and transcriptional activity in hypoxia. Cancers 12 2349
Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T and Chikamatsu K 2017 Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 8 8633–8647
Tang H, Zhang L, She YL, Su H and Chen C 2019 Surgical treatment of lung cancer patients with idiopathic pulmonary fibrosis. Zhonghua Wai Ke Za Zhi. 57 956–960
Tzouvelekis A, Gomatou G, Bouros E, Trigidou R, Tzilas V and Bouros D 2019 Common pathogenic mechanisms between idiopathic pulmonary fibrosis and lung cancer. Chest 156 383–391
Wang Z, Yang Q, Tan Y, Tang Y, Ye J, Yuan B and Yu W 2021 Cancer-associated fibroblasts suppress cancer development: the other side of the coin. Front. Cell Dev. Biol. 9 613534
Zhou W, He L, Dai Y, Zhang Y, Wang J and Liu B 2018 MicroRNA-124 inhibits cell proliferation, invasion and migration by targeting CAV1 in bladder cancer. Exp. Ther. Med. 16 2811–2820
Acknowledgements
We appreciate the Gene Expression Omnibus database and the Cancer Genome Atlas for the open data.
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: Sreenivas Chavali
Corresponding editor: Sreenivas Chavali
Rights and permissions
About this article

Cite this article
Yin, D., Qiu, J., Hu, S. et al. CAV1 is a prognostic predictor for patients with idiopathic pulmonary fibrosis and lung cancer. J Biosci 47, 13 (2022). https://doi.org/10.1007/s12038-021-00245-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-021-00245-4