
Abstract
Oxidative stress and apoptosis is involved in hypoxia-reoxygenation (H/R) induced myocardial injury. Increased expression of uncoupling protein 2 (UCP2), a cationic carrier protein, has protective effect against H/R injury. The present study aimed to find candidate drugs for H/R induced cardiac damage by identifying compounds regulating UCP2 expression. Here, among six natural compounds, ursolic acid (UA) had the most significant induction effect on UCP2 expression in H9c2 cells under H/R conditions. Subsequently, we found that UA significantly attenuated cell apoptosis and Caspase 3 activity, but increased nitric oxide (NO) release under H/R conditions. Additionally, UA pretreatment also decreased reactive oxygen species (ROS) production and malondialdehyde (MDA) content, but increased superoxide dismutase (SOD) activity. H/R caused a notable increase in the phosphorylation of p38, which was weakened by UA pretreatment. Moreover, p38 inhibitor (SB203580) showed the similar effects on H/R cells as UA pretreatment, while UCP2 knockdown had the reverse biological effects. More importantly, the effects of UA or p38 inhibitor exposure were partially rescued by UCP2 knockdown. Collectively, our data suggested the functions of UA on UCP2 expression and on the protection of H/R-stimulated H9c2 cells may be attributed to p38 signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Myocardial infarction is characterized by a sharp reduction or disruption of coronary blood supply, which leads to severe and persistent acute myocardial ischemia. Within 20–30 min after coronary occlusion, necrosis occurs in the myocardium, which may cause ventricular dysfunction, resulting in arrhythmia, decreased blood pressure, shock or heart failure (Keeley et al. 2003). The restoration of blood flow to the ischemic myocardium may induced myocardial injury. Oxidative stress, apoptosis and autophagy is involved in H/R-induced injury (Gustafsson and Gottlieb 2009; Seidlmayer et al. 2015). Besides, various signaling pathways, such as AMP-activated protein kinase (AMPK) (Gustafsson and Gottlieb 2009), mitogen-activated protein kinases (MAPKs, including JNK [c-Jun N-terminal kinase], ERK [extracellular signal-regulated kinase] and p38) (Anu et al. 2000; Engelbrecht et al. 2004; Capano and Crompton 2006) and Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway (Bolli et al. 2003), have been shown to play important roles during in H/R-induced injury.
Uncoupling protein 2 (UCP2), a cationic carrier protein, is located in the inner membrane of mitochondria. UCP2 is widely expressed in various tissues, such as liver, pancreas, adipose tissue, spleen, kidney, heart and the central nervous system (Fleury et al. 1997; Ricquier and Bouillaud 2000). UCP2 is able to uncouple the respiratory chain by dissipating the proton gradient, thus reducing the production of reactive oxygen species (ROS) (Garlid et al. 2000). Increasing evidence has shown that UCP2 is involved in atherosclerotic plaque formation (Blanc et al. 2003; Moukdar et al. 2009), immune response (Rousset et al. 2006; Rupprecht et al. 2012), metabolic diseases (Diano and Horvath 2012) and tumorigenesis (Derdák et al. 2006; Ayyasamy et al. 2011). Teshima Y et al. reported that UCP2 overexpression in cardiomyocytes could inhibit oxidative stress-induced mitochondrial death pathway (Teshima et al. 2003). McLeod CJ et al. also suggested the protective effects of UCP2 in H9c2 myocardial cells during hypoxia-reoxygenation injury (McLeod et al. 2005). Compounds which could increase UCP2 expression may be candidate drugs for relieving H/R induced cardiac damage. The present study aimed to find such natural compounds.
In this study, the effects of Ginsenoside Rg3 (Rg3), ursolic acid (UA), astragaloside (As), berberine (BB), ligustrazine (Lig) and ferulic acid (FA) on the cell viability and UCP2 mRNA expression in H9c2 cells under H/R conditions were investigated. The most significant increase of UCP2 expression was observed in UA-pretreated cells under H/R conditions. UA pretreatment significantly decreased H/R-increased cell apoptosis and ROS production. We also investigated the involvement of p38 signaling in the effects of UA.
2 Materials and methods
2.1 Cell culture and hypoxia/reoxygenation (H/R) injury model
Rat H9c2 cells were obtained from Cell Bank of Chinese Academy of Sciences (Shanghai, China) and cultured in high glucose Dulbecco’s modified Eagle medium (DMEM) (Hyclone, Logan, UT, USA) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA) and penicillin/streptomycin. The cells were maintained at 37°C in an atmosphere of 95% air/5% CO2.
H/R injury in vitro model was established as previously described (Shin et al. 2009). H9c2 cells were incubated in serum-free and low-glucose DMEM for 2 h under hypoxia condition (1% O2, 5% CO2, 94% N2), and then incubated in fresh high glucose DMEM containing 10% FBS for 6 h under normoxia (95% air/5% CO2). The control cells were maintained in normal conditions (95% air/5% CO2) throughout the experiments. At 24 h before the onset of hypoxia, H9c2 cells were randomly treated with Ginsenoside Rg3 (Rg3; Aladdin, G107710, Shanghai, China), ursolic acid (UA; Aladdin, U107242), astragaloside (As; Aladdin, A111275), berberine (BB; Aladdin, B139120), ligustrazine (Lig; Aladdin, L117992) and ferulic acid (FA; Aladdin, F103701), with dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO, USA) as a vehicle, respectively.
2.2 Cell viability assay
Cell viability was determined by the Cell Count Kit-8 (CCK-8; Beyotime Institute of Biotechnology, Jiangsu, China) assay. Briefly, H9c2 cells were plated in 96-well plates at a density of 3×103 cells/well. After the cells adhered, various concentrations of compounds were added and cultured for 24 h. The cells were then subjected to H/R as described above. After 6 h of reoxygenation, cells were incubated with CCK-8 for 1 h at 37°C and optical density at 450 nm (OD450nm) was measured using a microplate reader. The relative cell viability (%) was calculated by dividing OD450nm in the treated cells by OD450nm in the control cells.
2.3 RNA extraction and real-time PCR
Total RNA was isolated from H9c2 cells with TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. After treated with DNase, the total RNA was reverse-transcribed by using First Strand cDNA Synthesis Kit (Fermentas, Hanover, MD, USA) primed with oligodeoxythymidine (dT). Real-time PCR reactions were performed using diluted cDNA product and SYBR Green kit (Thermo Fisher Scientific, Rockford, IL,USA) according to the manufacturer’s instructions on ABI-Prism 7300 (Applied Biosystems, Foster City, CA, USA). Primers used for RT-PCR were: UCP2, 5′-TGTGGTAAAGGTCCGCTTCC-3′ (forward) and 5′-GCATTTCGGGCAACATTGGG-3′ (reverse); GAPDH, 5′-GGAGTCTACTGGCGTCTTCAC-3′ (forward), 5′-ATGAGCCCTTCCACGATGC-3′ (reverse). GAPDH was used as an internal control. The PCR conditions were 95°C for 10 min, and 40 cycles at 95°C for 15 s and 60°C for 45 s. All reactions were performed in triplicate.
2.4 Western blot analysis
After washing twice with ice-cold PBS, H9c2 cells were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime). The protein concentration was measured with bicinchoninic acid (BCA) assay. Equal amounts of protein were loaded onto 10% sodium dodecyl sulfate-polyacrylamindegelelectrophoresis (SDS-PAGE) gels and then transferred to nitrocellulose membranes, which were blocked with 5% skim milk in Tris-buffered saline/Tween 20 for 1 h at room temperature. The membranes were probed with anti-UCP2 (1:800; Abcam, Cambridge, MA, USA; Ab67241), anti-p-p38 (1:1000; Cell Signal Tech. #9211) or anti-p38 (1:1000; Cell Signal Technology, Danvers, MA, USA; #9212) at 4°C overnight. GAPDH, detected with rabbit monoclonal antibody (1:2000; Cell Signal Tech. #5174), was used as a loading control. Then, the membranes were incubated with the horseradish peroxidase labeled secondary antibody for 1 h at room temperature. The signals were visualized using an enhanced chemiluminescent detection kit (Millipore, Bredford, MA, USA). The relative expression of proteins were quantified by Image J software (http://rsb.info.nih.gov/ij/, Bethesda, MD, USA) and calculated relative to GAPDH.
2.5 Cell apoptosis assay
Cell apoptosis was detected using Annexin V-FITC apoptosis analysis kit (Beyotime) according to the manufacturer’s protocol. In brief, cells were collected, washing twice with ice-cold phosphate-buffered saline (PBS), and incubated with Annexin V-FITC and propidium iodide (PI) for 15 min in the dark. Samples were then analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA). The percentage of early apoptotic cells (Annexin V+PI−) in each group was compared.
2.6 Caspase 3 activity assay
Caspase 3 activity was detected with Caspase 3 colorimetric assay kit (Kengen Biotech., Nanjing, China) per the manufacturer’s instructions. Briefly, cells were collected and lysed with the provided lysis buffer. Protein concentration was determined with BCA assay. After incubation with Caspase 3 Substrate at 37°C for 4 h in the dark, absorbance values were read at 400 nm.
2.7 Nitric oxide (NO) production
NO production was determined by the Griess reaction method. Cultured medium collected from H9c2 cells were incubated with equal volume of Griess reagent (Jiancheng) at room temperature for 10 min in the dark. The absorbance was measured at 550 nm.
2.8 Measurement of ROS production in H9c2 Cells
ROS production was detected by flow cytometry using the fluorescent probe 2′,7′-dichlorofluorescein-diacetate (DCFH-DA; Beyoime). The cells were collected, washing twice with ice-cold PBS, and incubated with DCFH-DA at 37°C for 20 min in the dark. Samples were then analyzed by flow cytometry (excitation 480 nm and emission 525 nm).
2.9 Superoxide dismutase (SOD) activity and Malondialdehyde (MDA) assay
SOD activity and MDA level were determined with commercial available kits according to the manufacturer’s protocols (Jiancheng). The absorbance was read at 550 nm and 532 nm, respectively.
2.10 siRNA Transfection
UCP2 siRNA (siUCP2, 5′-CCCGAAAUGCCAUUGUCAA-3′) or control siRNA (siNC) were designed and synthesized by Genepharma (Shanghai, China). H9C2 cells were transfected with siRNAs using Lipofectamine 2000 (Invitrogen). All experiments were performed 48 h after transfection.
2.11 Statistical analysis
Data were analyzed using GraphPad Prism 6 (San Diego, CA, USA). All experiments were repeated at least three times and the data are presented as mean ± SD. One-way ANOVA was applied for statistical comparisons. P-values less than 0.05 were considered significant.
3 Results
3.1 Effects of six natural compounds on UCP2 expression in H9c2 cells under H/R injury
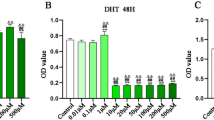
To test the effects of six natural compounds on H/R injury, cell viability assays were performed on H9c2 cells pretreated with various concentrations of Ginsenoside Rg3 (Rg3; 10, 50 and 100 μM (He et al. 2009)), ursolic acid (UA; 0.5, 2 and 4 μg/ml (Yu et al. 2009)), astragaloside (As; 25, 100 and 200 μM (Shi-Guang et al. 2015)), berberine (BB; 2, 10 and 20 μM (Pires et al. 2014)), ligustrazine (Lig; 2, 10 and 50 μg/ml (Luo et al. 1997; Wu et al. 2012)) and ferulic acid (FA; 20, 40 and 80 μM (Zhi-Juan et al. 2014)), with DMSO as a vehicle, respectively. As shown in figure 1, H/R significantly decreased cell viability relative to normoxic conditions (Control). Treatment with these six compounds induced a great increase in cell viability after H/R. The most significant enhanced effects of Rg3, UA, As, BB, Lig and FA on cell viability were observed at the concentrations of 100 μM, 4μg/mL, 100 μM, 10 μM, 50 μg/mL and 40 μM, respectively, which were chosen for the following assays.

Effects of six natural compounds on cardiomyocyte viability. H9c2 cells were pretreated with various concentrations of Ginsenoside Rg3 (Rg3; 10, 50 and 100 μM), ursolic acid (UA; 0.5, 2 and 4 μg/mL), astragaloside (As; 25, 100 and 200 μM), berberine (BB; 2, 10 and 20 μM), ligustrazine (Lig; 2, 10 and 50 μg/mL) and ferulic acid (FA; 20, 40 and 80 μM), with DMSO as a vehicle, respectively. After 24 h of pretreatment, cells were subjected to Hypoxia/Reoxygenation (H/R) injury as described in the section on materials and methods. Cell viability was assessed by CCK-8 assay after 6 h of Reoxygenation. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs H/R cells with vehicle treatment.
Real-time PCR was performed to detected UCP2 mRNA expression after H/R. As showed in figure 2A, challenge of H9C2 cells with H/R induced the increased of UCP2 mRNA expression. Pretreated with UA (4 μg/mL), Lig (50 μg/mL) and FA (40 μM) significantly increased UCP2 mRNA expression under H/R, while BB (10 μM) pretreatment had reverse effects. Similar results were achieved at protein level (figure 2B). Because the most significant increase was observed in UA (4 μg/mL), we then further investigated the protective effects of UA against H/R injury.

Effects of six natural compounds on UCP2 expression in H9c2 cells under H/R injury. H9c2 cells were treated with Rg3, UA, As, BB, Lig and FA on cell viability at the concentrations of 100 μM, 4μg/mL, 100 μM, 10 μM, 50 μg/mL and 40 μM, respectively. After 24 h of pretreatment, cells were subjected to H/R injury. UCP2 mRNA (A) and protein (B) levels were analyzed by real-time PCR and Western blot respectively after 6 h of Reoxygenation. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 vs H/R cells with vehicle treatment.
3.2 UA inhibited apoptosis and NO production induced by H/R injury
Cell apoptosis and Caspase 3 activity were examined in H9c2 cells exposed to H/R with or without UA pretreatment. Annexin V/PI staining (figure 3A) and colorimetric assay (figure 3B) clearly showed that H/R induced cell apoptosis and Caspase 3 activity, respectively. UA pretreatment reduced the ratio of early apoptosis and Caspase 3 activity in H/R cells in a dose-dependent manner as compared to H/R cells.

UA inhibited apoptosis and NO production induced by H/R injury. H9c2 cells were treated with various concentrations of UA with DMSO as a vehicle for 24 h. Then, the cells were subjected to H/R injury. (A) Cell apoptosis was analyzed by Annexin V/PI staining. (B) Caspase 3 activity was estimated by Caspase 3 colorimetric assay kit. (C) NO release was analyzed. **P<0.01, ****P<0.0001 vs H/R cells with vehicle treatment.
NO production plays an important regulatory role in cell apoptosis during myocardial ischemia-reperfusion (Gao et al. 2002). Thus, we assessed NO release. The results showed that H/R stimulation notably decreased NO production, and such effects were dose-dependently rescued by UA pretreatment (figure 3C).
3.3 Effects of UA on ROS, SOD and MDA in H/R cells
Furthermore, cells subjected to H/R displayed a significant increase in ROS production compared with control cells. UA pretreatment also caused a gradual decrease in ROS production in H/R cells (figure 4A).

Effects of UA on ROS, SOD and MDA in H/R cells. H9c2 cells were treated with various concentrations of UA with DMSO as a vehicle for 24 h. Then, the cells were subjected to H/R injury. ROS production (A), SOD activity (B) and MDA content (C) were evaluated. *P<0.05, ***P<0.001, ****P<0.0001 vs H/R cells with vehicle treatment.
Notable reduction in SOD activity was observed after H/R stimulation. Pretreatment with UA significantly elevated SOD activity (figure 4B). The content of MDA increased significantly in H/R cells compared with control cells. UA pretreatment significantly reduced the level of lipid peroxidation in a dose dependent manner (Figure 4C).
3.4 p38 Signaling pathway was involved in effects of UA/UCP2 on H/R cells
It has been reported that p38 is involved in UCP2 regulation (Emre et al. 2007; Selimovic et al. 2008). The total and phosphorylated forms of p38 were then detected by western blot. H/R caused a notable increase in the phosphorylation of p38, which was weakened by UA pretreatment (figure 5A). The total protein of p38 was not changed under different conditions.

p38 signaling pathway was involved in effects of UA/UCP2 on H/R cells. (A) The effects of UA on the levels of UCP2, p-p38 and p38 were assessed by western blot. **P<0.01, ****P<0.0001 vs H/R cells with vehicle treatment. (B–H) The H9c2 cells were divided into 8 groups: Group 1 (Control), cells were cultured without any treatment under normoxia; Group 2-Group 8, cells were subjected to H/R injury. Before H/R injury, cells in Group 2 were cultured for 24 h with DMSO; cells in Group 3 were transfected with siNC for 24 h, then cultured for 24 h with DMSO; cells in Group 4 were transfected with siNC for 24 h, then cultured for 24 h with 4 μg/mL UA; cells in Group 5 were transfected with siNC for 47 h, then cultured for 1 h with 10 μM SB203580 (SB; Selleck; Houston, TX, USA); cells in Group 6 were transfected with siUCP2 for 24 h, then cultured for 24 h with DMSO; cells in Group 7 were transfected with siUCP2 for 24 h, then cultured for 24 h with 4 μg/mL UA; cells in Group 8 were transfected with siUCP2 for 47 h, then cultured for 1 h with SB. The protein levels of UCP2, p-p38 and p38 (B), cell apoptosis (C), Caspase 3 activity (D), NO production (E), intracellular ROS (F), SOD activity (G) and MDA content (H) were evaluated. ***P<0.001, ****P<0.0001 vs control cells; +++P<0.001, ++++P<0.0001 vs H/R cells with vehicle treatment; $$$$P<0.0001 vs H/R cells with siUCP2 and vehicle treatment; ####P<0.0001 with siNC and UA treatment; &&&P<0.001, &&&&P<0.0001 vs H/R cells with siNC and SB treatment.
To determine the molecular mechanism of the protected effects of UA on H/R cells, H9c2 cells were transfected with UCP2 siRNA/control siRNA, treated with UA/vehicle/p38 inhibitor (SB203580), and then subjected to H/R injury. As shown in Figure 5B, H/R stimulation enhanced the levels of p-p38, which were significantly suppressed by UA and SB treatment. UCP2 siRNA transfection successfully knocked down its expression and had no effect on p-p38. Additionally, UCP2 knockdown in H/R cells promoted cell apoptosis (figure 5C), Caspase activity (figure 5D), ROS production (figure 5F) and lipid peroxidation (Figure 5H), but inhibited NO production (figure 5E) and SOD activity (figure 5G), while p38 inhibitor had reverse effects. More importantly, UCP2 siRNA partially rescued the effects of UA and SB. These data suggested that UA protected H9c2 cells against H/R injury via increasing the expression of UCP2. Inhibition of p38 signaling had the same effects as UA.
4 Discussion
It is well-known that oxidative stress and apoptosis plays a key role in H/R-induced injury. UCP2 can reduce the production of ROS and protect cardiomyocytes from oxidative stress-induced apoptosis and H/R injury (Teshima et al. 2003; McLeod et al. 2005). p38, a redox-sensitive protein kinase, is involved in UCP2 regulation (Emre et al. 2007; Selimovic et al. 2008). Studies have linked p38 activity with cardioprotection during H/R injury (Engelbrecht et al. 2004; Capano and Crompton 2006). Numerous natural compound from Chinese medicines, such as salidroside, tyrosol (Sun et al. 2012), quercetin (Angeloni et al. 2007) and Rutin (Jeong et al. 2009), exerts protective functions against H/R injury. In this study, we identified that ursolic acid (UA), a triterpene acid, can inhibit p38 phosphorylation, increase UCP2 expression and decrease H/R-induced injury.
Firstly, pretreatment with Ginsenoside Rg3, ursolic acid (UA), astragaloside, berberine, ligustrazine and ferulic acid significantly improved cell viability of H9c2 cells under H/R conditions (figure 1). In addition, UA had the most significant induction effect on UCP2 expression under H/R conditions (figure 2), which prompt us to study the functions of UA in H/R cardiomyocytes. UA, a natural compound presented in numerous classes of medicinal plants (Li et al. 2011), possesses anti-oxidative, anti-inflammatory, and anti-cancer effects (Ikeda et al. 2008; Tsai and Yin 2008; Kashyap et al. 2016). Our subsequent data suggested that UA significantly attenuated H/R-induced apoptosis (Figure 3A and 3B) and ROS production (figure 4A), which were consistent with previous findings in other cells. It is known that increased NO production contributed to the anti-apoptosis effects of insulin during myocardial ischemia-reperfusion (Gao et al. 2002). Here, H/R injury significantly decreased NO release, which was increased by UA pretreatment (figure 3C). These data suggested that UA may increase NO release and then exerted anti-apoptotic functions in H/R cardiomyocytes. The content of MDA, marker of lipid peroxidation (Uchida 1999) and the activity of SOD, an antioxidant enzyme (McCord and Edeas 2005) were also studied. The changes of MDA (figure 4C) and SOD (figure 4B) further indicated the anti-oxidative effects of UA in H/R cardiomyocytes. Thus, our data suggested that UA possess anti-apoptosis and anti-oxidative activities in H/R-stimulated cardiomyocytes.
Previous work has suggested that p38 mediated ischemia-induced intrinsic apoptosis pathway (Capano and Crompton 2006). The present study showed that UA markedly inhibited H/R-induced p38 activation (figure 5A), which was in line with previous findings in lipopolysaccharide -treated mouse brain (Wang et al. 2011). p38 signaling pathway has been linked to down-regulation of UCP2 in Taxol-treated melanoma cell lines (Selimovic et al. 2008) and lipopolysaccharide-induced macrophages (Emre et al. 2007). Here, p38 inhibitor treatment markedly decreased p38 phosphorylation and induced UCP2 expression (figure 5B). UCP2 knockdown had the reverse biological effects on H/R cells as compared to UA and p38 inhibitor. More importantly, UCP2 siRNA partially blocked the effects of UA or p38 inhibitor exposure (figure 5C–H). These data suggested that UCP2 mediated the functions of UA on H/R-stimulated H9c2 cells. p38 signaling pathway may be involved in the regulation of UA on UCP2 expression.
In conclusion, our findings demonstrated that UA increased UCP2 expression in H/R-treated cardiomyocytes, and that UA inhibited H/R-induced cardiomyocyte apoptosis and ROS production. p38 signaling pathway was involved in this process although the detailed mechanisms were not clear. Our study suggest the potential therapeutic application of UA although it should be further tested in animal models.
References
Angeloni C, Spencer J, Leoncini E, Biagi P and Hrelia S 2007 Role of quercetin and its in vivo metabolites in protecting H9c2 cells against oxidative stress. Biochimie 89 73–82
Anu P, Mockridge JW, Farooqui S, Marber MS and Richard J 2000 Sustained activation of p42/p44 mitogen-activated protein kinase during recovery from simulated ischaemia mediates adaptive cytoprotection in cardiomyocytes. Biochem. J. 350 891–899
Ayyasamy V, Owens KM, Desouki MM, Liang P, Bakin A, Thangaraj K, Buchsbaum DJ, LoBuglio AF, et al. 2011 Cellular model of Warburg effect identifies tumor promoting function of UCP2 in breast cancer and its suppression by genipin. PLoS ONE 6 e24792
Blanc J, Alves-Guerra M, Esposito B, Rousset S, Gourdy P, Ricquier D, Tedgui A, Miroux B, et al. 2003 Protective role of uncoupling protein 2 in atherosclerosis. Circulation 107 388–390
Bolli R, Dawn B and Xuan Y-T 2003 Role of the JAK–STAT pathway in protection against myocardial ischemia/reperfusion injury. Trends Cardiovasc. Med. 13 72–79
Capano M and Crompton M 2006 Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem. J. 395 57–64
Derdák Z, Fülöp P, Sabo E, Tavares R, Berthiaume EP, Resnick MB, Paragh G, Wands JR, et al. 2006 Enhanced colon tumor induction in uncoupling protein-2 deficient mice is associated with NF-κB activation and oxidative stress. Carcinogenesis 27 956–961
Diano S and Horvath TL 2012 Mitochondrial uncoupling protein 2 (UCP2) in glucose and lipid metabolism. Trends Mol. Med. 18 52–58
Emre Y, Hurtaud C, Nübel T, Criscuolo F, Ricquier D and Cassard-Doulcier A-M 2007 Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem. J. 402 271–278
Engelbrecht A-M, Niesler C, Page C and Lochner A 2004 p38 and JNK have distinct regulatory functions on the development of apoptosis during simulated ischaemia and reperfusion in neonatal cardiomyocytes. Basic Res. Cardiol. 99 338–350
Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, Levi-Meyrueis C, Bouillaud F, Seldin MF, et al. 1997 Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat. Genet. 15 269–272
Gao F, Gao E, Yue T-L, Ohlstein EH, Lopez BL, Christopher TA and Ma X-L 2002 Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion. Circulation 105 1497–1502
Garlid KD, Jabůrek M, Ježek P and Vařecha M 2000 How do uncoupling proteins uncouple? Biochim. Biophys. Acta Bioenerg. 1459 383–389
Gustafsson ÅB and Gottlieb RA 2009 Autophagy in ischemic heart disease. Circ. Res. 104 150–158
He B, Chen P, Yang L, Zhang X, Ji F and Shen Z 2009 Protective effects of 20 (R)-ginsenoside Rg3 on human umbilical vein endothelial cell injury induced by LPS. Chin. Pharmaceut. J. 44 1703–1707
Ikeda Y, Murakami A and Ohigashi H 2008 Ursolic acid: An anti‐and pro‐inflammatory triterpenoid. Mol. Nutr. Food Res. 52 26–42
Jeong JJ, Ha YM, Jin YC, Lee EJ, Kim JS, Kim HJ, Seo HG, Lee JH, et al. 2009 Rutin from Lonicera japonica inhibits myocardial ischemia/reperfusion-induced apoptosis in vivo and protects H9c2 cells against hydrogen peroxide-mediated injury via ERK1/2 and PI3K/Akt signals in vitro. Food Chem. Toxicol. 47 1569–1576
Kashyap D, Tuli HS and Sharma AK 2016 Ursolic acid (UA): a metabolite with promising therapeutic potential. Life Sci. 146 201–213
Keeley EC, Boura JA and Grines CL 2003 Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet 361 13–20
Li G, Zhang X, You J, Song C, Sun Z, Xia L and Suo Y 2011 Highly sensitive and selective pre-column derivatization high-performance liquid chromatography approach for rapid determination of triterpenes oleanolic and ursolic acids and application to Swertia species: optimization of triterpenic acids extraction and pre-column derivatization using response surface methodology. Anal Chim. Acta 688 208–218
Luo H, Yuan S, Xiao J and Guo H 1997 Experimental study on the effect of ligustrazine on cultured vascular smooth muscle cells proliferation. West China J. Pharmaceut. Sci. 12 6–7
McCord JM and Edeas MA 2005 SOD, oxidative stress and human pathologies: a brief history and a future vision. Biomed. Pharmacother. 59 139–142
McLeod CJ, Aziz A, Hoyt RF, McCoy JP and Sack MN 2005 Uncoupling proteins 2 and 3 function in concert to augment tolerance to cardiac ischemia. J. Biol. Chem. 280 33470–33476
Moukdar F, Robidoux J, Lyght O, Pi J, Daniel KW and Collins S 2009 Reduced antioxidant capacity and diet-induced atherosclerosis in uncoupling protein-2-deficient mice. J. Lipid Res. 50 59–70
Pires ENS, Frozza RL, Hoppe JB, de Melo Menezes B and Salbego CG 2014 Berberine was neuroprotective against an in vitro model of brain ischemia: survival and apoptosis pathways involved. Brain Res. 1557 26–33
Ricquier D and Bouillaud F 2000 The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem. J. 345 161–179
Rousset S, Emre Y, Join-Lambert O, Hurtaud C, Ricquier D and Cassard-Doulcier A-M 2006 The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine 35 135–142
Rupprecht A, Bräuer AU, Smorodchenko A, Goyn J, Hilse KE, Shabalina IG, Infante-Duarte C and Pohl EE 2012 Quantification of uncoupling protein 2 reveals its main expression in immune cells and selective up-regulation during T-cell proliferation. PLoS ONE 7 e41406
Seidlmayer LK, Juettner VV, Kettlewell S, Pavlov EV, Blatter LA and Dedkova EN 2015 Distinct mPTP activation mechanisms in ischaemia-reperfusion: contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 106 237–248
Selimovic D, Hassan M, Haikel Y and Hengge UR 2008 Taxol-induced mitochondrial stress in melanoma cells is mediated by activation of c-Jun N-terminal kinase (JNK) and p38 pathways via uncoupling protein 2. Cell Signal 20 311–322
Shi-Guang W, Yan X, Hao X, Wei W and Xiao-Hu C 2015 Astragaloside IV prevents lipopolysaccharide-induced injury in H9C2 cardiomyocytes. Chin. J. Nat. Med. 13 127–132
Shin EJ, Schram K, Zheng Xl and Sweeney G 2009 Leptin attenuates hypoxia/reoxygenation‐induced activation of the intrinsic pathway of apoptosis in rat H9c2 cells. J. Cell Physiol. 221 490–497
Sun, L, Isaak CK, Zhou Y, Petkau JC, Karmin O, Liu Y and Siow YL 2012 Salidroside and tyrosol from Rhodiola protect H9c2 cells from ischemia/reperfusion-induced apoptosis. Life Sci. 91 151–158
Teshima Y, Akao M, Jones SP and Marbán E 2003 Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 93 192–200
Tsai SJ and Yin MC 2008 Antioxidative and anti-inflammatory protection of oleanolic acid and ursolic acid in PC12 cells. J. Food Sci. 73 H174–H178
Uchida K 1999 Current status of acrolein as a lipid peroxidation product. Trends Cardiovasc. Med. 9 109–113
Wang Y-J, Lu J, Wu D-M, Zheng Z-H, Zheng Y-L, Wang X-H, Ruan J, Sun X, et al. 2011 Ursolic acid attenuates lipopolysaccharide-induced cognitive deficits in mouse brain through suppressing p38/NF-κB mediated inflammatory pathways. Neurobiol. Learn. Mem. 96 156–165
Wu H-J, Hao J, Wang S-Q, Jin B-L and Chen X-B 2012 Protective effects of ligustrazine on TNF-α-induced endothelial dysfunction. Eur. J. Pharmacol. 674 365–369
Yu H-H, Hur J-M, Seo S-J, Moon H-D, Kim H-J, Park R-K and You Y-O 2009 Protective effect of ursolic acid from Cornus officinalis on the hydrogen peroxide-induced damage of HEI-OC1 auditory cells. Am. J. Chin. Med. 37 735–746
Zhi-Juan WU, Jing YU, Wang RX, Fang QJ and Lin MJ 2014 Protective effect of ferulic acid on doxorubicin induced cellular injury in H9c2 myocardial cells. Chin. Pharmacol. Bull. 30 1059–1065
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Sorab Dalal.
Corresponding editor: Sorab Dalal
Rights and permissions
About this article

Cite this article
Chen, M., Wang, X., Hu, B. et al. Ursolic acid stimulates UCP2 expression and protects H9c2 cells from hypoxia-reoxygenation injury via p38 signaling. J Biosci 43, 857–865 (2018). https://doi.org/10.1007/s12038-018-9801-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12038-018-9801-2