
Abstract
SUMOylation is a post-translational modification that attaches a small ubiquitin-like modifier (SUMO) group to a target protein via SUMO ligases, while deSUMOylation refers to the removal of this SUMO group by sentrin-specific proteases (SENPs). Although the functions of these processes have been well described in the nucleus, the role of SUMOylation and deSUMOylation in regulating ion channels is emerging as a novel area of study. Despite this, their contributions to pain signaling remain less clear. Therefore, this review consolidates the current evidence on the link(s) between SUMOylation, deSUMOylation, and pain, with a specific focus on ion channels expressed in the sensory system. Additionally, we explore the role of SUMOylation in the expression and function of kinases, vesicle proteins, and transcription factors, which result in the modulation of certain ion channels contributing to pain. Altogether, this review aims to highlight the relationship between SUMOylation and deSUMOylation in the modulation of ion channels, ultimately exploring the potential therapeutic role of these processes in chronic pain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Neuropathic pain is a pathological condition resulting from a lesion or disease in the somatosensory nervous system. This condition affects 7–10% of the general population [1, 2] and is characterized by abnormal pain responses, numbness, burning, stabbing, or electric shock-like pain and tingling, among other symptoms. Notably, people suffering from neuropathic pain also experience anxiety, depression, and sleep disturbances, significantly reducing their quality of life [2]. Despite efforts to understand the cellular and molecular mechanisms underlying neuropathic pain, current pharmacological treatments are effective in less than 50% of patients and induce adverse effects that limit their clinical utility [2]. Thus, there is an urgent need to explore the mechanisms implicated in the pathophysiology of neuropathic pain to identify and validate new treatment targets.
Primary sensory neurons play a key role in processing and transmitting nociceptive information from the periphery to the central nervous system through the activation of ion channels and receptors [3]. Specifically, the electrical activity in sensory neurons is mainly controlled by different ion channels such as voltage-gated sodium, calcium, and potassium channels. Moreover, these ion channels can be modulated by post-translational modifications (PTMs), which influence their turnover rates and localization in the plasma membrane [4, 5], thereby affecting neuronal excitability. In this context, SUMOylation is a PTM that enables rapid cellular responses to dynamic changes in various cell types, including neurons through the addition of small ubiquitin-like modifier (SUMO) proteins. Although some targets of SUMO proteins (i.e., nuclear proteins and transcription factors) have been well established, the SUMOylation of ion channels is an emerging field in pain research. This review focuses on understanding the patterns of SUMOylation and deSUMOylation. It discusses the impact of increased SUMOylation on ion channels in chronic pain conditions and highlights the role of sentrin-specific proteases (SENPs) in regulating ion channel SUMOylation.
SUMO Isoforms and Function
SUMOylation is a reversible PTM that involves the covalent binding of an ~11-kilodalton SUMO protein to lysine residues in target proteins [6]. This modification modulates the activity and stability of the target protein by promoting or preventing protein-protein interactions [7, 8]. In mammals, four SUMO members have been identified and designated as SUMO-1 to SUMO-4 [6]. SUMO-1 to -3 are ubiquitously expressed, whereas SUMO-4 expression is restricted to specific tissues such as the spleen, kidney, and lymphatic nodes [9]. SUMO-2 (Smt3A) is 92 amino acids, and SUMO-3 (Smt3B) is 93 amino acids long. Due to their close homology, SUMO-2 and SUMO-3 are frequently referred to as SUMO-2/3 since they share ~97% sequence identity in humans [6]. Notably, SUMO-2 and SUMO-3 can form poly-SUMOylation chains that are involved in various cellular processes including replication, protein recycling, mitosis, and meiosis. SUMO-2 is more abundant than SUMO-3 and plays an important role in synaptic plasticity [10,11,12]. Moreover, SUMO-2 null mice present developmental impairments and are not viable beyond embryonic day 10.5, as result of poor proliferation and increased cell death [10]. In contrast, SUMO-1 null mice are viable since this deficiency can be compensated by SUMO-2/3 [9, 13]. SUMO-1 shares only ~48% and 46% sequence identity with SUMO-2 and SUMO-3, respectively [14, 15]. On the other hand, SUMO-4 contains a unique proline residue (P90) that prevents the efficient maturation required for E2 conjugation [16]. Furthermore, SUMO-4 has been associated with type 1 and type 2 diabetes [17]. Table 1 lists the proteins involved in each step of the SUMOylation cycle.
SUMO-Activating, Conjugating, and Ligating Enzymes
SUMOylation involves a series of enzymatic reactions, which start with the cleavage of the inactive precursor form of SUMO by the SENP enzyme, exposing a conserved di-Glycine motif at the C-terminus. Then, the di-Glycine motif of SUMO is activated by the heterodimer SUMO-activating enzyme subunit 1 and 2 (SAE1/2) in an ATP-dependent process [18]. This activation results in the formation of a SUMO adenylate intermediate, followed by the SUMO-thioester bond with E1, which precedes the SUMO-thioester bond with SAE2. After that, SUMO is transferred to a cysteine residue on ubiquitin-conjugating 9 (Ubc9) [18]. Finally, either E3 ligase or Ubc9 covalently forms an isopeptide bond between SUMO and the lysine residues on the target protein, following a Ψ-K-X-E pattern, where Ψ is a large amino acid in the target protein [19] (Figure 1). It is well established that the SUMOylation state in a protein can either inhibit or promote interactions with other proteins, triggering the assembly of complexes. Therefore, the SUMOylation is reversible and effectively controlled by distinct mechanisms [20].

SUMOylation and deSUMOylation process. SUMOylation and deSUMOylation are dynamically balanced processes under physiological conditions. (1) SUMOylation starts with the maturation of SUMO proteins through proteolytic cleaved by SENP enzyme. (2) The next step involves the activation of mature SUMO by the SUMO E1 enzyme (SAE1/SAE2) in an ATP-dependent process. (3) Once activated, SUMO proteins are conjugated to the cysteine residue of the E2 enzyme Ubc9, (4) while SUMO E3 ligase catalyzes the ligation of SUMO to a specific residue in the target protein. (5) Finally, SUMO proteins are cleaved off from their target proteins by SENPs in a process known as deSUMOylation, resulting in free SUMO proteins available for another catalytic cycle. ATP, adenosine triphosphate; SUMO, small ubiquitin-like modifier; SAE1, SUMO-activating enzyme subunit 1; SAE2, SUMO-activating enzyme subunit 2; Ubc9, ubiquitin-conjugating enzyme 9. Figure was created using Biorender.com
SENP Family
SUMO-specific proteases (SENPs) are cysteine proteases that cleave inactive SUMO forms. Furthermore, SENPs also catalyze the deconjugation of SUMO proteins from their substrates, which is an important activity for maintaining the balance between SUMOylated and deSUMOylated proteins [21]. In mammals, there are six SENP members (SENP1–3 and SENP5–7) that exhibit differences in their cellular distribution, specificity for different SUMO members, and regulation of SUMO maturation [22,23,24,25]. SENPs have both endopeptidase and isopeptidase activities [26]. Endopeptidase activity involves cleaving the SUMO precursor or pro-SUMO at the C-terminal, while isopeptidase activity deconjugates SUMO proteins from the target protein (Figure 1) [26]. It has been demonstrated that SENP1 prefers pro-SUMO-1 > pro-SUMO-2 > pro-SUMO-3, whereas SENP2/5 prefers pro-SUMO-2 > pro-SUMO-1 > proSUMO-3. This preference is linked to amino acid sequences at the C-terminal [27]. Notably, SENP6 and SENP7 exhibit poor endopeptidase activity, cleaving pro-SUMO-1, -2, or -3 at a low detectable rate [28]. On the other hand, the isopeptidase activity of SENP1 shows a preference for SUMO-2 > SUMO-3 > SUMO-1, while SENP2/5 has similar activity towards SUMO-2 and SUMO-3 [29]. Finally, SENP6 and SENP7 show notable isopeptidase activity for di-SUMO-2, di-SUMO-3, and poly SUMO chains [28, 30, 31]. In addition to the protective role of SUMOylation in a plethora of cellular processes in eukaryotes, including chromosome cohesion, mitosis, and transcription [32, 33]. In the central nervous system, SUMO-2, -3, and E2 SUMO-conjugating enzyme Ubc9 colocalize with synaptophysin and PSD95, suggesting their expression in pre- and post-synaptic neurons [34,35,36,37]. The expression of SUMO-1 in presynaptic sites has been controversial due to the lack of effective antibodies [34]. However, the development of a nanochannel device highly sensitive to the last 12 amino acids in the C-terminal of mature SUMO-1 allows its detection by immunofluorescence imaging [38]. Furthermore, SENP1, SENP6, and SENP7 colocalize with synaptophysin and drebrin, a postsynaptic marker in hippocampal neurons of mice [22]. These findings suggest a potential role of deSUMOylation machinery in presynaptic sites. On the other hand, activation of the metabotropic glutamate receptor (mGluR5) leads to the accumulation of SENP1 and Ubc9 in dendritic spines, resulting in the modulation of synaptic transmission and plasticity [39]. Additionally, mGluR1 activation prevents SENP1 accumulation in the synapses [40]. The accumulation of SENP1 mediated by mGluR5 and mGluR1 depends on protein kinase C (PKC) and Ca2+/calmodulin protein kinase II (CaMKII) activity [40]. Indeed, PKC phosphorylates mGluR7, facilitating SUMOylation by Ubc9 and deSUMOylation by SENP1, which results in its internalization in hippocampal neurons [41]. On the other hand, it has been reported that the SUMOylation is linked to cellular stress as heat shock [33], DNA damage [42], cancer [43, 44], and oxidative stress [45]. In this sense, oxidative stress influences SUMOylation through various mechanisms. For instance, high concentrations of H2O2 increase protein SUMOylation, while low concentrations induce protein deSUMOylation by inducing a disulfide bond between the catalytic cysteines of the SUMO E1 and E2 enzymes [46]. Moreover, H2O2 can also activate deSUMOylation machinery, by reversibly oxidizing a cysteine motif in the SENP3 regulates its stability and localization protecting this protease from proteasomal degradation [47, 48]. On the other hand, ROS inactivate SENPs, either reversibly through disulfide bond-induced dimerization or irreversibly by overoxidation of their catalytic cysteine [49]. These findings show the dynamic nature of the SUMOylation/deSUMOylation processes under oxidative stress conditions.
Expression of SUMOylation and DeSUMOylation Machinery in the Nervous System
The expression of the SUMOylation and deSUMOylation machinery has been reported in the sensory system. For instance, Ubc9, SUMO-1, and SENP1 are expressed in the spinal cord and in small-, medium-, and large-sized DRG neurons [50, 51]. Additionally, databases of deep RNA-sequencing show that Ubc9, SUMO-1, and SENP1 genes are expressed across different subpopulations of DRG neurons, including peptidergic, C-low threshold mechanoreceptors (LTM), non-peptidergic, Aβ- rapidly adapting (RA) + Aδ-LTMRs, and others [52]. Under pain conditions, alterations in the SUMOylation and deSUMOylation machinery become evident. Carrageenan-induced inflammatory pain increases the levels of SUMO-1-conjugated proteins in DRGs [50, 53]. Similarly, it has been reported that nerve injury increases the expression of Ubc9 in DRG neurons in mice [52], suggesting that the changes in the expression of SUMOylation and deSUMOylation machinery play an important role in pain signaling. While data for SENP expression in the DRG neurons has been limited due to its relatively understudied role in pain, available evidence suggests that SENP expression is directly proportional to the chronicity of the neuropathic pain [52], which could imply a compensatory mechanism to counteract prolonged SUMOylation effects. These findings, thus far, suggest that SUMOylation and deSUMOylation processes may play an essential role in modulating cellular processes in pain pathways. However, their specific contribution to the regulation of ion channels during chronic pain remains to be fully elucidated. Therefore, the following sections will focus on the current evidence on SUMOylation/deSUMOylation processes in ion channels and their potential implications in pain signaling.
Pain Modulation Through SUMOylation and DeSUMOylation of Voltage-Gated Sodium Channels
NaV1.7 Channels
The VGSC family comprises nine members (NaV1.1–1.9), with at least 3 subtypes (NaV1.7, 1.8, and 1.9) being highly expressed in nociceptors, where they control action potential (AP) generation and frequency of firing [3, 54]. Specifically, NaV1.7 is a tetrodotoxin (TTX)-sensitive channel encoded by the gene SCN9A [55]. NaV1.7 channels are expressed in relevant sites for nociceptive transmission, including primary sensory neurons of dorsal root ganglia (DRG) and trigeminal ganglion (TG) [56,57,58] and the superficial lamina of the spinal dorsal horn [59]. We have reported that NaV1.7 trafficking, localization, and conductance are strongly regulated by a variety of post-translational modifications, including SUMOylation. In the context of pain, NaV1.7 turnover is tightly regulated by the SUMOylation state of its interacting protein, the collapsin response mediator protein 2 (CRMP2). The addition of SUMO-1 to CRMP2 increases NaV1.7 membrane localization and channel activity [60, 61]. In contrast, increasing the expression and activity of SENP1 promotes CRMP2 deSUMOylation resulting in reduced NaV1.7 channel expression [60]. CRMP2 is phosphorylated at Ser522 by cyclin-dependent kinase 5 (Cdk5) and then SUMOylated at lysine 374 (K374) by Ubc9 [61,62,63,64]. Disruption of the Ubc9-CRMP2 interaction with a decoy peptide reversed the nociceptive behavior in a neuropathic pain model [64, 65]. Additionally, CRMP2 lacking SUMOylation recruits the endocytic complex (Numb, Eps15, and Nedd4-2) to promote clathrin-mediated endocytosis of NaV1.7 in female mice [62, 66, 67]. These findings suggest that preventing the SUMO conjugation of CRMP2 by Ubc9 decreases NaV1.7 expression and conductance, resulting in an antinociceptive effect in rodents. Interestingly, compound 194 disrupts the CRMP2-Ubc9 interaction by targeting the SUMOylation site in CRMP2 (K374) and reduces NaV1.7 currents and neuronal excitability in several pain models [64,65,66, 68,69,70]. Furthermore, CRMP2 SUMO deficiency in sensory neurons of TG results in decreased NaV1.7 currents and excitability [69]. In this context, intranasal delivery of compound 194 alleviated pain caused by chronic constriction injury of the infraorbital nerve in rats [69], supporting the notion that inhibiting SUMO conjugation of CRMP2 by Ubc9 with compound 194 exerts a beneficial effect in chronic pain conditions. Accordingly, we reported that reducing CRMP2 SUMOylation by replacing K374 with alanine reversed and prevented neuropathic pain-induced mechanical allodynia, suggesting that the SUMOylation of CRMP2 represents a promising strategy for pain treatment [70, 71]. Additionally, we found that increasing the isopeptidase activity of SENP1 through CRISPR/Cas9 mediated SENP1 overexpression which disrupts the interaction between CRMP2 and NaV1.7 by inducing CRMP2 deSUMOylation and NaV1.7 clathrin-mediated endocytosis [60]. Collectively, these findings show that the SUMOylation/deSUMOylation process impacts NaV1.7 plasma membrane expression, current density, and neuronal excitability, which converge to regulate nociceptive behaviors during chronic pain conditions.
Pain Modulation Through SUMOylation and DeSUMOylation of Voltage-Gated Calcium Channels
CaV1.2 Channels
VGCCs are grouped into two subfamilies according to their activation thresholds: high voltage-activated (HVA) and low voltage-activated (LVA). The HVA subfamily encompasses CaV1.1-1.4 (L-type), CaV2.1, CaV2.2, and CaV2.3 (P/Q-, N-, and R-types, respectively) channels, while the LVA subfamily comprises CaV3.1, CaV3.2, and CaV3.3 channels (T-type) [72]. The SUMOylation of collapsin response mediator protein 4 (CRMP4) increases calcium influx through CaV1.2 channels, resulting in heightened thermal pain sensitivity [73]. These findings show that CRMP4 interacts with CaV1.2 to regulate its function and signal transmission during pain. The SUMOylation site in CRMP4 has been identified as K374 residues [63]: a GST-pulldown assay reported that CRMP4 interacts with SUMO-1 to -3, while mutation of K374 residues abolished this interaction, which confirmed that K374 mediates the SUMOylation of CRMP4. In contrast, a study reported that overexpression of deSUMOylated CRMP4 induces thermal allodynia compared to wild-type CRMP4 [73]. Considering that deSUMOylation of CRMP4 is linked with neurite outgrowth [73], it is possible that aberrant neurite outgrowth explains why deSUMOylated CRMP4 increases pain in naïve animals. However, further research is needed to determine the precise role of deSUMOylation of CRMP4 in the pathophysiology of neuropathic pain, where the neurite outgrowth not only contributes to nerve injury but also recovery.
CaV2.2 Channels
The neuronal channels (N-type) CaV2.2 are mainly located in the synapse and form complex interactions with other proteins such as soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins, syntaxin-1 and synaptosomal-associated protein 25 kDa (SNAP-25), calcium/calmodulin-dependent serine protein kinase (CASK), CRMP-2, CaMKII, and Rab3-interacting molecule 1α (RIM1) [74]. CaV2.2 channels play an important role in regulating intracellular calcium concentrations and neurotransmitter release into the synaptic cleft. We have reported that phosphorylation of CRMP2 at serine 522 (S522) leads to the interaction between CaV2.2 channels and CRMP2 [75]. In this regard, the administration of the small molecule S-lacosamide inhibits cyclin-dependent kinase 5 (Cdk5)-dependent phosphorylation of CRMP2, resulting in a reduction of the membrane localization of the CaV2.2 channel [76]. We also reported that CRMP2-CaV2.2 interaction is disrupted by a 15-amino acid peptide derived from CRMP2 and linked to the cell-penetrating peptide TAT. Our findings show the 15 amino acid peptides decreased CaV2.2 plasma membrane expression, Ca2+ current density, and excitatory synaptic transmission in vitro and in vivo [77]. Additionally, the application of this peptide on spinal cord slices significantly decreased the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) with no changes in the amplitude, suggesting that the 15-amino acid domain of CRMP2 does not exert a postsynaptic effect in the spinal dorsal horn [77]. Remarkably, the interaction of CRMP2-CaV2.2 is disrupted by the systemic administration of CBD3063, a peptidomimetic small molecule. CBD3063 selectively ameliorates N-type Ca2+ currents in DRG neurons, resulting in pain relief in inflammatory and neuropathic pain models [78]. While SUMOylation of CRMP2 regulates the trafficking and activity of NaV1.7 channels in DRG neurons, it remains elusive whether CaV2.2 channels are directly SUMOylated under physiological or pathological conditions. In a heterologous system, the co-expression of SUMO-1, but not the SUMO conjugation-deficient mutant lacking the double-glycine motif (SUMO-1ΔGG), increased CaV2.2 Ca2+ current density in the presence of Ubc9 [79]. However, a single mutation of K394, located in the I–II loop, resulted in the loss of function of CaV2.2 channels [80]. Furthermore, SUMO-1 and Ubc9 produced a hyperpolarizing shift in the midpoint (V1/2) of activation. However, SUMO-1 or SENP1 alone did not affect N-type currents, suggesting that levels of endogenous SUMO-1 are not enough to be conjugated with CaV2.2 by Ubc9 or removed by SENP-1 [79]. On the other hand, we demonstrated that DRG neurons transfected with CRMP2 show a lower intracellular calcium concentration ([Ca2+]i), while transfection with CRMP2AAA, a SUMO resistant mutant, did not induce any change in ([Ca2+]i [80]. These results suggest that CRMP2 SUMOylation is inversely correlated with Ca2+ influx [80]. Interestingly, we found that SENP1 overexpression did not change [Ca2+]i or the surface expression of CaV2.2 channels in DRG neurons [60]. In line with these findings, a study reported that superior cervical ganglion neurons transfected with SUMO-1 show an increase in the paired excitatory postsynaptic potential (EPSP) ratio and reduced the inter-stimuli intervals, implicating presynaptic transmitter release likely involving CaV2.2 channels [79]. Collectively, our findings and those of others support the notion that other SENPs could effectively modulate CRMP2 SUMOylation and consequently the function and expression of CaV2.2 channels. However, further research is required to explore whether CRMP2 deSUMOylation by SENPs could represent a strategy to reduce neuronal excitability. We have demonstrated that CRMP2 phosphorylation at S522 is required for its interaction with CaV2.2 channels under neuropathic pain conditions, where the CaV2.2 channels are upregulated [81]. Furthermore, phosphorylation of S522 is required for the addition of a SUMO group to CRMP2 in K374, which upregulates NaV1.7 activity and expression in DRG neurons [62, 64]. However, whether CaV2.2 channels expressed in sensory neurons are directly SUMOylated remains unknown. We have identified that the first loop in both NaV1.7 and CaV2.2 channels is a site of interaction with CRMP2. CRMP2 interacts with the first intracellular loop (706–720 a. a.) of the hNaV1.7 channel with a KD ~1 µM [82]. On the other hand, CRMP2 binds to the distal C-terminal region of the CaV2.2 channel with a KD of 0.3 µM and with lower affinity to the first loop with a KD of ~24 µM [83]. The region in CRMP2 interacting with CaV2.2 is limited to 15 a. a. (484_ARSRLAELRGVPRGL_498), located towards its C-terminus [83]. In chronic pain, NaV1.7 and CaV2.2 channels are highly active due to their overexpression along the sensory neurons. NaV1.7 mainly contributes to the neuronal excitability and CaV2.2 to the transmitter release. It is possible that CRMP2 localization, patterns of PTM may facilitate a preferent interaction for a specific channel depending on the insult type. Moreover, in chronic pain states, a large variety of ion channels (see below) are dysregulated, contributing to neuronal excitability.
CaV3.2 Channels
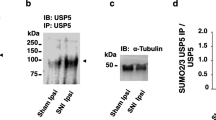
Low-voltage–activated T-type calcium channels regulate neuronal excitability and are involved in exocytosis [84, 85]. In pain conditions, T-type channels contribute to neurotransmitter release in the spinal dorsal horn [86]. Specifically, the CaV3.2 isoform has been reported to play a significant role in pain signaling as silencing or inhibiting CaV3.2 channels produce a pain-free phenotype characterized by an increase in mechanical and thermal thresholds [87, 88]. CaV3.2 channels are ubiquitinated by the WW domain-containing E3 ubiquitin protein ligase 1 (E3 ubiquitin ligase WWP1) in the III–IV intracellular linker [89]. Knockdown of ubiquitin-specific peptidase 5 (USP5) increases CaV3.2 ubiquitination and decreases CaV3.2 channel activity, producing analgesia in inflammatory and peripheral injury models [89]. Another study reported that SUMOylation regulates the interaction between CaV3.2 and USP5, with residue K113 being the target site for USP5 SUMO-1/2 conjugation [90]. Notably, the interaction between CaV3.2 and the SUMO-resistant USP5-K113R mutant was strong, suggesting that SUMO modification of USP5 reduces its affinity for CaV3.2 channels [90]. The study revealed that peripheral nerve damage reduces the SUMOylation of USP5, which in turn increases its interaction with CaV3.2, causing the channel to remain active and present in the plasma membrane for an extended period [90]. Therefore, it should be noted that inhibiting deSUMOylation of USP5 could be a potential strategy to enhance USP5 interactions with the CaV3.2 channel and consequently promote antinociceptive effects. It is still unclear as to whether the SUMOylation of USP5 post-nerve injury is due to a decrease in SUMO ligase activity or an increase in SENP activity. Regardless, it is essential to emphasize that the processes of SUMOylation and de-SUMOylation greatly influence the functionality of CaV channels.
Pain Modulation Through SUMOylation and DeSUMOylation of Potassium Channels
Kir7.1 Channels
The potassium (K+) channel family comprises approximately 80 members, divided into voltage-gated (KV), two-pore (K2P), calcium-activated (KCa), and inward-rectifying (Kir) channels [91]. It has been demonstrated that dysfunction in K+ channels is associated with the development and maintenance of pathological conditions like neuropathic pain [92]. Specifically, Kir7.1 channels are expressed in the neurons of the spinal cord [93]. In naïve animals, genetic silencing or pharmacological blockade of Kir7.1 contributes to the development of mechanical allodynia, suggesting that Kir7.1 downregulation promotes pain behaviors [94]. SUMO-1 and Kir7.1 colocalize in the spinal cord of neuropathic animals, implying that SUMO-1 may regulate the function or expression of Kir7.1 in pain conditions [94]. Administration of GA, ubiquitin-activating enzyme (E1) inhibitor, or 2-D08, a Ubc9 inhibitor, prevented Kir7.1-SUMOylation and increased the expression of Kir7.1 in the plasma membrane of the spinal cord in neuropathic animals [94]. The research suggests that inhibiting Kir7.1 SUMOylation could reduce sensory neuron excitability by amplifying K+ currents and affecting action potential duration or resting membrane potential. However, it is important to conduct additional studies to identify the target amino acids for SUMOylation in Kir7.1 and investigate if deSUMOylation via specific SENPs contributes to pain.
Kv2.1 Channels
Voltage-gated potassium channel subunit 2.1 (KV2.1) plays a crucial role in the nervous system by controlling activity-dependent excitability [95]. SUMO proteins modulate KV2.1 function through reversible, enzyme-mediated attachment to specific lysine residues [96]. SUMOylation of KV2.1 in hippocampal neurons influences neuronal firing by altering the half-maximal activation voltage of the channels. Moreover, SUMO proteins and KV2.1 channel interact both within and outside channel clusters on the neuronal surface. Heterologous expression of KV2.1 channels revealed that residue K470 undergoes SUMOylation [96]. Although KV2.1 channels are composed of four subunits, no more than two non-adjacent subunits are simultaneously modified by SUMO [96, 97]. Indeed, SUMOylation at one site shifts half-maximal activation voltage by 15 mV, while modification at two sites results in a full response [96]. Collectively, these findings suggest that SUMOylation exerts control over neuronal excitability by modulating KV2.1 channels at the cell surface. The widespread presence of the SUMO pathway, combined with the extensive tissue expression and physiological significance of KV2.1 channels, suggests that the regulation of excitability observed in hippocampal neurons might also occur in other structures, such as DRG neurons. Indeed, it has been reported that KV2.1 channels are highly expressed in medium- and large-diameter DRG neurons [98]. However, it is important to highlight that the effects of SUMOylation on the function of KV2.1 channels in sensory neurons are still largely unknown. Abnormal regulation of KV2.1 leads to potassium ions outflow, triggering apoptotic cascades, mitochondrial dysfunction, and inflammation responses, contributing to the pathological process of diabetic periphery neuropathy [99, 100]. Therefore, it is likely that the SUMOylation of KV2.1 has a broad impact on pain signaling at the DRG level. However, further research is still required to test this.
KV4.2 Channels
The regulation of the function and expression of voltage-gated potassium channels subunit 4.2 (Kv4.2) by SUMOylation has been studied in heterologous systems [101,102,103,104]. In HEK cells, KV4.2 is SUMOylated at residues K437 and K579, which are conserved mouse, rat, and human [101]. Notably, SUMOylation at K579 decreases the current amplitude (IA) of Kv4.2, while SUMOylation at K437 increases the surface expression of KV4.2 by reducing its internalization [102]. These findings suggest that SUMOylation can independently regulate Kv4.2 surface expression and IA to promote opposing effects. Moreover, KV4.2 channels are modified by SUMO-2/3, but not by SUMO-1, in hippocampal neurons [102]. These results are particularly relevant considering that KV4 channels are expressed in excitatory interneurons in spinal lamina II and KV4.2 knockout increases the excitability of dorsal spinal neurons, resulting in mechanical and thermal hypersensitivity in mice [101, 105]. However, it is important to note that the effects of SUMOylation or deSUMOylation on the function and expression of KV4 channels in sensory neurons remain largely unknow.
K2P1 Channels
The potassium two-pore domain (K2P) channels are constitutively open, allowing K+ ions to leak outward. These channels help sustain the hyperpolarized resting membrane potential and impact the cell’s electrical characteristics. SUMOylation of K2P1 has been shown to reduce K+ currents in oocytes [104]. In contrast, a mutant K2P1 channel in which the K274 was replaced with glutamate produced large K+ currents. Additionally, the removal of SUMO from K274 by SENP1, but not by the inactive C603S-SENP1 enzyme, results in intact K+ currents through K2P1. These findings suggest that SUMOylation in K274 is responsible for the silencing of K2P1 channels [104]. The expression and function of K2P1 channels are altered during inflammatory and neuropathic pain conditions [106,107,108]. However, whether SUMOylation plays a role in the mechanisms underlying the altered function of K2P1 in sensory neurons during chronic pain conditions remains unknown.
Pain Modulation Through SUMOylation and DeSUMOylation of Other Ion Channels
TRPV1 Channels
Transient receptor potential vanilloid 1 (TRPV1) is a calcium-permeable nonselective ion channel activated by capsaicin, noxious heat (> 43 °C), pH (<5.9), and noxious stimuli [109, 110]. TRPV1 channels are abundantly expressed in small sensory C-fibers in DRG [111] and TG neurons [112]. In the spinal cord, TRPV1 is localized in both pre- and post-synaptic neurons in lamina I and II, as well as in astrocytes [113]. Beyond the spinal level, the TRPV1 receptor is also found at supraspinal levels such as the periaqueductal grey and medial prefrontal cortex, contributing to the descending modulation of nociception [114]. In sensory neurons, TRPV1 channels undergo SUMOylation by Ubc9 [51] and deSUMOylation by SENP1 [50]. Acute and chronic peripheral inflammation increases TRPV1 SUMOylation in DRGs [50]. Conditional deletion of Ubc9 in DRG neurons decreases capsaicin-evoked Ca2+ transients, Ca2+ currents, and deactivation time of TRPV1 channels, while the expression of deSUMOylated TRPV1 reduces nocifensive responses induced by intraplantar capsaicin injection [51]. Additionally, knockout of SENP1 or SUMOylation of TRPV1 has been linked to increased thermal hyperalgesia in an inflammatory pain model [50]. In contrast, another study reported that TRPV1 SUMOylation specifically reduces TRPV1 activation induced by heat, but not by capsaicin, protons, and voltage [112]. It has been reported that replacing residue K822 with an arginine reduces the response to heat but does not modify the membrane expression of TRPV1 channels or affect their voltage dependence, capsaicin sensitivity, or proton-induced currents [50]. Conversely, the mutation of residue K324 to an alanine decreases the sensitivity to capsaicin [51]. Collectively, these findings highlight that the SUMOylation of specific residues of TRPV channels regulates different functions of the channel. Indeed, in vivo studies have reported that nocifensive responses induced by intraplantar capsaicin are decreased in sensory neurons from (SNS)-TRPV1K325A knock-in mice compared to controls [51]. Similarly, TRPV1K822R expression in wild-type mice significantly reduces the thermal hypersensitivity induced by carrageenan injection [50]. These data emphasize that preventing TRPV1 SUMOylation in inflammatory pain models results in an antinociceptive effect. In contrast, it has been reported that increased deSUMOylation processes are associated with the loss of epidermal nerve fibers in chronic stages of diabetic peripheral neuropathy (DPN), which is linked with the development of mechanical and thermal hypoalgesia [115]. Furthermore, SNS-TRPV1K325A mice fail to develop hypoalgesia in the late phases of DPN, without changes in the peripheral nerve fiber density [51]. These findings suggest that during DPN, SUMOylation increases the function of TRPV1, while deSUMOylation of metabolic enzymes induces the loss of sensory neurons by increasing oxidative stress, thereby hastening DPN [51].
TRPA1 Channels
Transient receptor potential ankyrin 1 (TRPA1) is a nonselective cation channel activated by chemical irritants such as mustard oil and allicin or noxious cold temperatures (10–20 °C) [116]. In the sensory system, TRPA1 channels are expressed in DRG neurons, nerve roots, intestinal neurons, spinal cord neurons, and skin basal keratinocytes [117, 118]. High expression of TRPA1 has been linked with cold hypersensitivity in inflammatory, neuropathy, and cancer-induced pain [119]. Heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) is a member of the RNA binding protein family involved in RNA stability regulation [120, 121]. Indeed, it has been reported that hnRNPA1 can regulate the expression of TRPA1 [118], through various PTMs including phosphorylation, SUMOylation, and glycosylation [121, 122]. Specifically, SUMOylation of hnRNPA1 at K113 and K183 regulates extracellular vesicle sorting and mRNA translation, respectively [123, 124]. SUMOylation of hnRNPA1 prolongs the half-life of TRPA1 mRNA by reducing its degradation, thereby increasing the TRPA1 mRNA stability via the coding sequence (CDS) rather than through its interaction with 5̲′ or 3̲′ untranslated regions (UTRs), suggesting that SUMO-1 stabilizes TRPA1 mRNA [118]. Moreover, overexpression of SUMO-1 increased the interaction between hnRNPA1 and TRPA1 mRNA, while hnRNPA1 knockdown prevented this effect [118]. In this context, CFA-induced inflammatory pain increases hnRNPA1 SUMOylation in DRGs, correlating with the development of cold hypersensitivity [118]. Moreover, overexpression of hnRNPA1 in DRGs facilitated CFA-induced increased TRPA1 protein expression at the plasma membrane, an effect absent when expressing the hnRNAPA1 SUMO-null mutant K3R, the SUMOylation site that promotes hnRNPA1 cytoplasmic localization [118]. These findings were consistent with a higher response of TRPA1 to allyl isothiocyanate in HEK cells overexpressing hnRNAP1 in the presence of SUMO-1 compared to the K3R mutants [118]. Taken together, these findings suggest that hnRNAP1 SUMOylation enhances TRPA1 channel activity in inflammatory pain, contributing to the maintenance of CFA-induced cold hypersensitivity.
HCN Channels
The hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are homo- or heterotetramers assembled from four isoforms (HCN1–4), with each subunit being composed of six transmembrane segments [125]. HCN channels mediate an inward cation current (Ih), allowing the influx of Na+ and K+ ions after membrane hyperpolarization, which is regulated by cyclic nucleotides, particularly cyclic adenosine monophosphate (cAMP) [125, 126]. It has been reported that HCN2 channels are SUMOylated by both SUMO-1 and SUMO-2/3 in rodent brains [127]. Furthermore, SUMOylation of HCN2 at K669 increases its surface expression and the hyperpolarization-activated current (Ih) in heterologous expression systems [127]. Inflammatory pain increases SUMO-2/3-induced SUMOylation of the HCN2 channel in small neurons from the L5 nerve, while SUMO-1-induced SUMOylation of HCN2 is reduced in medium and large neurons from the L5 nerve 1 day post CFA-injection. These findings suggest that the SUMOylation of these channels could be contributing to the hyperalgesia observed after CFA injection [128]. Conversely, another study reported that CFA injection increases HCN2 SUMOylation in medium and large cells at day 1 in the lumbar nerve L6, but not in the L4 nerve [129]. Therefore, these findings suggest that inflammation may induce differential expression of SUMO forms, which could have varying impacts on the function of ion channels during inflammation-induced pain. The discrepancies between these studies could be attributed to the fact that sensory fibers from the L5 and L6 nerves mostly project to the hind paw and visceral organs, respectively [129], while sensory fibers from the L4 nerve project to the hindlimb, knee, and hip joint [129]. This implies that DRG neurons may exhibit a unique and dynamic pattern of HCN2 SUMOylation. Another explanation could be the specificity of SENPs and E3 ligases for different SUMO isoforms [29, 130]. Considering these factors, it is likely that multiple mechanisms regulate the function and expression of HCN2 during CFA-induced inflammation.
Pain Modulation Through the SUMOylation of Kinases and RIM1
Protein kinase C (PKC) is a family of serine/threonine kinases divided into classic (α, βI, βII, and γ), novel (δ, ε, θ, and Ƞ) and typical (ζ and ι/λ) based on their calcium and diacylglycerol dependence [131]. Genetic [132] and pharmacological [133] studies have demonstrated that these different PKC isoenzymes play important roles in several cellular functions such as proliferation, apoptosis, neuronal excitability, and neurotransmitter release [134, 135]. It has been reported that the activation of kainate, but not AMPA or NMDA receptors, induces glycine receptor endocytosis in a calcium- and PKC-dependent manner, resulting in reduced glycine receptor activity in the spinal cord [136]. Interestingly, kainate treatment decreased the expression of glycine receptors in neurons overexpressing SENP1, but not in SENP1 knockout neurons. Furthermore, the substitution of K465 with arginine in PKC prevents its SUMOylation and abolishes kainate-induced glycine receptor endocytosis, suggesting that K465 is the key site of PKC SUMOylation in this kinase [136]. Collectively, these findings suggest that kainate-induced glycine receptor endocytosis requires PKC activation in response to intracellular Ca2+ elevations and the subsequent SENP1-dependent deSUMOylation of PKC. Several studies have demonstrated the role of PKCε in inflammation-induced hyperalgesia [136]. Notably, PKCε SUMOylation increases in response to peripheral inflammation [53, 135]. It has been reported that phosphorylation of PKCε at serine 729 (S729) promotes its SUMOylation at K534, resulting in the interaction between PKCε and TRPV1 [53]. In this context, expression of SUMO-resistant PKCε failed to induce inflammatory-induced thermal hyperalgesia [53]. Furthermore, TRPV1 knockout mice injected with SUMO-resistant PKCε exhibited blunted thermal hyperalgesia compared to wild-type mice, suggesting that PKC SUMOylation modulates thermal hyperalgesia through TRPV1 [53].
Extracellular signal–regulated kinases (ERK), a member of the family of mitogen-activated protein kinases (MAPK), is another important kinase in pain signaling [137]. ERK is expressed in the spinal cord, astrocytes, microglia, and DRG neurons after the peripheral noxious stimulation [138]. Phosphorylation of ERK has been related to the activation and inflammatory response of microglia, as well as increased nociceptive behaviors in neuropathic pain conditions [138]. The activation of spinal C-C chemokine receptor type 1 (CCR1) increases the levels of phosphorylated ERK, leading to the SUMOylation of DiGeorge syndrome critical region 8 (DGCR8) in microglia after exposure to LPS [139]. DGCR8 SUMOylation induced by SUMO-1 stabilizes its expression and reduces its ubiquitin-dependent degradation, resulting in the release of proinflammatory cytokines such as TNF-α, IL-6, and IL-1B [139]. Interestingly, SUMOylation of DGCR8 was prevented by CCR1 knockdown or an ERK-inhibitor, suggesting that CCR1/p-ERK/DGCR8 axis plays an important role in neuropathic pain–induced microglial activation [139]. While this study underscores the significance of SUMOylation in glial cells, it also opens up new research directions. These include determining the SUMOylation site in DGCR8 and assessing the impact of overexpressed SENPs on microglial activation and inflammation triggered by neuropathic pain.
RIM1
It is well known that neurotransmitter release is mediated by synaptic vesicles, which are strictly dependent on Ca2+ domains in the vicinity of the presynaptic terminal [140]. The role of Rab3-interacting molecule 1α (RIM1α) in the active zone is essential for presynaptic function. In this context, the increase in [Ca2+]i levels induces vesicle priming, where RIM1α interacts with Munc13-1. Then, RIM1α engages Rab3α in vesicle docking and synaptotagmin during exocytosis [141]. In cortical neurons, RIM1α is SUMOylated at K502, facilitating its interaction with CaV2.1 through its PDZ domain. This interaction promotes Ca2+ influx that precedes the fast phase of vesicle exocytosis of the readily releasable pool. In contrast, deSUMOylation of RIM1α by SENP1 reduces its binding to the CaV2.1 channel [141]. In the absence of SUMOylation, RIM1α still facilitates the docking/priming of synaptic vesicles and maintains the active zone structure, suggesting that SUMOylation acts as a molecular switch in the active zone, controlling interactions and defining the function of different protein pools [141]. Although this evidence demonstrates the potential role of SUMOylated RIM1α in the function or expression of CaV2.1 channels located in the synaptic terminals of DRG neurons, further studies are needed to explore the precise role of RIM1α SUMOylation in the context of pain.
Conclusions and Future Perspectives
SUMOylation and deSUMOylation are dynamic post-translational modifications that significantly impact the function, expression, and localization of several ion channels. (Figure 2). These processes influence downstream signaling pathways by modifying the activity of kinases and other signaling molecules that interact with ion channels, thereby affecting pain signaling. The dynamic balance between SUMOylation and deSUMOylation provides a mechanism for fine-tuning pain sensitivity. Therefore, small changes in the SUMOylation status of ion channels can have significant effects on pain perception, ranging from hypersensitivity to reduced pain sensitivity. While current investigations have shed light on these processes, it is necessary to acknowledge the existing gaps in our understanding, particularly in the discovery and development of novel therapeutic strategies specifically targeting the SUMOylation and deSUMOylation modifications to modulate neuronal excitability and pain behaviors in different models. To advance our understanding of SUMOylation and deSUMOylation in pain mechanisms, it is essential to deepen our comprehension of these processes and their implications for pain management. Investigating the precise sites of SUMOylation on ion channels, receptors, kinases, and others, elucidating the role of specific SENPs in modulating SUMOylation dynamics and exploring the crosstalk between SUMOylation and other post-translational modifications will be crucial steps in defining the potential therapeutic applications of SUMOylation/deSUMOylation processes. Furthermore, studies examining the effects of pharmacological interventions targeting SUMOylation pathways in preclinical pain models will provide valuable insights into the therapeutic potential of these strategies and guide the development of tailored therapeutic interventions. Some clinical studies have reported that SUMO-1 and Ubc9 are upregulated in the human skin of patients with diabetic peripheral neuropathy [142]. Furthermore, patients with carpal tunnel syndrome (entrapment neuropathy) present high levels of Ubc9 that decrease after carpal tunnel surgery [143]. These observations suggest that SUMOylation and associated machinery may play distinct and condition-specific roles in neuropathic pain conditions. This variability highlights the need for targeted therapeutic strategies tailored to the specific SUMOylation profiles associated with different pain types. Future research should focus on developing specific inhibitors or activators of SUMOylation pathways. Additionally, creating personalized pain management approaches based on individual SUMOylation patterns could enhance treatment efficacy and reduce adverse effects.

SUMOylation and deSUMOylation process new targets to modulate the excitability in sensory neurons. SUMOylation and deSUMOylation processes play a relevant role in the modulation of different ion channels, kinases, and other proteins implicated in noxious transmission in sensory neurons. In primary afferent neurons, SUMOylation of CRMP2 increases the trafficking, expression, and function of Nav1.7 channels, increasing neuronal excitability. In contrast, deSUMOylation of CRMP2 recruits the endocytic complex composed of Ub, Numb, Eps15, and Nedd4-2, leading to endocytosis of Nav1.7 channels. Another ion channel modulated by SUMOylation is the TRP family. Specifically, SUMOylation of hnRNAP1 prolongs the half-life of TRPA1 mRNA, resulting in increased expression of TRPA1 and increased pain sensitivity. Direct SUMOylation of TRPV1 increases its activity. Additionally, SUMOylation of PKC enhances the expression and currents of TRPV1, contributing to an increase in the excitability in sensory neurons. The SUMOylation process also indirectly regulates the function of some calcium channels. SUMOylation of CRMP4 by SUMO-1 leads to increased Ca2+ currents through Cav1.2 channels. Similarly, SUMOylation of CRMP2 results increases Cav2.2 currents in a heterologous system, though its effect on Cav2.2 in sensory neurons is unknown. Conversely, deSUMOylation of USP5 leads to strong interaction with Cav3.2, resulting in an increase of intracellular calcium in sensory neurons. At the spinal level, SUMOylation modulates ion channels implicated in the control of neuronal excitability. In this context, SUMOylation of potassium channels such as Kir7.1, Kv4.2, and K2P1 reduces K+ currents, increasing the excitability in spinal neurons. Furthermore, the activation of kainate receptors induces the phosphorylation of PKC, promoting its SUMOylation, which results in the endocytosis of glycine receptors in the spinal cord. Finally, the role of SUMOylation has been reported in glial cells. In microglia, the activation of ERK induces SUMOylation of DGCR8, leading to the transcription of genes related to pro-inflammatory cytokines such as TNF- α, IL-6, and IL-1 β. These pro-inflammatory cytokines can sensitize spinal neurons, contributing to increased neuronal excitability and pain. Dashed arrows represent mechanisms that remain unknown in the sensory neurons and require further research to determine their potential contribution to pain signaling. Cav2.2, voltage-gated calcium channel subunit alpha1B; Cav3.2, voltage-gated calcium channel subunit alpha1H; CRMP2, collapsin response mediator protein 2; CRMP4, collapsin response mediator protein 4; DGCR8, DiGeorge syndrome critical region gene 8; Eps15, endocytic adaptor epidermal growth factor receptor substrate 15; ERK, extracellular signal-regulated kinase; hnRNAP1, heterogeneous nuclear ribonucleoprotein A1; IL-1β, interleukin-1 Beta; IL-6, interleukin-6; K+, potassium ion; K2P1, two-pore domain potassium channel TREK-1; Kir7.1, inwardly rectifying potassium channel 7.1; Kv4.2, potassium voltage-gated channel subfamily D member 2; Nav1.7, voltage-gated sodium channel alpha subunit 7; Nedd4-2, neural precursor cell expressed developmentally down-regulated protein 4-2; Numb, Numb protein; PKC, protein kinase C; TNF-α, tumor necrosis factor Alpha; TRPA1, transient receptor potential ankyrin 1; TRPV1, transient receptor potential vanilloid 1; Ub, ubiquitin; USP5, ubiquitin-specific protease 5. Figure was created using Biorender.com
Inhibitors of the SUMOylation machinery including SUMO-E1, -E2, -E3, and SENP1 inhibitors have been reported [144]. A small molecule inhibitor of Ubc9, named 2-DO8, has been found to block SUMOylation, but also cause neuronal damage [145]. This adverse effect could be attributed to Ubc9’s ability to conjugate SUMO to certain substrates in the absence of an E3 ligase [146]. Excessive or inappropriate SUMOylation of neuronal proteins could impair their function and lead to neuronal damage. This suggests that targeting Ubc9 requires a more refined approach to avoid unwanted side effects on neuronal cells. In this regard, selectively inhibiting the SUMOylation of specific proteins by targeting particular lysine residues, as demonstrated with compound 194, has shown greater efficacy and minimized unwanted side effects [147].
Overall, these findings clearly suggest that SUMOylation and deSUMOylation are promising targets to treat pain that require further translational validation.
Data Availability
No data was generated for this study. Figures were generated using BioRender.com.
References
Rice ASC, Smith BH, Blyth FM (2016) Pain and the global burden of disease. Pain 157(4):791–796. https://doi.org/10.1097/j.pain.0000000000000454
Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, Freeman R, Truini A, Attal N, Finnerup NB, Eccleston C, Kalso E, Bennett DL, Dworkin RH, Raja SN (2017) Neuropathic pain. Nat Rev Dis Primers 3:17002. https://doi.org/10.1038/nrdp.2017.2
Dib-Hajj SD, Waxman SG (2019) Sodium channels in human pain disorders: genetics and pharmacogenomics. Annu Rev Neurosci 42:87–106. https://doi.org/10.1146/annurev-neuro-070918-050144
Laedermann CJ, Abriel H, Decosterd I (2015) Post-translational modifications of voltage-gated sodium channels in chronic pain syndromes. Front Pharmacol 6:263. https://doi.org/10.3389/fphar.2015.00263
Cai S, Gomez K, Moutal A, Khanna R (2021) Targeting T-type/CaV3.2 channels for chronic pain. Transl Res 234:20–30. https://doi.org/10.1016/j.trsl.2021.01.002
Henley JM, Craig TJ, Wilkinson KA (2014) Neuronal SUMOylation: mechanisms, physiology, and roles in neuronal dysfunction. Physiol Rev 94(4):1249–1285. https://doi.org/10.1152/physrev.00008.2014
Ulrich HD (2008) The fast-growing business of SUMO chains. Mol Cell 32(3):301–305. https://doi.org/10.1016/j.molcel.2008.10.010
Vertegaal AC (2010) SUMO chains: polymeric signals. Biochem Soc Trans 38(Pt 1):46–49. https://doi.org/10.1042/BST0380046
Zhang FP, Mikkonen L, Toppari J, Palvimo JJ, Thesleff I, Jänne OA (2008) Sumo-1 function is dispensable in normal mouse development. Mol Cell Biol 28(17):5381–5390. https://doi.org/10.1128/mcb.00651-08
Wang L, Wansleeben C, Zhao S, Miao P, Paschen W, Yang W (2014) SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep 15(8):878–885. https://doi.org/10.15252/embr.201438534
Yu S, Galeffi F, Rodriguiz RM, Wang Z, Shen Y, Lyu J, Li R, Bernstock JD, Johnson KR, Liu S, Sheng H, Turner DA, Wetsel WC, Paschen W, Yang W (2020) Small ubiquitin-like modifier 2 (SUMO2) is critical for memory processes in mice. FASEB J 34(11):14750–14767. https://doi.org/10.1096/fj.202000850RR
Suk TR, Nguyen TT, Fisk ZA, Mitkovski M, Geertsma HM, Parmasad JA, Heer MM, Callaghan SM, Benseler F, Brose N, Tirard M, Rousseaux MWC (2023) Characterizing the differential distribution and targets of Sumo1 and Sumo2 in the mouse brain. Science 26(4):106350. https://doi.org/10.1016/j.isci.2023.106350
Evdokimov E, Sharma P, Lockett SJ, Lualdi M, Kuehn MR (2008) Loss of SUMO1 in mice affects RanGAP1 localization and formation of PML nuclear bodies, but is not lethal as it can be compensated by SUMO2 or SUMO3. J Cell Sci 121(Pt 24):4106–4113. https://doi.org/10.1242/jcs.038570
Saitoh H, Hinchey J (2000) Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem 275(9):6252–6258. https://doi.org/10.1074/jbc.275.9.6252
Wang W, Matunis MJ (2023) Paralogue-specific roles of SUMO1 and SUMO2/3 in protein quality control and associated diseases. Cells 13(1):8. https://doi.org/10.3390/cells13010008
Owerbach D, McKay EM, Yeh ET, Gabbay KH, Bohren KM (2005) A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem Biophys Res Commun 337(2):517–520. https://doi.org/10.1016/j.bbrc.2005.09.090
Bohren KM, Nadkarni V, Song JH, Gabbay KH, Owerbach D (2004) A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J Biol Chem 279(26):27233–27238. https://doi.org/10.1074/jbc.M402273200
Hay RT (2007) SUMO-specific proteases: a twist in the tail. Trends Cell Biol 17(8):370–376. https://doi.org/10.1016/j.tcb.2007.08.002
Reverter D, Lima CD (2004) A basis for SUMO protease specificity provided by analysis of human Senp2 and a Senp2-SUMO complex. Structure 12(8):1519–1531. https://doi.org/10.1016/j.str.2004.05.023
Raman N, Nayak A, Muller S (2013) The SUMO system: a master organizer of nuclear protein assemblies. Chromosoma 122(6):475–485. https://doi.org/10.1007/s00412-013-0429-6
Wilkinson KA, Henley JM (2010) Mechanisms, regulation and consequences of protein SUMOylation. Biochem J 428(2):133–145. https://doi.org/10.1042/bj20100158
Colnaghi L, Conz A, Russo L, Musi CA, Fioriti L, Borsello T, Salmona M (2020) Neuronal localization of SENP proteins with super resolution microscopy. Brain Sci 10(11):778. https://doi.org/10.3390/brainsci10110778
Hay RT (2005) SUMO: a history of modification. Mol Cell 18(1):1–12. https://doi.org/10.1016/j.molcel.2005.03.012
Li SJ, Hochstrasser M (1999) A new protease required for cell-cycle progression in yeast. Nature 398(6724):246–251. https://doi.org/10.1038/18457
Li SJ, Hochstrasser M (2000) The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol Cell Biol 20(7):2367–2377. https://doi.org/10.1128/MCB.20.7.2367-2377.2000
Mikolajczyk J, Drag M, Békés M, Cao JT, Ze Ronai, Salvesen GS (2007) Small ubiquitin-related modifier (SUMO)-specific proteases: profiling the specificities and activities of human SENPs*. J Biol Chem 282(36):26217–26224. https://doi.org/10.1074/jbc.M702444200
Xu Z, Au SW (2005) Mapping residues of SUMO precursors essential in differential maturation by SUMO-specific protease, SENP1. Biochem J 386(Pt 2):325–330. https://doi.org/10.1042/BJ20041210
Lima CD, Reverter D (2008) Structure of the human SENP7 catalytic domain and poly-SUMO deconjugation activities for SENP6 and SENP7. J Biol Chem 283(46):32045–32055. https://doi.org/10.1074/jbc.M805655200
Kunz K, Piller T, Müller S (2018) SUMO-specific proteases and isopeptidases of the SENP family at a glance. J Cell Sci 131(6):jcs211904. https://doi.org/10.1242/jcs.211904
Bylebyl GR, Belichenko I, Johnson ES (2003) The SUMO isopeptidase Ulp2 prevents accumulation of SUMO chains in yeast. J Biol Chem 278(45):44113–44120. https://doi.org/10.1074/jbc.M308357200
Shen LN, Geoffroy MC, Jaffray EG, Hay RT (2009) Characterization of SENP7, a SUMO-2/3-specific isopeptidase. Biochem J 421(2):223–230. https://doi.org/10.1042/BJ20090246
Vertegaal ACO (2022) Signalling mechanisms and cellular functions of SUMO. Nat Rev Mol Cell Biol 23(11):715–731. https://doi.org/10.1038/s41580-022-00500-y
Enserink JM (2015) Sumo and the cellular stress response. Cell Div 10:4. https://doi.org/10.1186/s13008-015-0010-1
Daniel JA, Cooper BH, Palvimo JJ, Zhang FP, Brose N, Tirard M (2017) Analysis of SUMO1-conjugation at synapses. Elife 6:e26338. https://doi.org/10.7554/eLife.26338
Wilkinson KA, Martin S, Tyagarajan SK, Arancio O, Craig TJ, Guo C, Fraser PE, Goldstein SAN, Henley JM (2017) Commentary: Analysis of SUMO1-conjugation at synapses. Front Cell Neurosci 11:345. https://doi.org/10.3389/fncel.2017.00345
Daniel JA, Cooper BH, Palvimo JJ, Zhang FP, Brose N, Tirard M (2018) Response: Commentary: Analysis of SUMO1-conjugation at synapses. Front Cell Neurosci 12:117. https://doi.org/10.3389/fncel.2018.00117
Colnaghi L, Russo L, Natale C, Restelli E, Cagnotto A, Salmona M, Chiesa R, Fioriti L (2019) Super resolution microscopy of SUMO proteins in neurons. Front Cell Neurosci 13:486. https://doi.org/10.3389/fncel.2019.00486
Qin Y, Zhang X, Song Y, Zhong B, Liu L, Wang D, Zhang Y, Lu W, Zhao X, Jia Z, Li M, Zhang L, Qing G (2023) A highly sensitive nanochannel device for the detection of SUMO1 peptides. Chem Sci 14(31):8360–8368. https://doi.org/10.1039/d3sc02140h
Schorova L, Pronot M, Poupon G, Prieto M, Folci A, Khayachi A, Brau F, Cassé F, Gwizdek C, Martin S (2019) The synaptic balance between sumoylation and desumoylation is maintained by the activation of metabotropic mGlu5 receptors. Cell Mol Life Sci 76(15):3019–3031. https://doi.org/10.1007/s00018-019-03075-8
Pronot M, Poupon G, Pizzamiglio L, Prieto M, Chato-Astrain I, Lacagne I, Schorova L, Folci A, Brau F, Martin S (2022) Bidirectional regulation of synaptic SUMOylation by Group 1 metabotropic glutamate receptors. Cell Mol Life Sci 79(7):378. https://doi.org/10.1007/s00018-022-04405-z
Choi JH, Park JY, Park SP, Lee H, Han S, Park KH, Suh YH (2016) Regulation of mGluR7 trafficking by SUMOylation in neurons. Neuropharmacology 102:229–235. https://doi.org/10.1016/j.neuropharm.2015.11.021
Bhachoo JS, Garvin AJ (2024) SUMO and the DNA damage response. Biochem Soc Trans 52(2):773–792. https://doi.org/10.1042/bst20230862
Han ZJ, Feng YH, Gu BH, Li YM, Chen H (2018) The post-translational modification, SUMOylation, and cancer (Review). Int J Oncol 52(4):1081–1094. https://doi.org/10.3892/ijo.2018.4280
Li X, Ding Z, Tong Y (2024) Identification of SUMOylation-related biomarkers in papillary thyroid carcinoma. Cancer Cell Int 24(1):149. https://doi.org/10.1186/s12935-024-03323-3
Shrivastava V, Pekar M, Grosser E, Im J, Vigodner M (2010) SUMO proteins are involved in the stress response during spermatogenesis and are localized to DNA double-strand breaks in germ cells. Reproduction 139(6):999–1010. https://doi.org/10.1530/rep-09-0492
Bossis G, Melchior F (2006) Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol Cell 21(3):349–357. https://doi.org/10.1016/j.molcel.2005.12.019
Yan S, Sun X, Xiang B, Cang H, Kang X, Chen Y, Li H, Shi G, Yeh ET, Wang B, Wang X, Yi J (2010) Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. Embo J 29(22):3773–3786. https://doi.org/10.1038/emboj.2010.245
Huang C, Han Y, Wang Y, Sun X, Yan S, Yeh ET, Chen Y, Cang H, Li H, Shi G, Cheng J, Tang X, Yi J (2009) SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. Embo J 28(18):2748–2762. https://doi.org/10.1038/emboj.2009.210
Xu Z, Lam LS, Lam LH, Chau SF, Ng TB, Au SW (2008) Molecular basis of the redox regulation of SUMO proteases: a protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. Faseb J 22(1):127–137. https://doi.org/10.1096/fj.06-7871com
Wang Y, Gao Y, Tian Q, Deng Q, Wang Y, Zhou T, Liu Q, Mei K, Wang Y, Liu H, Ma R, Ding Y, Rong W, Cheng J, Yao J, Xu TL, Zhu MX, Li Y (2018) TRPV1 SUMOylation regulates nociceptive signaling in models of inflammatory pain. Nat Commun 9(1):1529. https://doi.org/10.1038/s41467-018-03974-7
Agarwal N, Taberner FJ, Rangel Rojas D, Moroni M, Omberbasic D, Njoo C, Andrieux A, Gupta P, Bali KK, Herpel E, Faghihi F, Fleming T, Dejean A, Lechner SG, Nawroth PP, Lewin GR, Kuner R (2020) SUMOylation of enzymes and ion channels in sensory neurons protects against metabolic dysfunction, neuropathy, and sensory loss in diabetes. Neuron 107(6):1141-1159e1147. https://doi.org/10.1016/j.neuron.2020.06.037
Barry AM, Zhao N, Yang X, Bennett DL, Baskozos G (2023) Deep RNA-seq of male and female murine sensory neuron subtypes after nerve injury. Pain 164(10):2196–2215. https://doi.org/10.1097/j.pain.0000000000002934
Zhao X, Xia B, Cheng J, Zhu MX, Li Y (2020) PKCepsilon SUMOylation is required for mediating the nociceptive signaling of inflammatory pain. Cell Rep 33(1):108191. https://doi.org/10.1016/j.celrep.2020.108191
Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD (2019) The role of voltage-gated sodium channels in pain signaling. Physiol Rev 99(2):1079–1151. https://doi.org/10.1152/physrev.00052.2017
Cummins TR, Howe JR, Waxman SG (1998) Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci 18(23):9607–9619. https://doi.org/10.1523/JNEUROSCI.18-23-09607.1998
Dib-Hajj SD, Yang Y, Black JA, Waxman SG (2013) The Na(V)1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14(1):49–62. https://doi.org/10.1038/nrn3404
Sangameswaran L, Fish LM, Koch BD, Rabert DK, Delgado SG, Ilnicka M, Jakeman LB, Novakovic S, Wong K, Sze P, Tzoumaka E, Stewart GR, Herman RC, Chan H, Eglen RM, Hunter JC (1997) A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J Biol Chem 272(23):14805–14809. https://doi.org/10.1074/jbc.272.23.14805
Djouhri L, Newton R, Levinson SR, Berry CM, Carruthers B, Lawson SN (2003) Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel alpha subunit protein. J Physiol 546(Pt 2):565–576. https://doi.org/10.1113/jphysiol.2002.026559
Higerd-Rusli GP, Tyagi S, Liu S, Dib-Hajj FB, Waxman SG, Dib-Hajj SD (2023) The fates of internalized Na. J Biol Chem 299(1):102816. https://doi.org/10.1016/j.jbc.2022.102816
Gomez K, Allen HN, Duran P, Loya-Lopez S, Calderon-Rivera A, Moutal A, Tang C, Nelson TS, Perez-Miller S, Khanna R (2024) Targeted transcriptional upregulation of SENP1 by CRISPR activation enhances deSUMOylation pathways to elicit antinociception in the spinal nerve ligation model of neuropathic pain. Pain 165(4):866–883. https://doi.org/10.1097/j.pain.0000000000003080
Dustrude ET, Perez-Miller S, Francois-Moutal L, Moutal A, Khanna M, Khanna R (2017) A single structurally conserved SUMOylation site in CRMP2 controls NaV1.7 function. Channels (Austin) 11(4):316–328. https://doi.org/10.1080/19336950.2017.1299838
Dustrude ET, Moutal A, Yang X, Wang Y, Khanna M, Khanna R (2016) Hierarchical CRMP2 posttranslational modifications control NaV1.7 function. Proc Natl Acad Sci U S A 113(52):E8443–E8452. https://doi.org/10.1073/pnas.1610531113
Dustrude ET, Wilson SM, Ju W, Xiao Y, Khanna R (2013) CRMP2 protein SUMOylation modulates NaV1.7 channel trafficking. J Biol Chem 288(34):24316–24331. https://doi.org/10.1074/jbc.M113.474924
Francois-Moutal L, Dustrude ET, Wang Y, Brustovetsky T, Dorame A, Ju W, Moutal A, Perez-Miller S, Brustovetsky N, Gokhale V, Khanna M, Khanna R (2018) Inhibition of the Ubc9 E2 SUMO-conjugating enzyme-CRMP2 interaction decreases NaV1.7 currents and reverses experimental neuropathic pain. Pain 159(10):2115–2127. https://doi.org/10.1097/j.pain.0000000000001294
Li J, Stratton HJ, Lorca SA, Grace PM, Khanna R (2022) Small molecule targeting NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces pain in chronic constriction injury (CCI) rats. Channels (Austin) 16(1):1–8. https://doi.org/10.1080/19336950.2021.2023383
Gomez K, Ran D, Madura CL, Moutal A, Khanna R (2021) Non-SUMOylated CRMP2 decreases Na. Mol Brain 14(1):20. https://doi.org/10.1186/s13041-020-00714-1
Gomez K, Ran D, Madura CL, Moutal A, Khanna R (2021) Non-SUMOylated CRMP2 decreases NaV1.7 currents via the endocytic proteins Numb, Nedd4-2 and Eps15. Mol Brain 14(1):20. https://doi.org/10.1186/s13041-020-00714-1
Braden K, Stratton HJ, Salvemini D, Khanna R (2022) Small molecule targeting NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces and prevents pain chronification in a mouse model of oxaliplatin-induced neuropathic pain. Neurobiol Pain 11:100082. https://doi.org/10.1016/j.ynpai.2021.100082
Loya-Lopez SI, Allen HN, Duran P, Calderon-Rivera A, Gomez K, Kumar U, Shields R, Zeng R, Dwivedi A, Saurabh S, Korczeniewska OA, Khanna R (2024) Intranasal CRMP2-Ubc9 inhibitor regulates Na V 1.7 to alleviate trigeminal neuropathic pain. Pain 165(3):573–588. https://doi.org/10.1097/j.pain.0000000000003053
Moutal A, Dustrude ET, Largent-Milnes TM, Vanderah TW, Khanna M, Khanna R (2018) Blocking CRMP2 SUMOylation reverses neuropathic pain. Mol Psychiatry 23(11):2119–2121. https://doi.org/10.1038/mp.2017.117
Moutal A, Cai S, Yu J, Stratton HJ, Chefdeville A, Gomez K, Ran D, Madura CL, Boinon L, Soto M, Zhou Y, Shan Z, Chew LA, Rodgers KE, Khanna R (2020) Studies on CRMP2 SUMOylation-deficient transgenic mice identify sex-specific Nav1.7 regulation in the pathogenesis of chronic neuropathic pain. Pain 161(11):2629–2651. https://doi.org/10.1097/j.pain.0000000000001951
Dolphin AC, Lee A (2020) Presynaptic calcium channels: specialized control of synaptic neurotransmitter release. Nat Rev Neurosci 21(4):213–229. https://doi.org/10.1038/s41583-020-0278-2
Lai S, Pan M, Liao H, Chen J, Jiang Y, Li Y (2021) The impact of CRMP4 SUMOylation on the Cav1.2 interaction, neurite outgrowth and thermal pain sensitivity. J Integr Neurosci 20(3):595-603. https://doi.org/10.31083/j.jin2003063
Muller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B, Schulte U (2010) Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A 107(34):14950–14957. https://doi.org/10.1073/pnas.1005940107
Brittain JM, Wang Y, Eruvwetere O, Khanna R (2012) Cdk5-mediated phosphorylation of CRMP-2 enhances its interaction with CaV2.2. FEBS Lett 586(21):3813–3818. https://doi.org/10.1016/j.febslet.2012.09.022
Moutal A, White KA, Chefdeville A, Laufmann RN, Vitiello PF, Feinstein D, Weimer JM, Khanna R (2019) Dysregulation of CRMP2 post-translational modifications drive its pathological functions. Mol Neurobiol 56(10):6736–6755. https://doi.org/10.1007/s12035-019-1568-4
Wilson SM, Brittain JM, Piekarz AD, Ballard CJ, Ripsch MS, Cummins TR, Hurley JH, Khanna M, Hammes NM, Samuels BC, White FA, Khanna R (2011) Further insights into the antinociceptive potential of a peptide disrupting the N-type calcium channel-CRMP-2 signaling complex. Channels (Austin) 5(5):449–456. https://doi.org/10.4161/chan.5.5.17363
Gomez K, Santiago U, Nelson TS, Allen HN, Calderon-Rivera A, Hestehave S, Rodríguez Palma EJ, Zhou Y, Duran P, Loya-Lopez S, Zhu E, Kumar U, Shields R, Koseli E, McKiver B, Giuvelis D, Zuo W, Inyang KE, Dorame A, Chefdeville A, Ran D, Perez-Miller S, Lu Y, Liu X, Handoko Arora PS, Patek M, Moutal A, Khanna M, Hu H, Laumet G, King T, Wang J, Damaj MI, Korczeniewska OA, Camacho CJ, Khanna R (2023) A peptidomimetic modulator of the CaV2.2 N-type calcium channel for chronic pain. Proc Natl Acad Sci U S A 120(47):e2305215120. https://doi.org/10.1073/pnas.2305215120
Silveirinha VC, Lin H, Tanifuji S, Mochida S, Cottrell GS, Cimarosti H, Stephens GJ (2021) CaV2.2 (N-type) voltage-gated calcium channels are activated by SUMOylation pathways. Cell Calcium 93:102326. https://doi.org/10.1016/j.ceca.2020.102326
Ju W, Li Q, Wilson SM, Brittain JM, Meroueh L, Khanna R (2013) SUMOylation alters CRMP2 regulation of calcium influx in sensory neurons. Channels (Austin) 7(3):153–159. https://doi.org/10.4161/chan.24224
Moutal A, Luo S, Largent-Milnes TM, Vanderah TW, Khanna R (2019) Cdk5-mediated CRMP2 phosphorylation is necessary and sufficient for peripheral neuropathic pain. Neurobiol Pain 5:100022. https://doi.org/10.1016/j.ynpai.2018.07.003
Gomez K, Stratton HJ, Duran P, Loya S, Tang C, Calderon-Rivera A, Francois-Moutal L, Khanna M, Madura CL, Luo S, McKiver B, Choi E, Ran D, Boinon L, Perez-Miller S, Damaj MI, Moutal A, Khanna R (2023) Identification and targeting of a unique Na(V)1.7 domain driving chronic pain. Proc Natl Acad Sci U S A 120(32):e2217800120. https://doi.org/10.1073/pnas.2217800120
Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R (2009) An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem 284(45):31375–31390. https://doi.org/10.1074/jbc.M109.009951
Chemin J, Monteil A, Perez-Reyes E, Bourinet E, Nargeot J, Lory P (2002) Specific contribution of human T-type calcium channel isotypes (alpha(1G), alpha(1H) and alpha(1I)) to neuronal excitability. J Physiol 540(Pt 1):3–14. https://doi.org/10.1113/jphysiol.2001.013269
Weiss N, Hameed S, Fernandez-Fernandez JM, Fablet K, Karmazinova M, Poillot C, Proft J, Chen L, Bidaud I, Monteil A, Huc-Brandt S, Lacinova L, Lory P, Zamponi GW, De Waard M (2012) A Ca(v)3.2/syntaxin-1A signaling complex controls T-type channel activity and low-threshold exocytosis. J Biol Chem 287(4):2810–2818. https://doi.org/10.1074/jbc.M111.290882
Jacus MO, Uebele VN, Renger JJ, Todorovic SM (2012) Presynaptic Cav3.2 channels regulate excitatory neurotransmission in nociceptive dorsal horn neurons. J Neurosci 32(27):9374–9382. https://doi.org/10.1523/JNEUROSCI.0068-12.2012
Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, Nargeot J (2005) Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 24(2):315–324. https://doi.org/10.1038/sj.emboj.7600515
Francois A, Kerckhove N, Meleine M, Alloui A, Barrere C, Gelot A, Uebele VN, Renger JJ, Eschalier A, Ardid D, Bourinet E (2013) State-dependent properties of a new T-type calcium channel blocker enhance Ca(V)3.2 selectivity and support analgesic effects. Pain 154(2):283–293. https://doi.org/10.1016/j.pain.2012.10.023
García-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, Bladen C, Chen L, Hamid J, Pizzoccaro A, Deage M, François A, Bourinet E, Zamponi GW (2014) The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 83(5):1144–1158. https://doi.org/10.1016/j.neuron.2014.07.036
Garcia-Caballero A, Zhang FX, Chen L, M’Dahoma S, Huang J, Zamponi GW (2019) SUMOylation regulates USP5-Cav3.2 calcium channel interactions. Mol Brain 12(1):73. https://doi.org/10.1186/s13041-019-0493-9
Taura J, Kircher DM, Gameiro-Ros I, Slesinger PA (2021) Comparison of K+ Channel Families. Handb Exp Pharmacol 267:1–49. https://doi.org/10.1007/164_2021_460
Tsantoulas C (2015) Emerging potassium channel targets for the treatment of pain. Curr Opin Support Palliat Care 9(2):147–154. https://doi.org/10.1097/SPC.0000000000000131
Krapivinsky G, Medina I, Eng L, Krapivinsky L, Yang Y, Clapham DE (1998) A novel inward rectifier K+ channel with unique pore properties. Neuron 20(5):995–1005. https://doi.org/10.1016/s0896-6273(00)80480-8
Lv YY, Wang H, Fan HT, Xu T, Xin WJ, Guo RX (2022) SUMOylation of Kir7.1 participates in neuropathic pain through regulating its membrane expression in spinal cord neurons. CNS Neurosci Ther 28(8):1259–1267. https://doi.org/10.1111/cns.13871
Misonou H, Mohapatra DP, Menegola M, Trimmer JS (2005) Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J Neurosci 25(48):11184–11193. https://doi.org/10.1523/jneurosci.3370-05.2005
Plant LD, Dowdell EJ, Dementieva IS, Marks JD, Goldstein SA (2011) SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J Gen Physiol 137(5):441–454. https://doi.org/10.1085/jgp.201110604
Misonou H, Mohapatra DP, Trimmer JS (2005) Kv2.1: a voltage-gated k+ channel critical to dynamic control of neuronal excitability. Neurotoxicology 26(5):743–752. https://doi.org/10.1016/j.neuro.2005.02.003
Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS (2001) Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A 98(23):13373–13378. https://doi.org/10.1073/pnas.231376298
Ramírez A, Vázquez-Sánchez AY, Carrión-Robalino N, Camacho J (2016) Ion channels and oxidative stress as a potential link for the diagnosis or treatment of liver diseases. Oxid Med Cell Longev 2016:3928714. https://doi.org/10.1155/2016/3928714
Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E (2003) Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci 23(12):4798–4802. https://doi.org/10.1523/jneurosci.23-12-04798.2003
Welch MA, Jansen LR, Baro DJ (2021) SUMOylation of the Kv4.2 ternary complex increases surface expression and current amplitude by reducing internalization in HEK 293 cells. Front Mol Neurosci 14:757278. https://doi.org/10.3389/fnmol.2021.757278
Welch MA, Forster LA, Atlas SI, Baro DJ (2019) SUMOylating two distinct sites on the A-type potassium channel, Kv4.2, increases surface expression and decreases current amplitude. Front Mol Neurosci 12:144. https://doi.org/10.3389/fnmol.2019.00144
Steffensen AB, Andersen MN, Mutsaers N, Mujezinovic A, Schmitt N (2018) SUMO co-expression modifies KV 11.1 channel activity. Acta Physiol (Oxf) 222(3). https://doi.org/10.1111/apha.12974
Rajan S, Plant LD, Rabin ML, Butler MH, Goldstein SA (2005) Sumoylation silences the plasma membrane leak K+ channel K2P1. Cell 121(1):37–47. https://doi.org/10.1016/j.cell.2005.01.019
Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, Gereau RW (2006) The kv4.2 potassium channel subunit is required for pain plasticity. Neuron 50(1):89–100. https://doi.org/10.1016/j.neuron.2006.03.010
Marsh B, Acosta C, Djouhri L, Lawson SN (2012) Leak K⁺ channel mRNAs in dorsal root ganglia: relation to inflammation and spontaneous pain behaviour. Mol Cell Neurosci 49(3):375–386. https://doi.org/10.1016/j.mcn.2012.01.002
Pollema-Mays SL, Centeno MV, Ashford CJ, Apkarian AV, Martina M (2013) Expression of background potassium channels in rat DRG is cell-specific and down-regulated in a neuropathic pain model. Mol Cell Neurosci 57:1–9. https://doi.org/10.1016/j.mcn.2013.08.002
Tulleuda A, Cokic B, Callejo G, Saiani B, Serra J, Gasull X (2011) TRESK channel contribution to nociceptive sensory neurons excitability: modulation by nerve injury. Mol Pain 7:30. https://doi.org/10.1186/1744-8069-7-30
Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389(6653):816–824. https://doi.org/10.1038/39807
Aneiros E, Cao L, Papakosta M, Stevens EB, Phillips S, Grimm C (2011) The biophysical and molecular basis of TRPV1 proton gating. The EMBO J 30(6):994–1002. https://doi.org/10.1038/emboj.2011.19
Guo A, Vulchanova L, Wang J, Li X, Elde R (1999) Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci 11(3):946–958. https://doi.org/10.1046/j.1460-9568.1999.00503.x
Gao Y, Ma R, Weng W, Zhang H, Wang Y, Guo R, Gu X, Yang Y, Yang F, Zhou A, Cheng J, Chen ZY, Zhu MX, Li Y (2022) TRPV1 SUMOylation suppresses itch by inhibiting TRPV1 interaction with H1 receptors. Cell Rep 39(11):110972. https://doi.org/10.1016/j.celrep.2022.110972
Doly S, Fischer J, Salio C, Conrath M (2004) The vanilloid receptor-1 is expressed in rat spinal dorsal horn astrocytes. Neurosci Lett 357(2):123–126. https://doi.org/10.1016/j.neulet.2003.12.051
Maione S, Starowicz K, Cristino L, Guida F, Palazzo E, Luongo L, Rossi F, Marabese I, de Novellis V, Di Marzo V (2009) Functional interaction between TRPV1 and mu-opioid receptors in the descending antinociceptive pathway activates glutamate transmission and induces analgesia. J Neurophysiol 101(5):2411–2422. https://doi.org/10.1152/jn.91225.2008
Agarwal N, Taberner FJ, Rangel Rojas D, Moroni M, Omberbasic D, Njoo C, Andrieux A, Gupta P, Bali KK, Herpel E, Faghihi F, Fleming T, Dejean A, Lechner SG, Nawroth PP, Lewin GR, Kuner R (2020) SUMOylation of enzymes and ion channels in sensory neurons protects against metabolic dysfunction, neuropathy, and sensory loss in diabetes. Neuron 107(6):1141-1159.e1147. https://doi.org/10.1016/j.neuron.2020.06.037
Karashima Y, Talavera K, Everaerts W, Janssens A, Kwan KY, Vennekens R, Nilius B, Voets T (2009) TRPA1 acts as a cold sensor in vitro and in vivo. Proc Natl Acad Sci U S A 106(4):1273–1278. https://doi.org/10.1073/pnas.0808487106
Anand U, Otto WR, Facer P, Zebda N, Selmer I, Gunthorpe MJ, Chessell IP, Sinisi M, Birch R, Anand P (2008) TRPA1 receptor localisation in the human peripheral nervous system and functional studies in cultured human and rat sensory neurons. Neurosci Lett 438(2):221–227. https://doi.org/10.1016/j.neulet.2008.04.007
Zhang Q, Weng W, Gu X, Xiang J, Yang Y, Zhu MX, Gu W, He Z, Li Y (2023) hnRNPA1 SUMOylation promotes cold hypersensitivity in chronic inflammatory pain by stabilizing TRPA1 mRNA. Cell Rep 42(11):113401. https://doi.org/10.1016/j.celrep.2023.113401
Monteiro Souza, de Araujo D, Nassini R, Geppetti P, De Logu F (2020) TRPA1 as a therapeutic target for nociceptive pain. Expert Opin Ther Targets 24(10):997–1008. https://doi.org/10.1080/14728222.2020.1815191
Wang H, Han L, Zhao G, Shen H, Wang P, Sun Z, Xu C, Su Y, Li G, Tong T, Chen J (2016) hnRNP A1 antagonizes cellular senescence and senescence-associated secretory phenotype via regulation of SIRT1 mRNA stability. Aging Cell 15(6):1063–1073. https://doi.org/10.1111/acel.12511
Roy R, Huang Y, Seckl MJ, Pardo OE (2017) Emerging roles of hnRNPA1 in modulating malignant transformation. Wiley Interdiscip Rev RNA 8(6). https://doi.org/10.1002/wrna.1431
Clarke JP, Thibault PA, Salapa HE, Levin MC (2021) A comprehensive analysis of the role of hnRNP A1 function and dysfunction in the pathogenesis of neurodegenerative disease. Front Mol Biosci 8:659610. https://doi.org/10.3389/fmolb.2021.659610
Li T, Evdokimov E, Shen RF, Chao CC, Tekle E, Wang T, Stadtman ER, Yang DC, Chock PB (2004) Sumoylation of heterogeneous nuclear ribonucleoproteins, zinc finger proteins, and nuclear pore complex proteins: a proteomic analysis. Proc Natl Acad Sci U S A 101(23):8551–8556. https://doi.org/10.1073/pnas.0402889101
Roy R, Durie D, Li H, Liu BQ, Skehel JM, Mauri F, Cuorvo LV, Barbareschi M, Guo L, Holcik M, Seckl MJ, Pardo OE (2014) hnRNPA1 couples nuclear export and translation of specific mRNAs downstream of FGF-2/S6K2 signalling. Nucleic Acids Res 42(20):12483–12497. https://doi.org/10.1093/nar/gku953
Sartiani L, Mannaioni G, Masi A, Novella Romanelli M, Cerbai E (2017) The hyperpolarization-activated cyclic nucleotide-gated channels: from biophysics to pharmacology of a unique family of ion channels. Pharmacol Rev 69(4):354–395. https://doi.org/10.1124/pr.117.014035
Mayer ML, Westbrook GL (1983) A voltage-clamp analysis of inward (anomalous) rectification in mouse spinal sensory ganglion neurones. J Physiol 340:19–45. https://doi.org/10.1113/jphysiol.1983.sp014747
Parker AR, Welch MA, Forster LA, Tasneem SM, Dubhashi JA, Baro DJ (2016) SUMOylation of the hyperpolarization-activated cyclic nucleotide-gated channel 2 increases surface expression and the maximal conductance of the hyperpolarization-activated current. Front Mol Neurosci 9:168. https://doi.org/10.3389/fnmol.2016.00168
Forster LA, Jansen LR, Rubaharan M, Murphy AZ, Baro DJ (2020) Alterations in SUMOylation of the hyperpolarization-activated cyclic nucleotide-gated ion channel 2 during persistent inflammation. Eur J Pain 24(8):1517–1536. https://doi.org/10.1002/ejp.1606
Jansen LR, Forster LA, Smith XL, Rubaharan M, Murphy AZ, Baro DJ (2021) Changes in peripheral HCN2 channels during persistent inflammation. Channels (Austin) 15(1):165–179. https://doi.org/10.1080/19336950.2020.1870086
Koidl S, Eisenhardt N, Fatouros C, Droescher M, Chaugule VK, Pichler A (2016) The SUMO2/3 specific E3 ligase ZNF451-1 regulates PML stability. Int J Biochem Cell Biol 79:478–487. https://doi.org/10.1016/j.biocel.2016.06.011
Newton AC (1995) Protein kinase C: structure, function, and regulation. J Biol Chem 270(48):28495–28498. https://doi.org/10.1074/jbc.270.48.28495
Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO (1999) A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron 24(1):253–260. https://doi.org/10.1016/s0896-6273(00)80837-5
Dina OA, Chen X, Reichling D, Levine JD (2001) Role of protein kinase C epsilon and protein kinase A in a model of paclitaxel-induced painful peripheral neuropathy in the rat. Neuroscience 108(3):507–515. https://doi.org/10.1016/s0306-4522(01)00425-0
Aley KO, Messing RO, Mochly-Rosen D, Levine JD (2000) Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci 20(12):4680–4685. https://doi.org/10.1523/JNEUROSCI.20-12-04680.2000
Zhou Y, Li GD, Zhao ZQ (2003) State-dependent phosphorylation of epsilon-isozyme of protein kinase C in adult rat dorsal root ganglia after inflammation and nerve injury. J Neurochem 85(3):571–580. https://doi.org/10.1046/j.1471-4159.2003.01675.x
Sun H, Lu L, Zuo Y, Wang Y, Jiao Y, Zeng WZ, Huang C, Zhu MX, Zamponi GW, Zhou T, Xu TL, Cheng J, Li Y (2014) Kainate receptor activation induces glycine receptor endocytosis through PKC deSUMOylation. Nat Commun 5:4980. https://doi.org/10.1038/ncomms5980
Kondo M, Shibuta I (2020) Extracellular signal-regulated kinases (ERK) 1 and 2 as a key molecule in pain research. J Oral Sci 62(2):147–149. https://doi.org/10.2334/josnusd.19-0470
Zhuang Z-Y, Gerner P, Woolf CJ, Ji R-R (2005) ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 114(1):149–159. https://doi.org/10.1016/j.pain.2004.12.022
Shi C, Jin J, Xu H, Ma J, Li T, Xie Y, Li Z (2022) CCR1 enhances SUMOylation of DGCR8 by up-regulating ERK phosphorylation to promote spinal nerve ligation-induced neuropathic pain. Gene Ther 29(6):379–389. https://doi.org/10.1038/s41434-021-00285-3
Stanley EF (2016) The nanophysiology of fast transmitter release. Trends Neurosci 39(3):183–197. https://doi.org/10.1016/j.tins.2016.01.005
Girach F, Craig TJ, Rocca DL, Henley JM (2013) RIM1α SUMOylation is required for fast synaptic vesicle exocytosis. Cell Rep 5(5):1294–1301. https://doi.org/10.1016/j.celrep.2013.10.039
Baskozos G, Themistocleous AC, Hebert HL, Pascal MMV, John J, Callaghan BC, Laycock H, Granovsky Y, Crombez G, Yarnitsky D, Rice ASC, Smith BH, Bennett DLH (2022) Classification of painful or painless diabetic peripheral neuropathy and identification of the most powerful predictors using machine learning models in large cross-sectional cohorts. BMC Med Inform Decis Mak 22(1):144. https://doi.org/10.1186/s12911-022-01890-x
Baskozos G, Sandy-Hindmarch O, Clark AJ, Windsor K, Karlsson P, Weir GA, McDermott LA, Burchall J, Wiberg A, Furniss D, Bennett DLH, Schmid AB (2020) Molecular and cellular correlates of human nerve regeneration: ADCYAP1/PACAP enhance nerve outgrowth. Brain 143(7):2009–2026. https://doi.org/10.1093/brain/awaa163
Brackett CM, Blagg BSJ (2021) Current status of SUMOylation inhibitors. Curr Med Chem 28(20):3892–3912. https://doi.org/10.2174/0929867327666200810135039
Fan YP, Lou JS, Jin MR, Zhou CH, Shen HH, Fu CY, Mao XJ, Chen YY, Zhong JJ, Wang LL, Wu JS (2024) UBC9-mediated SUMOylation of lamin B1 enhances DNA-damage-induced nuclear DNA leakage and autophagy after spinal cord injury. J Cell Physiol 239(5):e31213. https://doi.org/10.1002/jcp.31213
Gareau JR, Lima CD (2010) The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11(12):861–871. https://doi.org/10.1038/nrm3011
Cai S, Moutal A, Yu J, Chew LA, Isensee J, Chawla R, Gomez K, Luo S, Zhou Y, Chefdeville A, Madura C, Perez-Miller S, Bellampalli SS, Dorame A, Scott DD, François-Moutal L, Shan Z, Woodward T, Gokhale V, Hohmann AG, Vanderah TW, Patek M, Khanna M, Hucho T, Khanna R (2021) Selective targeting of NaV1.7 via inhibition of the CRMP2-Ubc9 interaction reduces pain in rodents. Sci Transl Med 13(619):eabh1314. https://doi.org/10.1126/scitranslmed.abh1314
Funding
The work was funded by an award from the National Institutes of Health: National Institute of Neurological Disorders and Stroke, K99NS134965 to KG; National Institute of Neurological Disorders and Stroke, NS098772, NS120663 to RK; and National Institute on Drug Abuse, DA042852 to RK.
Author information
Authors and Affiliations
Contributions
ACR, KG, and RK conceived the idea for the article. ACR performed the literature search and wrote the first draft with help from KG. ACR and EJR-P created the figures using BioRender.com. EJR-P edited the article. All authors critically revised the work..
Corresponding author
Ethics declarations
Ethics Approval
This is a review paper. The University of Florida Research Ethics Committee has confirmed that no ethical approval is required.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Calderon-Rivera, A., Gomez, K., Rodríguez-Palma, E.J. et al. SUMOylation and DeSUMOylation: Tug of War of Pain Signaling. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04478-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04478-w