
Abstract
Neuroinflammation is a pivotal factor in the progression of both age-related and acute neurodegenerative disorders, including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, multiple sclerosis, and stroke. Mitochondria, essential for neuronal health due to their roles in energy production, calcium buffering, and oxidative stress regulation, become increasingly susceptible to dysfunction under conditions of metabolic stress, aging, or injury. Impaired mitophagy in aged or injured neurons leads to the accumulation of dysfunctional mitochondria, which release mitochondrial-derived damage-associated molecular patterns (mtDAMPs). These mtDAMPs act as immune checkpoints, activating pattern recognition receptors (PRRs) and triggering innate immune signaling pathways. This activation initiates inflammatory responses in neurons and brain-resident immune cells, releasing cytokines and chemokines that damage adjacent healthy neurons and recruit peripheral immune cells, further amplifying neuroinflammation and neurodegeneration. Long-term mitochondrial dysfunction perpetuates a chronic inflammatory state, exacerbating neuronal injury and contributing additional immunogenic components to the extracellular environment. Emerging evidence highlights the critical role of mtDAMPs in initiating and sustaining neuroinflammation, with circulating levels of these molecules potentially serving as biomarkers for disease progression. This review explores the mechanisms of mtDAMP release due to mitochondrial dysfunction, their interaction with PRRs, and the subsequent activation of inflammatory pathways. We also discuss the role of mtDAMP-triggered innate immune responses in exacerbating both acute and chronic neuroinflammation and neurodegeneration. Targeting dysfunctional mitochondria and mtDAMPs with pharmacological agents presents a promising strategy for mitigating the initiation and progression of neuropathological conditions.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria are bioenergetic and biosynthetic organelles involved in adenosine triphosphate (ATP) production, carbohydrate metabolism, lipid metabolism, amino acid metabolism, ketogenesis, heme synthesis, calcium (Ca2+) buffering, and redox balance [1]. Due to their high bioenergetic demands, neurons compel their mitochondria to increase oxygen consumption, leading to elevated reactive oxygen species (ROS) generation as a byproduct, ultimately resulting in mitochondrial damage and dysfunction. Misfolded protein accumulation due to aging is another risk factor for mitochondrial dysfunction [2]. Damaged and dysfunctional mitochondria release several mitochondrial components, termed mitochondrial damage-associated molecular patterns (mtDAMPs), such as cytochrome c (Cytc), Ca2+, ROS, heme, ATP, succinate, cardiolipin, mitochondrial transcription factor A (TFAM), N-formyl peptides (NFPs), mitochondrial (mt)DNA, mitochondrial double-stranded (mtds)RNA, and others [3]. Due to the mitochondrial origin from alpha-proteobacteria, mtDAMPs retain foreign signatures and, when released into the cytosol or extracellular milieu, are recognized by one or more pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), RIG-I-like receptors (RLRs), C-type lectin receptors (CLRs), receptor for advanced glycation end products (RAGE), formyl peptide receptors (FPRs), purinergic receptors, and DNA sensors present on glial and neuronal cells [4, 5]. This event triggers several intracellular signaling cascades, such as inflammasome, nuclear factor kappa (NFκ)B, and others, leading to gliosis (microgliosis and astrogliosis), which produces a variety of proinflammatory cytokines, chemokines, adhesion molecules, and neurotoxic substances such as ROS that damage neuronal cells, ultimately causing sterile neuroinflammation, neurodegeneration, and related pathologies [3, 6]. Persistent mtDAMPs release and neuroinflammation cause blood‒brain barrier (BBB) disruption, resulting in the infiltration of peripheral immune cells such as neutrophils, monocytes, T cells, and B cells, further aggravating neuroinflammation and neurodegeneration [7]. Several reports have suggested that mtDAMPs-mediated sterile neuroinflammation is a bona fide driver of chronic and acute neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Amyotrophic lateral sclerosis (ALS), Multiple sclerosis (MS), and Stroke [3, 8].
This comprehensive review discusses the mechanisms underlying the release of mtDAMPs and analyzes recent evidence of their role in triggering intricate inflammatory signaling pathways. Furthermore, we explore their involvement in both acute and chronic neuropathological conditions, along with potential therapeutic strategies to mitigate their harmful effects on neurological health.
mtDAMPs: Release and Spread
Reduced integrity of the mitochondrial outer membrane (MOM) and mitochondrial inner membrane (MIM) facilitates the release of mtDAMPs into the cytosol. Once in the cytosol, mtDAMPs activate several inflammatory signaling cascades and facilitate the rupture of the cellular membrane, ultimately escaping into the extracellular milieu. Even with an intact cellular membrane, mtDAMPs can still be transferred outside with the help of vesicles. The escaped mtDAMPs translocate and spread from one location to another via extracellular fluids [9, 10].
Mitochondrial Outer Membrane Permeabilization
The MOM is permeable to small molecules (≤ 5 kDa) due to the presence of voltage-dependent anion channels (VDACs), also known as porins, while larger molecules such as Cytc (~ 12 kDa) require the translocase of the outer mitochondrial membrane (TOM) for entry. MOM also contains stress-sensitive apoptosis proteins, such as B-cell lymphoma (Bcl)2 family proteins, which include proapoptotic Bcl2 homology (BH)3-only proteins, proapoptotic pore-forming proteins (e.g., Bax and Bak), and antiapoptotic proteins (e.g., Bcl2, Bcl-XL, Bcl-W, and Mcl1). BH3-only proteins are categorized into BH3 sensitizers (e.g., Bik, Bad, and Noxa) and BH3 activators (e.g., Bim, Bid, and Puma). Under physiological conditions, the Bax:Bak ratio is strictly maintained between the cytosol and MOM [11]. Additionally, antiapoptotic proteins such as Bcl2 bind to the hydrophobic groove of activated Bax and Bak, as well as BH3-only proteins (both sensitizers and activators), rendering them inactive [12]. Bcl2 also interacts with and closes VDACs, thereby regulating Ca2+ and ROS within the mitochondria. Diverse cellular stresses such as Ca2+ overload, excessive ROS production, and faulty protein accumulation, disrupt cellular homeostasis, leading to the transcriptional upregulation and post-translational activation of several BH3-only proteins. Caspase 8-mediated Bid cleavage to form truncated (t)Bid and p53-mediated Puma upregulation are examples of post-translational activation of BH3-only proteins [13]. The increased levels or activation of BH3-only proteins outcompete the antiapoptotic Bcl2 proteins. BH3 sensitizer proteins neutralize antiapoptotic proteins, liberating BH3 activator proteins, which subsequently bind to the hydrophobic groove of Bax and Bak, causing their conformational changes and activation [14]. Moreover, Bax has a second BH3-binding site, enabling its allosteric conformational change and further activation [15]. Activated Bax and Bak homodimerize, subsequently oligomerize, and form small proteinaceous pores (~ 5 nm in diameter) in the MOM, leading to MOM permeabilization (MOMP), which is sufficient to release ~ 12 kDa Cytc from the intermembrane space (IMS) to the cytosol [16] (Fig. 1). For further activation, activated Bax and Bak do not require BH3-only protein stimulation because, in their activated state, they have their own BH3 domains [17]. Over time, the concentration of activated Bax and Bak increases in small proteinaceous pores, leading to the formation of macropores (~ 100–160 nm in diameter), which can release larger IMS proteins, such as the ~ 27 kDa second mitochondria-derived activator of caspase (SMAC; also known as DIABLO) and the ~ 49 kDa OMI (also known as high-temperature requirement protein A2, HtrA2) [18]. Additionally, activated Bax and Bak interact with VDAC1 and 2, causing their opening, which facilitates Ca2+ mobilization into the mitochondria and further release of IMS proteins into the cytosol [19]. Interestingly, Bax and Bak also interact with OMA1 and dynamin-related protein (Drp)1, causing optic atrophy (OPA)1 cleavage, resulting in mitochondrial cristae remodeling and cristae junction widening, ultimately leading to IMS protein release. This process can efficiently release Cytc because a higher amount of Cytc is entrapped within the cristae [20,21,22]. Bok is another proapoptotic protein of the Bcl2 family that can also induce MOMP in the absence of Bax, Bak, and BH3-only proteins. Its activity is not affected by the presence of Bcl2 [23]. Furthermore, some non-Bcl2 family proteins can also induce MOMP, such as gasdermin E (GSDME), which is a cleaved product of caspase 3 [24].

Molecular mechanisms involved in mitochondrial permeabilization and mtDAMPs release. Left: Various cellular stressors or disease conditions disrupt the balance of Bcl2 and BH3-only proteins, facilitating the formation and accumulation of Bax-Bak pores on the MOM. This drives mitochondrial cristae remodeling and the release of IMS proteins such as Cytc, SMAC, and OMI into the cytosol, leading to caspase-mediated apoptosis. Additionally, MOMP exposes the MIM, promoting water influx into the mitochondrial matrix, causing mitochondrial swelling, and subsequently, MIM herniation and rupture. Right: The Bax-Bak pore also activates VDAC, promoting mitochondrial Ca2+ overload and subsequent ROS accumulation. Elevated mtROS directly damages the MIM and enhances Bax-Bak pore assembly on the MOM. Both Ca2+ and ROS activate CypD, triggering the opening of the mPTP and leading to the release of mtDAMPs, including mtDNA fragments, from the mitochondrial matrix. The mPTP also facilitates the transfer of cytosolic fluid into the mitochondrial matrix, promoting mitochondrial swelling and rupture, which results in the extensive release of mtDAMPs into the cytosol. Released mtDAMPs can directly damage cellular membranes or indirectly activate inflammatory signaling, ultimately driving necrosis and the release of DAMPs into the extracellular milieu
Released IMS proteins such as Cytc, SMAC, and OMI facilitate the intrinsic apoptosis cascade either by forming the apoptosome or by antagonizing inhibitors of apoptosis proteins (IAPs) [25]. Cytc interacts with inactive Apaf1 in the cytosol, causing its conformational change and subsequent hydrolysis of bound deoxy (d)ATP/ATP into dADP/ADP. This change facilitates the assembly of the apoptosome, which provides a platform for the binding and activation of procaspase 9. Activated caspase 9 cleaves and activates executioner caspases such as caspase 3 and 7, subsequently leading to enzymatic cell demolition [26]. Physiologically, this apoptotic process is tightly regulated by antiapoptotic proteins such as X-linked inhibitor of apoptosis protein (XIAP), which inhibits caspases 3, 7, and 9, thereby preventing the apoptosis cascade. However, the release of SMAC and OMI (a serine protease) into the cytosol inhibits XIAP activity, enabling caspase-mediated apoptosis [27, 28] (Fig. 1). Even in the absence of caspases and Apaf1, MOMP can still induce cell death, due to a reduction in mitochondrial metabolism, such as ATP production. Therefore, MOMP is considered a point of no return (also termed an all-or-nothing event) in apoptosis [29, 30]. Additionally, under conditions of caspase inhibition, SMAC promotes inflammation by preventing IAP-mediated degradation of NFκB inducing kinase (NIK). This leads to the activation and nuclear translocation of NFκB, resulting in the secretion of proinflammatory cytokines, including TNFα [31].
Notably, MOMP is a rapid and complete event in which all IMS proteins, such as Cytc, SMAC, and OMI, are released within ~ 5 min into the cytosol, ultimately triggering the apoptotic cascade but not inflammation [32, 33]. Thus, MOMP induces programmed cell death with an intact cell membrane, preventing mtDAMPs release into the extracellular space and subsequent immune cell activation. Several studies have shown that Cytc released from dysfunctional mitochondria induces excessive apoptosis of neuronal and glial cells, contributing to the progression of various neurodegenerative disorders, such as AD and PD [34, 35].
Mitochondrial Inner Membrane Permeabilization
Unlike the porous MOM, the MIM does not contain VDACs, rendering it impermeable to ions and molecules. The MIM contains the anionic phospholipid cardiolipin, which plays a crucial role in maintaining membrane dynamics, stability, morphology, strength, and impermeability [36]. However, due to MOMP, the IMS contents are washed away, exposing the MIM to the cytosol. This exposure leads to water transfer from the cytosol to the mitochondrial matrix due to the osmotic gradient, either directly via the MIM or via aquaporins present on it, ultimately causing matrix dilution. This increase in matrix volume builds high pressure, facilitating MIM extrusion along with the matrix through macropores into the cytosol, a process known as mitochondrial herniation. Over time, the MOM and MIM are unable to resist high fluid tension, leading to MIM permeabilization (MIMP) and mitochondrial rupture, resulting in the release of matrix content into the cytosol. The released matrix contains several mtDAMPs, such as Cytc, Ca2+, ROS, heme, ATP, succinate, cardiolipin, TFAM, NFPs, mtDNA, and mtdsRNA [9, 30, 37,38,39] (Fig. 1).
Furthermore, other studies have demonstrated that mitochondrial Ca2+ overload and excessive ROS initially induce MIMP, followed by MOMP, facilitating the release of mtDAMPs into the cytosol [40, 41]. The endoplasmic reticulum (ER) releases large amounts of Ca2+ that enter mitochondria via VDACs and the mitochondrial Ca2+ uniporter (MCU), which are located on the MOM and MIM, respectively. Under normal conditions, positively charged Cytc binds to negatively charged cardiolipin on the outer leaflet of the MIM, which is essential for efficient ATP production and maintaining membrane integrity. However, excessive free Ca2+ competes with and displaces Cytc from cardiolipin, thereby compromising ATP production and membrane integrity [42]. Dysregulated Ca2+ levels also enhance ROS production by stimulating excessive mitochondrial metabolism and activating calmodulin, nitric oxide synthase (NOS), and mitochondrial NADPH oxidase (NOX) [43]. The produced ROS oxidize and damage various mitochondrial components, including mtDNA, proteins, and lipid membranes. In particular, cardiolipin is highly susceptible to peroxidation due to its unsaturated fatty acids, such as docosanoic and linoleic acid, which further reduce ATP generation and membrane integrity. Peroxidized cardiolipin promotes the opening of the mitochondrial permeability transition pore (mPTP) by stabilizing and activating one of its structural components, adenine nucleotide translocator (ANT) [44]. Additionally, Ca2+ and ROS synergistically induce mPTP opening either by activating cyclophilin D (CypD), a regulator of mPTP opening, or by other mechanisms [45,46,47]. The sustained opening of the mPTP (with a pore size of ~ 1.5–3 nm in diameter) renders the otherwise impermeable MIM freely permeable. This allows the passive diffusion of mitochondrial matrix components smaller than 3 nm, such as Ca2+, ROS, Cytc, ATP, succinate, heme, cardiolipin, TFAM, NFPs, mtdsRNA, and mtDNA fragments, but not larger components such as intact mtDNA or mtDNA nucleoids, into the cytosol [48,49,50,51]. Concurrently, the influx of cytosolic water into the mitochondrial matrix leads to mitochondrial swelling, a reduction in the mitochondrial membrane potential (ΔΨm), and decreased ATP production, ultimately resulting in mitochondrial rupture and the release of the remaining mitochondrial components [52, 53]. The released mtDAMPs initiate various intracellular signaling pathways, leading to cellular damage. For instance, iron from heme and Ca2+ increase cytoplasmic ROS levels, causing peroxidation of cellular biomolecules, particularly lipid membranes. Additionally, mitochondrial dysfunction results in ATP deficiency, promoting cell lysis and death through necrosis, allowing mtDAMPs to escape into the extracellular fluid [51, 52, 54] (Fig. 1). The distinctions between MOMP and MIMP events are summarized in Table 1. Further studies are necessary to fully elucidate the exact and sequential phenomena of MOMP and MIMP, enabling targeted interventions to preserve mitochondrial function and promote cell survival.
Translocation and Spread of mtDAMPs
mtDAMPs exhibit remarkable stability in extracellular environments. For instance, mtDNA is significantly more resistant to nuclease degradation than nuclear DNA. Following cellular membrane rupture, mtDAMPs are passively released into various biological fluids, including cerebrospinal fluid (CSF) and plasma [55, 56]. These fluids facilitate the dissemination of mtDAMPs and their associated pathologies throughout the body, affecting compartments such as the central nervous system (CNS) and peripheral tissues. The precise mechanisms underlying the transfer of mtDAMPs from the brain to CSF and plasma remain under investigation. It is hypothesized that neuroinflammation induced by mtDAMPs may cause ependymal wall and blood‒brain barrier (BBB) leakage, allowing mtDAMPs to escape from the brain parenchyma into the CSF and plasma. Elevated levels of Cytc have been observed in pediatric CSF following traumatic brain injury, and cardiolipin has been detected in human serum [55, 57]. Increased concentrations of circulating cell-free (ccf)-mtDNA have also been identified in the CSF of patients with AD, PD, and MS, as well as in the serum of AD and PD patients [56, 58,59,60]. Therefore, mtDAMP concentrations in CSF and plasma could serve as valuable biomarkers for detecting stages of neuroinflammation and neurodegenerative disorders.
In intact cells, mtDAMPs can be actively transferred from one cell to another via exosomes, which are small extracellular vesicles (30–150 nm in diameter). Stressed or dysfunctional mitochondria secrete damaged components and proteins packaged into 70–100 nm vesicles known as mitochondrial-derived vesicles (MDVs) or mitovesicles. MDVs accumulate in late endosomes, also known as multivesicular bodies (MVBs), within the cytoplasm. These MVBs typically fuse with lysosomes to form the endolysosomal system for intravesicular content degradation. However, high trafficking rates can lead to MVB escape from this regulatory pathway, resulting in their fusion with the cell membrane and the release of mtDAMP-containing vesicles as exosomes (Fig. 2). These circulating exosomes can release mtDAMPs into extracellular fluids or deliver them into neuronal and glial cells via membrane fusion, endocytosis, and ligand-receptor interactions, thereby activating inflammatory signaling cascades. Additionally, exosomes can be expressed directly on cell surfaces for antigen presentation and immune activation [61,62,63]. Exosomes containing mtDAMPs such as mtDNA, TFAM, Cytc, and mtDNA-RNA have been isolated from various neuropathological conditions [63, 64]. Elevated levels of mitochondrial protein-containing exosomes have been detected in the serum of geriatric PD patients [65]. Notably, 148 mitochondria-related proteins were detected in neuron-derived exosomes from AD patients, and 279 mitochondrial proteins, with a high proportion of cardiolipin, were found in exosomes derived from the brains of healthy mice [63, 66, 67]. Collectively, these studies highlight exosomes as major drivers of mtDAMP spread and neuroinflammation.

Transfer and spread of mtDAMPs across biological compartments. Mitochondrial leakage releases mtDAMPs into the cytosol, triggering inflammatory cascades that result in cell membrane rupture and necrosis. This process releases cellular contents, including mtDAMPs and inflammatory proteins, into the extracellular space. Both mtDAMPs and inflammatory proteins can be transferred to adjacent healthy cells via intercellular gap junctions and to distant cells via MVs. It remains unclear whether these cytotoxic proteins are transferred via TNTs or whether mtDAMPs are exchanged between mitochondria through mtNTs. Additionally, dysfunctional intact mitochondria release mtDAMPs within MDVs, which subsequently fuse with MVBs. Cytosolic mtDAMPs and activated signaling proteins also enter MVBs via endocytosis. Increased numbers of MVBs escape lysosomal fusion and instead fuse with the cell membrane, leading to the release of vesicles containing mtDAMPs and activated signaling proteins as exosomes into the extracellular space. These circulating exosomes deliver their contents to other cells through several mechanisms, including fusion, endocytosis, and ligand‒receptor interactions. Extracellular mtDAMPs can exit the brain parenchyma and enter other biological fluids, spreading pathology. mtDAMPs-mediated neuroinflammation weakens both the ependymal wall and the BBB, facilitating the transfer of mtDAMPs and activated signaling proteins from brain interstitial fluids to the CSF and blood
Mitochondrial nanotunnels (mtNTs) represent another mode of mitochondrial communication. However, it remains unclear whether damaged proteins or mtDAMPs are exchanged via this route to propagate mitochondrial stress or dysfunction [68, 69]. Some studies suggest that entire mitochondria can be transferred between cells via microvesicles (MVs), similar to exosomes [70]. The transfer of healthy mitochondria between cells by tunneling nanotubes (TNTs) is well documented and is known to improve cell viability and functionality. However, the potential exchange of damaged mitochondria through TNTs, which contributes to neuroinflammation and neurodegeneration, warrants further investigation [71]. Recent reports indicate that dysfunctional mitochondria and mtDAMP-mediated inflammatory signaling proteins, such as connexin 36 and 43, can be transferred from damaged to healthy cells through intercellular gap junctions [72, 73] (Fig. 2). Further research in these areas will enhance our understanding of mtDAMP spread and associated pathologies.
mtDAMPs Mediated Inflammatory Cascades
Extracellular mtDAMPs are recognized by PRRs, which can be located on the surface, within endosomes, or in the cytosol of immune cells. To access endosomal or cytosolic PRRs, mtDAMPs enter immune cells through various mechanisms, including passive diffusion, active transport, receptor-mediated endocytosis, and phagocytosis. Once inside immune cells, mtDAMPs bind to and activate specific PRRs, initiating inflammatory signaling cascades that alter gene expression and immune cell phenotypes. Activated inflammatory immune cells secrete a variety of substances, such as cytokines and chemokines, which facilitate immune cell survival, differentiation, proliferation, and recruitment to the site of injury. These secreted substances are cytotoxic, causing damage to cellular components and promoting cell death. Additionally, damaged or dead cells release DAMPs, which further activate inflammatory immune cells, perpetuating the inflammatory cycle. Recent evidence suggests that mtDAMPs can activate CNS-resident immune cells, including microglia and astrocytes, by stimulating several signaling pathways, such as TLRs, RAGE, Inflammasomes, cGAS-STING, RIG-I/MDA5-MAVS, FPRs, and RIPK1-RPK3-MLKL [74, 75]. Activation of these pathways may lead to neuroinflammation and neurodegeneration.
Toll-like Receptors
The TLR family comprises several isoforms, with TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 expressed on the cell surface, while TLR3, TLR7, TLR8, and TLR9 are located within the endosomes of neuronal and non-neuronal cells. Their expression levels are elevated under neuroinflammatory conditions [76]. Each TLR isoform is specialized for detecting specific DAMPs. For example, TLR4 recognizes mitochondrial (mt)ROS and recruits the adaptor protein myeloid differentiation primary response protein 88 (MyD88). In mouse macrophages, MyD88 post-transcriptionally primes NLRP3 via its deubiquitination [77]. Exogenous or inducible H2O2 in immune cells has also been reported to activate NFκB and mitogen-activated protein kinase (MAPK) family proteins, such as extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38, in a MyD88-dependent manner. This activation leads to immune cell activation and the production of proinflammatory cytokines, ultimately resulting in neuroinflammation and neuronal death [78]. Extracellular Cytc has been shown to activate microglia via the TLR4 and JNK signaling cascades. These activated microglia secrete nitric oxide (NO), enhancing the cytotoxicity of monocytes, while inhibition of JNK reduces NO secretion from microglia [79]. Additionally, extracellular Cytc activates astrocytes through TLR4, promoting the secretion of granulocyte–macrophage colony-stimulating factor (GM-CSF), interleukin (IL)1β, and IL12 (Fig. 3). These activated astrocytes are highly toxic to human neuroblastoma cells, and inhibition of TLR4 in astrocytes blocks the secretion of GM-CSF and IL1β [80]. Therefore, inhibition of TLR4 may attenuate Cytc-induced neuroinflammation and neuronal death in neurodegenerative disorders, although in vivo studies are required for further clarification. Intra-articular injection of recombinant Cytc has been shown to promote synovitis in mice by increasing the activation and infiltration of neutrophils and monocytes via NFκB [81]. Extracellular ATP also acts as a DAMP, activating microglia through the TLR4-NLRP3 pathway and leading to increased secretion of proinflammatory cytokines such as IL1β, ultimately promoting neuroinflammation [82]. Extracellular heme, which is primarily derived from red blood cells with a small amount from mitochondria, is another ligand for TLR4. Extracellular heme activates microglia through the TLR4-MyD88/TRIF pathways, resulting in increased production of proinflammatory cytokines (TNFα, IL1β, and IL6), likely due to NFκB activation, leading to neuroinflammation and neuronal injury. Additionally, inhibition of extracellular heme release attenuates microglial activation and associated inflammatory neuronal injury [83, 84].

Effect of mtDAMPs on TLR and RAGE signaling. Extracellular mtDAMPs such as mtROS, Cytc, ATP, heme, and cardiolipin activate TLR4 expressed on glial cells, facilitating MyD88-mediated IRF7, MAPK, and NFκB signaling. Extracellular mtDNA also initiates the same inflammatory signaling pathway by binding to TLR9. Ox-mtDNA binds to TLR9 and promotes IRF1-mediated shelf synthesis, further enhancing TLR9 activation and inflammation. Additionally, mtdsRNA activates TLR3 and drives TRIF-mediated IRF3 activation. Both IRF3 and IRF7 promote IFN-I expression, facilitating antiviral signaling. Furthermore, heme, TFAM, and mtDNA activate RAGE, leading to the nuclear translocation of AP1 and NFκB. AP1 promotes the expression of proteins involved in immune cell survival, differentiation, proliferation, and apoptosis. Moreover, NFκB increases the expression of proinflammatory cytokines and chemokines, which recruit additional immune cells and further facilitate inflammation and cell death. Abbreviations: TRIF, TIR domain-containing adapter-inducing IFNβ; TRAF, TNF receptor-associated factor; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule
Interestingly, extracellular cardiolipin has been found to modulate the activity of microglia, reducing their neurotoxic and neuroinflammatory effects, such as the production of TNFα and NO, while enhancing their phagocytic activity, potentially through a TLR4-dependent mechanism. Cardiolipin also stimulates microglia to secrete various chemokines, including monocyte chemoattractant protein (MCP)1 and interferon gamma-induced protein (IP)10. Additionally, extracellular cardiolipin promotes the expression of neurotrophic factors in microglia, facilitating neuronal healing and differentiation following injury [85, 86]. However, conflicting evidence suggests that cardiolipin-TLR4-mediated activation of microglia can also increase NO production [86]. Cardiolipin has similar effects on astrocytes. It enhances the phagocytic activity of astrocytes, suppresses TLR4 expression, and facilitates TLR4-mediated MCP1 production. Additionally, cardiolipin shifts astrocytes from producing cytotoxins in response to lipopolysaccharide (LPS) to secreting IL1β. It also suppresses LPS-induced expression of glial fibrillary acidic protein (GFAP) in astrocytes [57]. Further studies, particularly in in vivo models, are needed to fully understand the role of extracellular cardiolipin in neuroinflammation and its potential therapeutic implications.
mtDNA, like bacterial DNA, is hypomethylated or unmethylated at cytosine–phosphate–guanine (CpG) motifs, rendering it highly immunogenic [87]. In the extracellular space, mtDNA can be found either naked or bound to proteins such as TFAM. Naked mtDNA is directly recognized by TLR9, while protein-bound mtDNA is engulfed by immune cells, subsequently detached from proteins, and then recognized in endosomes by TLR9. Once bound to mtDNA, TLR9 recruits MyD88, leading to the activation of the MAPK and NFκB signaling pathways and the secretion of proinflammatory cytokines, such as TNFα, IL1β, and IL6 [88,89,90]. Alternatively, TLR9-MyD88-mediated activation of interferon regulatory factor (IRF)7 can increase the secretion of type I interferons (IFN-I) [91]. Through this mechanism, mtDNA activates various immune cells, including neutrophils, dendritic cells, natural killer cells, and B cells; however, its activity in microglia and astrocytes remains to be elucidated [88, 89, 92]. The oxidation of mitochondrial polynucleotides in astrocytes leads to their activation and the production of MCP1, TNFα, IL1β, and IL6 [93]. Oxidized (Ox)-mtDNA binds to TLR9, causing MyD88/TRIF-mediated activation of IRF1. IRF1 upregulates the expression of mitochondrial UMP-CMP kinase 2, which synthesizes additional copies of Ox-mtDNA, further activating immune cells, including macrophages [94] (Fig. 3). Oxidized mtDNA induces a robust immune response and inflammation [94, 95]. Moreover, extracellular dsRNA has been reported to activate astrocytes by binding to TLR3, facilitating the release of IFN-I. This activation leads to the attenuation of neurite outgrowth and impairment of memory functions during brain development [96]. Whether mtdsRNAs induce similar effects remains unknown.
RAGE
RAGE is widely expressed in CNS resident cells, including microglia, astrocytes, and neurons. Upon activation, RAGE triggers intracellular signaling pathways involving MAPKs and NFκB, leading to the production of chemokines and cytokines, similar to TLRs. Recent evidence suggests that RAGE recognizes extracellular heme, resulting in the production of proinflammatory cytokines, such as TNFα and IL1β, via the ERK1/2 and Akt signaling pathways [97]. Another ligand for RAGE, TFAM, has been shown to activate microglia and microglia-like cells through the JNK signaling pathway. These activated immune cells secrete IL1β, IL6, and IL8, which are toxic to neuroblastoma cells [98] (Fig. 3). Injection of TFAM into the cisterna magna of rats increased the expression of TNFα, IL1β, IL6, and MCP1 in the hippocampus and frontal cortex, possibly by enhancing NFκB activity. Inhibition of RAGE reduced the secretion of MCP1, suggesting that TFAM may also be recognized by other PRRs [99]. Further studies revealed that TFAM augments the production of IFN-I induced by mtDNA via RAGE and TLR9-mediated activation of the phosphoinositide 3-kinase (PI3K), ERK, and NFκB signaling pathways [89]. Overall, RAGE plays a crucial role in mtDAMP-mediated neuroinflammation and neurodegeneration. Thus, inhibiting this receptor could potentially attenuate these pathologies.
Inflammasomes
NLRs are a group of cytosolic sensors for DAMPs. This family includes NOD, leucine-rich repeat (LRR) pyrin domain-containing 3 (NLRP3), NLR caspase activation and recruitment domain (CARD)-containing 4 (NLRC4), and absent in melanoma 2 (AIM2). Among these, NLRP3 is particularly significant, as it detects a broad range of DAMPs and PAMPs and plays a crucial role in several inflammatory diseases, including neuroinflammation and neurodegenerative disorders. NLRP3 is expressed in neurons and immune cells, such as microglia and astrocytes. Upon binding to DAMPs or PAMPs, NLRP3 undergoes activation and oligomerization with its adaptor protein, apoptosis-associated speck-like protein containing a CARD (ASC). The activated ASC then recruits the effector protein pro-caspase 1, leading to its cleavage into the p10 and p20 subunits. These subunits dimerize to form an active tetramer known as caspase 1. This highly organized cytosolic multiprotein complex, comprising sensor, adaptor, and effector proteins, is termed the NLRP3 inflammasome. Caspase 1 cleaves inactive pro-IL1β and pro-IL18 into their mature active forms, IL1β and IL18, respectively. Additionally, caspase 1 cleaves gasdermin D (GSDMD) into its N-terminal (GSDMD-N) and C-terminal (GSDMD-C) fragments. GSDMD-N self-oligomerizes to form pores in the cell plasma membrane, facilitating the release of cytosolic contents, including IL1β, IL18, and potassium ions (K+), into the extracellular space. A decrease in cytosolic K+ further enhances NLRP3 activation, while increased extracellular IL1β and IL18 levels promote immune activation and neuroinflammation. The GSDMD pores also allow extracellular fluids to enter the cytosol, causing cell swelling, membrane rupture, and cell death, a process known as pyroptosis [100, 101].
Several mtDAMPs have been shown to activate the NLRP3 inflammasome, thereby triggering inflammation-mediated cell death. For instance, the upregulation of mtROS via the inhibition of mitochondrial respiratory chain complexes I and III activates the NLRP3 inflammasome. Conversely, preventing mtROS release by blocking VDAC inhibits NLRP3 inflammasome activation [102]. Mechanistically, mtROS disrupts the binding of thioredoxin-interacting protein (TXNIP) to the antioxidant thioredoxin (TRX)2. Free TXNIP subsequently binds to and activates NLRP3, leading to inflammasome formation [103, 104] (Fig. 4). Additionally, cytosolic TRX1 has been reported to activate the NLRP3 inflammasome and promote IL1β secretion independently of TXNIP [105]. The released IL1β contributes to ROS accumulation and related pathologies by disrupting intracellular antioxidants such as superoxide dismutase (SOD) and catalase and by recruiting other inflammatory immune cells, thereby forming a continuous inflammatory loop (ROS-NLRP3-ROS). Furthermore, ROS enhance NLRP3 priming by promoting NFκB-mediated nuclear transcription [77, 106]. Inhibition of TXNIP has been shown to prevent mtROS-mediated NLRP3 inflammasome activation. Similarly, mitochondria-targeting antioxidants attenuate NLRP3 activation and subsequent IL1β secretion by reducing ROS levels [103]. ATP is another mtDAMP that can be passively released into the extracellular milieu from damaged or dead cells or actively transferred from activated cells via vesicles or channels such as pannexin 1, connexin, volume-regulated anion, and maxi-anion channels [107]. Its target receptors, purinergic receptors (P1 and P2), are abundantly expressed in neuronal and glial cells, including microglia, astrocytes, and oligodendrocytes. Extracellular ATP binds to several isoforms of ionotropic P2X and metabotropic P2Y receptors, with a particularly high affinity for the P2X7 isoform under neuroinflammatory conditions. Activation of the P2X7 ion channel leads to the exchange of intracellular K+ for extracellular Na+ and Ca2+ as well as for larger organic molecules, causing depolarization of glial cells [108]. A reduced cytosolic K+ concentration in glial cells activates the NLRP3 inflammasome, while Ca2+ overload exacerbates NLRP3 activity by upregulating ROS-generating enzymes such as inducible (i)NOS and NOX. Both ions compromise mitochondrial health [109,110,111]. ATP stimulation also induces the expression of mitochondrial antiviral signaling protein (MAVS), which recruits and activates the NLRP3 inflammasome in immune cells, leading to IL1β secretion [112]. Extracellular ATP activates NFκB, possibly through ROS production, resulting in the release of IL6, IL12, and IFNγ [113]. This activation promotes the activation, proliferation, differentiation, and trafficking of immune cells, including microglia, astrocytes, and oligodendrocytes [114]. By producing several toxic molecules, such as reactive oxygen and nitrogen species, proteases, proinflammatory cytokines, and chemokines, these activated immune cells contribute to various neuroinflammatory and neurodegenerative disorders. Blocking P2X7 ion channels has been shown to attenuate these pathologies [63].

Role of mtDAMPs in NLRP3 inflammasome activation and pyroptosis. TLR- and ROS-mediated nuclear translocation of NFκB increases the expression of NLRP3 and related proteins. The TLR adapter protein MyD88 also prevents the post-translational degradation of NLRP3 via ubiquitination. Various mtDAMPs, such as heme, mtDNA, and cardiolipin, bind to NLRP3, facilitating the formation of the NLRP3 inflammasome and activation of caspase 1. Caspase 1 cleaves pro-IL1β and pro-IL18, forming active IL1β and IL18, as well as generating GSDMD-N. In addition to caspase 1, GSDMD-N is also produced by caspase 4 and 5, which can be activated by heme and mtDNA. GSDMD-N aggregates and forms pores in both mitochondrial and cell membranes, facilitating the release of mtDAMPs, IL1β, and IL18 into the extracellular space. Additionally, GSDMD pores allow extracellular fluid to enter the cytosol, causing cell swelling and subsequent cell membrane rupture and the release of DAMPs into the extracellular space. Released DAMPs further promote NLRP3 expression and activation via TLR-NFκB signaling. Moreover, caspase 4 and 5 promote ATP release into the extracellular milieu by activating pannexin 1 channels. Extracellular ATP activates P2X7 receptors, reducing intracellular K+ and increasing Ca2+ concentrations. Increased intracellular Ca2+ promotes MAVS expression, which further reduces intracellular K+ levels, leading to activation of the NLRP3 inflammasome. Furthermore, mitochondrial Ca2+ overload enhances ROS accumulation, which disrupts the inhibition of TXNIP, further driving NLRP3 inflammasome formation, inflammation, and cell death
Extracellular heme has been reported to activate the NLRP3 inflammasome, leading to caspase 1-mediated production of IL1β by immune cells. Additionally, heme activates caspases 4 and 5, further enhancing IL1β secretion [115]. Cardiolipin, another mtDAMP, binds to NLRP3, facilitating inflammasome assembly and subsequent immune activation and neuroinflammation [63, 116]. Similarly, released mtDNA activates the NLRP3 inflammasome, resulting in the production of IL1β and IL18 [117]. Like ATP, mtDNA triggers caspase 1 activation through NLRP3 inflammasome assembly and can directly activate caspases 4 and 5 in its absence [118] (Fig. 4). Studies have demonstrated that a deficiency in autophagic proteins, such as LC3B and Beclin 1, leads to the accumulation of mtDNA and subsequent NLRP3 activation, resulting in increased secretion of IL1β and IL18 [119]. Moreover, compared with its non-oxidized form, oxidized or newly synthesized Ox-mtDNA has a greater affinity for NLRP3 activation. Activated NLRP3 also prevents the enzymatic degradation of released mtDNA in the cytosol, further exacerbating inflammation and cell death [94, 120]. Conversely, maintaining autophagy, deleting mtDNA, or inhibiting the synthesis of Ox-mtDNA has been shown to prevent NLRP3 activation and the secretion of proinflammatory cytokines [94, 121]. In addition to NLRP3 activation, extracellular mtDNA also activates other inflammasomes, such as NLRC4 and AIM2 [122, 123]. Unlike NLRP3 and NLRC4, which preferentially recognize Ox-mtDNA fragments, AIM2 predominantly senses large non-oxidized mtDNA fragments [94, 120, 122]. Furthermore, H2O2-induced damaged mtDNA and Ox-mtDNA activate astrocytes, leading to the production of proinflammatory cytokines such as TNFα, IL1β, IL6, and MCP1, thereby contributing to neuroinflammatory pathologies. The underlying mechanisms may involve the activation of the NFκB and inflammasome pathways [93]. Extracellular mtdsRNA can also activate NLRP3 directly or via TLR3, although this has yet to be fully elucidated [96].
The NLRP3 inflammasome also attenuates mitophagy via partially caspase 1-mediated Parkin cleavage, augmenting the release of mtDAMPs [124]. Additionally, the caspase-mediated active forms of GSDMD and GSDME perforate the MOM and MIM, facilitating the further release of mtDAMPs into the cytosol. GSDMA3/GSDMD form pores of 28–40 nm in the MOM and 10–18 nm in the MIM, allowing the release of mtDAMPs, including mtDNA fragments but not nucleoids, into the cytosol. Consequently, mtDAMPs-mediated activation of inflammasomes creates an inflammatory feedback loop that enhances inflammasome activity, potentially exacerbating neuroinflammation and neurodegeneration [24, 125, 126]. Further research is needed to better understand mtDAMPs-mediated inflammasome signaling cascades in specific neurodegenerative animal models.
cGAS-STING
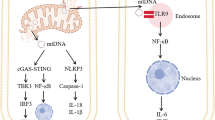
Cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS) is a cytosolic nucleic acid sensor that induces antiviral immune responses upon activation. In addition to its presence in the cytosol, cGAS has also been localized to the nucleus and the plasma membrane [127, 128]. It is constitutively expressed in neurons and glial cells, including microglia and astrocytes. cGAS has been shown to recognize both cytosolic and extracellular mtDNA [129]. Extracellular mtDNA, released from damaged or dying neuronal cells, can enter glial cells via endocytosis and subsequently be released into the cytosol. mtDNA binds to the long α-helix site of cGAS, causing a conformational change and activation of its enzymatic site [88, 130]. The sugar-phosphate backbone of mtDNA, rather than the nitrogenous bases, is responsible for detection by cGAS. Activated cGAS facilitates the binding of ATP and GTP to its enzymatic site, catalyzing the production of cyclic GMP-AMP (cGAMP), a second messenger [130, 131]. Notably, mtDNA fragments ≥ 18 base pairs in length or U-shaped/bent mtDNA (due to the presence of TFAM or other proteins) enhance the activation and catalytic activity of cGAS by promoting dimerization of the mtDNA-cGAS complex (2:2 mtDNA:cGAS). Moreover, dsDNA ≥ 40 base pairs in length can form a highly stable oligomeric complex comprising dsDNA helices bound to multiple copies of cGAS, further enhancing its catalytic activity. Collectively, these findings indicate that cGAS activation and cGAMP production depend on mtDNA length, not sequence [132,133,134]. cGAMP binds to and induces conformational changes in the ER-resident transmembrane protein stimulator of interferon genes (STING). This results in the tetramerization and translocation of STING to the ER-Golgi intermediate compartment (ERGIC) or the Golgi apparatus. The oligomeric assembly of STING recruits TANK-binding kinase (TBK)1 to its highly conserved C-terminal tail (CTT) motif, promoting TBK1 auto-transphosphorylation and activation [135, 136]. Activated TBK1 phosphorylates the CTT motif and recruits interferon regulatory factor (IRF)3 for phosphorylation. Phosphorylated IRF3 homodimerizes and translocates to the nucleus, where it induces the expression of IFN-I, including the α and β isoforms. Released IFN-I binds to its heterodimeric receptor, triggering the expression of interferon-stimulated genes and driving antiviral responses [137, 138]. Additionally, STING or TBK1 phosphorylates and activates the inhibitor of NFκB (IκB) kinase (IKK), leading to the proteasomal degradation of IκB and subsequent translocation of NFκB from the cytosol to the nucleus. This triggers the expression of proinflammatory cytokines such as TNFα, IL1β, and IL6, ultimately provoking inflammation-mediated cell death [129, 139] (Fig. 5).

mtDAMPs drive cGAS-STING, RIG-I/MDA5-MAVS, and RIPK1/RIPK3/MLKL signaling. mtDNA is recognized by cGAS, which cleaves GTP and ATP to form cGAMP. cGAMP acts as a second messenger, binding to the ER-resident protein STING. Activated STING tetramerizes (only three STING molecules are shown in the figure for simplicity) and translocates to the ERGIC or Golgi body. STING recruits TBK1 to its CTT motif, initiating TBK1 auto-transphosphorylation and activation. Activated TBK1 phosphorylates the CTT motif of STING, which subsequently recruits and phosphorylates IRF3. Phosphorylated IRF3 homodimerizes and translocates into the nucleus, leading to increased IFN-I expression. Additionally, TBK1 or STING phosphorylates IKK, facilitating NFκB nuclear translocation and the expression of proinflammatory cytokines. STING also upregulates the expression of RIPK3, while mtROS stimulate the autophosphorylation and activation of RIPK1, promoting necroptosis, DAMP release, and inflammation. Furthermore, RIG-I and MDA5 upon detecting mtROS and mtdsRNA, expose their CARD dimers to interact with the MAVS CARD. These interactions form MAVS CARD filaments, which recruit and activate TBK1 and IKKε, driving IRF3-, IRF7-, and NFκB-mediated expression of IFN-I and proinflammatory cytokines. Conversely, the NLRP3 inflammasome suppresses cGAS-STING signaling by promoting caspase 1-mediated cGAS degradation
Studies have found that mtDNA released into the circulation causes systemic inflammation and is linked to patient mortality [140, 141]. Stress or injury to mitochondria can release mtDNA, subsequently activating the cGAS-STING signaling pathway. mtDNA-mediated activation of the cGAS-STING pathway increases receptor-interacting protein kinase (RIPK)3 levels, promoting interferon and TNFα signaling and facilitating necroptosis [142]. Conversely, the NLRP3 inflammasome has been reported to prevent the activation of the cGAS-STING pathway by degrading cGAS via caspase 1 and promoting K+ efflux through GSDMD pores, leading to a reduction in IFN-I production [143, 144]. Blocking caspase 1 activity has been shown to reactivate the cGAS-STING pathway, which, interestingly, promotes inflammasome activation and IL1β secretion. This indicates that the cGAS-STING and inflammasome pathways regulate each other to prevent excessive and uncontrolled inflammation [145]. Further studies are required to better understand these mechanisms.
RIG-I/MDA5-MAVS
Retinoic acid-inducible gene (RIG)-I and melanoma differentiation-associated protein (MDA)5 are key RLRs, a group of cytosolic nucleic acid sensors that initiate antiviral immune responses and inflammation upon activation. Mechanistically, the binding of dsRNA to RIG-I or MDA5 induces conformational changes that expose their N-terminal CARDs. These CARD dimers oligomerize, becoming active and interacting with the CARD-containing adaptor protein MAVS, which is anchored in the MOM, mitochondrial-associated membranes, and peroxisomes. The noncovalent interaction between RIG-I/MDA5-2CARD and CARD-MAVS results in the formation of filaments, activating MAVS. Activated MAVS subsequently recruits and activates TBK1 and IKKε. TBK1 phosphorylates IRF3, while IKKε phosphorylates both IRF3 and IRF7, facilitating their translocation into the nucleus to promote the expression of IFN-I. Additionally, IKKε activates NFκB by phosphorylating and degrading IκB, leading to the expression of other inflammatory proteins [146, 147] (Fig. 5). mtdsRNA activates RIG-I and MDA5 receptors, leading to increased IFN-I expression [148, 149]. Normally, mtdsRNA is synthesized from mtDNA and rapidly cleared by the mitochondrial RNA degradosome, which contains the dimeric helicase Suv3 and trimeric polynucleotide phosphorylase. Mutation or inhibition of the RNA degradosome results in the accumulation of mtdsRNA, which is subsequently released into the cytoplasm, triggering the activation of the RIG-I and MDA5 receptors. Silencing Bax and Bak has been shown to attenuate IFN-I responses, indicating that Bax/Bak-mediated MOMP is responsible for the release of mtdsRNA into the cytoplasm [94, 149]. Additionally, mtROS has been found to facilitate the RIG-I-MAVS-IRF3 signaling pathway, leading to the induction of IFN-I responses [150]. However, the roles of mtdsRNA or mtROS in the activation of the RIG-I/MDA5-MAVS pathway in neuroinflammation and neurodegenerative disorders remain to be elucidated.
Formyl Peptide Receptors
FPRs are a group of G protein-coupled chemoattractant receptors predominantly expressed in phagocytic immune cells such as microglia, with lower expression in astrocytes and neurons [151]. FPRs recognize extracellular NFPs, leading to the activation and recruitment of immune cells to sites of injury. This process results in the secretion of inflammatory proteins and promotes cellular damage and death [151, 152]. Under normal conditions, NFPs reside in the mitochondrial matrix, but pathological conditions such as trauma or injury facilitate their release into the extracellular milieu [153]. Studies have reported that mitochondria-derived NFPs activate neutrophils and recruit them to injury sites, where they release ROS and IL8. This action is mediated via FPR1 activation, which subsequently triggers the activity of p38 MAPK and ERK1/2 [154]. Additionally, axonal mitochondria-derived NFPs have been shown to trigger inflammatory responses in Schwann cells following nerve injury, which is partially mediated by FPR2 expression [155]. NFPs also induce the FPR-mediated release of lysosomal enzymes, ROS, and proinflammatory cytokines from monocytes. Furthermore, the activation of FPR2 expressed on microglia induces pathological responses in the CNS similar to those observed in peripheral immune cells. Activated microglia secrete ROS, complement proteins, and proinflammatory cytokines, leading to neuroinflammation and neuronal death [156]. Therefore, blocking FPRs or preventing the release of NFPs from damaged mitochondria could be a potential therapeutic approach for treating neuroinflammation and neurodegeneration.
Others
mtDAMPs have been reported to induce inflammation and cell death by activating necroptosis and hypoxia-inducible factor (HIF)1α. Necroptosis, a regulated form of necrosis, can be triggered by the activation of various extracellular and intracellular PRRs and facilitated by the inhibition of caspase 8. This process leads to the autophosphorylation and activation of RIPK1, which subsequently binds and phosphorylates RIPK3. The complex formed by phosphorylated RIPK1 and RIPK3, known as the necrosome, further phosphorylates and activates mixed lineage kinase domain-like pseudokinase (MLKL). Phosphorylated MLKL oligomerizes and forms pores in the plasma membrane, leading to cell death and the release of cytosolic contents into the extracellular space. These released cytosolic DAMPs activate and recruit immune cells, thereby facilitating inflammation and cell death. Several studies have identified necroptosis as a major driver of neuroinflammation in neurodegenerative diseases, including AD and PD [157]. Notably, mtROS can trigger the autophosphorylation and activation of RIPK1, thereby promoting necrosome formation [158] (Fig. 5). However, further studies are needed to clarify the role of mtDAMPs in necrosome formation and their contribution to neuroinflammation and neurodegeneration. Additionally, hypoxia, injury, and inflammation can trigger the release of succinate, an intermediate metabolite of the Krebs cycle, from mitochondria into the cytosol and subsequently into extracellular fluids. Plasma succinate levels have been proposed to be a predictor of mortality in severely injured patients. Extracellular succinate can activate dendritic and other immune cells, possibly through the activation of HIF1α, leading to increased expression of proinflammatory cytokines. Succinate can also upregulate ROS production, thereby facilitating cellular damage and death [159,160,161]. Further studies exploring the release of succinate from neuronal mitochondria and its role in neuroinflammation and neurodegeneration would provide valuable insights.
Regulations of mtDAMP Release and Inflammatory Cascades
Several endogenous mechanisms, such as mitophagy, apoptosis, and oxeiptosis, prevent the release of mtDAMPs into the extracellular milieu. Mitophagy selectively removes damaged or dysfunctional mitochondria from the cytosol through an autophagic process. The key proteins involved in this process include PTEN-induced kinase (PINK)1, Parkin, and microtubule-associated protein 1 light chain (LC)3. Sublethal MOMP triggers mitophagy, leading to the elimination of permeabilized and defective mitochondria via autophagolysosomal degradation [162, 163]. This process prevents the release of mtDAMPs, either directly or through MDVs, thereby inhibiting the activation of PRRs such as TLR9, the NLRP3 inflammasome, and cGAS, and reducing the production of proinflammatory cytokines, including IFNβ [163,164,165,166]. Suppression of mitophagy/autophagy proteins, such as beclin 1, LC3B, PINK1, and Parkin, leads to the accumulation of defective mitochondria and mtDAMPs (such as mtROS and mtDNA), which activate the NLRP3 inflammasome and result in the production of proinflammatory cytokines, including IL1β and IL18 [124, 165, 167, 168]. To maintain cellular homeostasis, activated PRRs have been shown to prevent further immune stimulation and inflammation by activating mitophagy. Notably, the inflammasome, via engagement of NFκB, has been found to activate Parkin-mediated mitophagy to remove permeabilized mitochondria [166]. Parkin also ubiquitinates and inactivates BAK1 after MOMP to restore mitochondrial health and cellular homeostasis [162]. Similarly, cGAS promotes mitophagy by activating TBK1 and IKKα [169, 170]. However, persistent cellular stress or disease can compromise the mitophagy quality-control pathway, allowing defective mitochondria to release excess mtDAMPs into the cytosol. This leads to the activation of neuroinflammatory cascades and neuronal death [3].
Apoptosis is another protective mechanism akin to mitophagy, operating at the cellular level rather than solely at the mitochondrial level. It encapsulates cellular contents in apoptotic bodies, allowing their rapid degradation by phagocytic immune cells, a process known as efferocytosis. This mechanism prevents the release of mtDAMPs into the extracellular space, thereby preventing immune activation and inflammation [171, 172]. Failure to clear apoptotic bodies can lead to a switch to necrotic cell death, which releases immunogenic mtDAMPs into the extracellular space [173]. Apoptotic cells also release various immunosuppressive molecules, such as IL10, transforming growth factor β, and prostaglandin E2, creating an anti-inflammatory microenvironment that prevents undue inflammation [174]. Following widespread MOMP, caspases rapidly cleave and inactivate various activated inflammatory signaling proteins, further preventing the initiation and progression of inflammation. For example, caspases block IFN-I responses and NFκB signaling by directly cleaving and inactivating cGAS, MAVS, IRF3, NFκB essential modulator (NEMO), and IKKβ [175, 176]. Additionally, caspase 3 and caspase 7 cleave and inactivate IL33 [177]. Caspases also inhibit immunogenic protein synthesis by cleaving initiation factors such as eIF2α, eIF2B, and eIF4G [178]. Blocking caspase activity after MOMP has been shown to enhance IFN-I responses and NFκB signaling, potentially leading to increased inflammation and immunogenic cell death [31, 175, 179]. However, mice lacking caspase 3 or Apaf1 survive to adulthood without a hyperinflammatory phenotype, indicating that MOMP engages other mechanisms in addition to caspases to prevent inflammation [180, 181]. For instance, following MOMP, PNPT1, an IMS resident protein, is released and degrades global mRNA, including inflammatory transcripts [182]. In contrast to widespread MOMP, caspase activity is generally insufficient to degrade inflammatory signaling proteins or induce apoptosis following minor MOMP. Under these conditions, caspases activate DNases, including mitochondrial endonuclease G, causing DNA damage and promoting cell senescence. Accumulated damaged DNA can trigger inflammatory signaling and favor the production of proinflammatory cytokines, including IFN-I. Inhibiting caspase activity following minor MOMP has minimal effect on proinflammatory cytokine production, while other studies have shown improved genomic stability and recovered mitochondrial health, as well as the survival and proliferation of cells, including neurons [183,184,185,186]. However, further studies are needed to clarify the role of caspases in mtDAMP release and inflammation. Recently, a soluble serum protein, leucine-rich α-2 glycoprotein 1, was found to prevent inflammation and cellular death by promoting the phagocytosis of extracellular Cytc [187].
Oxeiptosis is another noninflammatory cell death pathway that prevents the release of cytosolic contents into the extracellular space. Unlike apoptosis, it is not mediated by caspases but depends on the activation of the MIM-resident serine-threonine phosphatase, commonly known as phosphoglycerate mutase (PGAM)5. Normally, PGAM5 is inhibited by kelch-like ECH-associated protein (KEAP)1, which also regulates the activities of nuclear factor erythroid 2-related factor (Nrf)2. KEAP1 acts as a major cytosolic sensor of oxidative stress and becomes oxidized at its C-terminal cysteine residues in a ROS-dependent manner. At low levels of cytosolic ROS, Nrf2 is released from the Nrf2-KEAP1-PGAM5 complex and translocates into the nucleus to promote the expression of cytoprotective antioxidant proteins. In contrast, high levels of accumulated ROS trigger the release of PGAM5 from the KEAP1 tripartite complex. Once released, PGAM5 enters the mitochondrial matrix and dephosphorylates apoptosis-inducing factor mitochondria-associated (AIFM)1 at the Ser116 residue. Dephosphorylated and activated AIFM1 then translocates from the mitochondria to the nucleus, where it causes chromatin condensation and DNA fragmentation, leading to oxeiptosis. Notably, exposure to ozone in PGAM5−/− mice has been reported to induce the production of proinflammatory cytokines, indicating that oxeiptosis negatively regulates ROS-mediated inflammation. This suggests that oxeiptosis plays a crucial role in mitigating inflammation by preventing excessive ROS-induced inflammatory responses [188, 189].
Role of mtDAMPs in Neurodegenerative Disorders
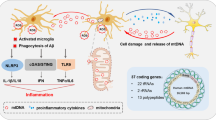
The deterioration of mitochondrial health and functionality in neuronal cells is an inherent aspect of the aging process. Nevertheless, pathological risk factors, including genetic predispositions, proteinopathies, physical injuries, free radical accumulation, and excitotoxicity, precipitate a more rapid decline in mitochondrial structural integrity within these cells. Extensive evidence indicates that compromised mitochondrial health and the subsequent release of mtDAMPs initiate a cascade of inflammatory responses, resulting in the activation of immune cells in the brain. This cascade leads to neuroinflammation, neuronal injury, and neuronal death, thereby contributing to the onset and progression of neurodegenerative disorders such as AD, PD, ALS, MS, and stroke [3, 190] (Table 2).
Alzheimer’s Disease
AD is the most prevalent neurodegenerative disorder and is characterized by impairments in short- and long-term memory, visual perception, language, complex attention, and executive functions. Neuroinflammation is a major pathological hallmark of AD, primarily arising from the accumulation of extracellular amyloid-beta peptide (Aβ) and intracellular hyperphosphorylated (p)-tau protein. The disease typically initiates in the allocortex, including the entorhinal cortex and hippocampus, and progresses to the neocortex, eventually affecting the basal ganglia in the terminal stages. Research has demonstrated that aggregated Aβ and p-tau not only damage neurons but also disrupt mitochondrial integrity [191]. Aβ has been shown to induce the overexpression of VDAC1 in cultured neurons. This pathology is similar to that observed in neurons adjacent to Aβ plaques in quintuple-transgenic familial (5 × F) AD mice. Pharmacological inhibition of VDAC1 has been shown to mitigate neurometabolic dysfunctions, neuroinflammation, neuronal death, and cognitive decline in these mice [192]. Furthermore, Aβ plaques damage microglial mitochondria and trigger microglial activation. Activated microglia communicate with astrocytes by releasing dysfunctional and fragmented mitochondrial components into the extracellular milieu. These mtDAMPs activate reactive astrocytes, triggering neuroinflammation and neurodegeneration. Inhibition of mitochondrial damage through either Fis1 inhibition or mitophagy has been suggested as a neuroprotective strategy against glial cell activation and neuroinflammation [8, 193]. Dysfunctional mitochondria in neuronal cells fail to buffer cytosolic Ca2+, leading to increased Aβ production and p-tau accumulation, further exacerbating AD pathology and cognitive decline. Presenilin 2, a transmembrane protein localized at mitochondria-associated ER membranes, has been shown to increase Ca2+ uptake into mitochondria by activating the mPTP, leading to excessive free radical generation, mitochondrial dysfunction, neuronal death, and impaired learning and memory in AD [194]. The knockdown of MCU in hippocampal neurons ameliorates astrogliosis and the production of proinflammatory cytokines (such as TNFα and IL1β) while restoring mitophagy, synaptic structure, and memory performance in APP/PS1/tau triple-transgenic AD mice [195]. Moreover, activated microglia and astrocytes have been shown to compromise mitochondrial health by reducing the ΔΨm and ATP levels, thus hindering the survival of cortical neurons. MCU knockdown in cortical neurons alleviates glial cell-induced impairments in mitochondrial health and bioenergetics [196]. Further studies in AD patients and mouse models revealed that Aβ accumulation triggers the activation of the NLRP3 inflammasome in microglia, leading to neuroinflammation and cognitive dysfunction. Elevated levels of IL1β have been detected in the serum, CSF, and brain tissue of AD patients [197]. Although it remains unclear whether Aβ directly activates the NLRP3 inflammasome or requires intermediary mechanisms, recent findings suggest that Aβ promotes intracellular Ca2+ overload via the activation of transient receptor potential melastatin 2 channels, impairing mitochondrial quality control, facilitating ROS production, and subsequently activating the NLRP3 inflammasome [198]. Activation of the NLRP3 inflammasome exacerbates Aβ plaque deposition, neuroinflammation, hippocampal neuronal damage, and cognitive deficits, while inhibition of NLRP3 signaling reverses these pathologies [197, 199]. Additionally, ROS have been shown to promote AD pathology through the activation of TLRs, such as TLR4 and TLR9. Preventing mitochondrial dysfunction and reducing ROS levels alleviates TLR4/NFκB signaling-mediated neuroinflammation, neurodegeneration, and memory impairment in AD mouse models [200,201,202].
Furthermore, extracellular Cytc has been shown to activate TLR4 expressed in microglia and astrocytes, contributing to neuroinflammation and neuronal death. Cytc-activated astrocytes have been reported to secrete IL1β, IL12, and GM-CSF via TLR4-mediated pathways, which are toxic to neuronal (SH-SY5Y) cells [80]. Additionally, serum amyloid A, Aβ, and p-tau have been reported to bind TLR4/TLR2, facilitating the priming of the P2X7 receptor on microglia. Upon recognizing extracellular ATP, the P2X7 receptor triggers NLRP3 inflammasome activation, leading to microgliosis, IL1β and TNFα secretion, as well as motor and memory deficits. Pharmacological blockade or reduction of P2X7 receptor expression mitigates microgliosis, IL1β secretion, senile plaque deposition, neuronal loss, motor and memory deficits, and anxious behaviors in both the early and advanced stages of AD [203,204,205,206]. Moreover, the restoration of mitochondrial quality has the potential to alleviate these pathologies, although this area requires further exploration. Cardiolipin, which is released from fragmented mitochondria, has been shown to activate the NLRP3 inflammasome, leading to increased production of IL1β, Aβ, and p-tau in cultured cortical neurons [207]. Conversely, exposure of microglia to Aβ results in the release of cardiolipin, which subsequently acts on neighboring glial cells (such as microglia, astrocytes, and oligodendrocytes) and facilitates Aβ phagocytosis [208]. Another study indicated that extracellular cardiolipin binds to TLR4 expressed on microglia, promoting Aβ phagocytosis. Additionally, extracellular cardiolipin stimulates resting microglia to secrete several cytokines, including MCP1 and interferon gamma-induced protein (IP)10 [86]. Further studies, especially in in vivo models, are necessary to better understand the role of extracellular cardiolipin in AD pathology. AD pathology is also associated with RAGE-immunoreactive microglia. Mechanistically, Aβ has been shown to bind RAGE and activate TXNIP, leading to microglial activation, cytokine secretion, and GFAP release in the hippocampus of 5 × FAD mice. TXNIP also facilitates the transport of Aβ from the cell surface to mitochondria, promoting mitochondrial dysfunction via Drp1 activation and subsequent NLRP3 inflammasome activation and IL1β secretion. Downregulating the RAGE-TXNIP axis ameliorates Aβ-mediated mitochondrial dysfunction, neuroinflammation, and GSDMD activation [209]. However, TFAM can also be released from dysfunctional mitochondria and might aggravate RAGE-mediated pathology and neuroinflammation in AD, although this hypothesis requires further validation in future studies. A recent study revealed that Aβ binds to FPR2, inducing astrocyte activation, which subsequently leads to neuroinflammation, cognitive decline, and increased p-tau levels [210]. Similarly, NFPs released from damaged mitochondria may also act on FPR2 and promote AD-related pathology. Studies have also shown that inflammatory mediators or TLR4 agonists, such as Aβ, increase the expression of FPR2 in microglia [211].
Aβ is also implicated in the activation of the cGAS-STING pathway in neuronal and glial cells, promoting IFN-I-mediated neuroinflammation, Aβ accumulation, and memory decline in AD patients and rodent models [212,213,214]. However, whether Aβ directly activates cGAS or through intermediary processes remains to be elucidated. Reduced levels of ccf-mtDNA in the CSF of asymptomatic patients suggest that mtDNA could serve as a potential biomarker for neuronal damage in AD [215]. Early-stage AD studies have detected ccf-mtDNA peripherally, with higher concentrations reported in the serum of mild AD patients, which is associated with mitochondrial dysfunction, neurodegeneration, and cognitive decline [60]. The efficiency of mtDNA repair decreases with age due to the reduced activity of mitochondrial 8-oxoguanine glycosylase 1, leading to the accumulation of mutated mtDNA in the cytoplasm and extracellular space, which triggers cGAS-STING pathway-mediated inflammation [216]. Postmortem reports have revealed elevated levels of STING in the brain endothelial and neuronal cells of AD patients. Inducing mitochondrial dysfunction in brain endothelial cells with palmitic acid has been shown to increase the amount of cytosolic DNA and the levels of cGAS-STING signaling proteins, including cGAS, TBK1, IRF3, and IFNβ [217]. Neutrophil infiltration into cerebral tissue is another pathological hallmark of AD. Aβ recruits neutrophils into cerebral tissue, promoting neuronal apoptosis and the release of mitochondria and mtDNA into the extracellular space. Together, Aβ and mtDNA synergistically promote neutrophil recruitment into the cerebrum. Infiltrated neutrophils induce neuroinflammation via the mtDNA-STING-NLRP3/IL1β axis. Blocking any protein involved in this signaling pathway alleviates neutrophil migration into cerebral tissue, neuroinflammation, and cognitive decline in AD [218].
In addition to cGAS-STING signaling, mtDNA activates other inflammatory pathways. The presence of ccf-mtDNA in serum has been identified as a significant factor for increasing inflammation, worsening composite gait, impairing cognitive function, and increasing mortality risk in older adults [219]. Elevated ccf-mtDNA levels correlate with increased inflammatory markers such as C-reactive protein, TNFα, and IL6, serving as potential predictive biomarkers for physical decline in older adults. Studies have shown that mtDNA levels begin to increase after the fifth decade of life, with the concentrations of proinflammatory cytokines/chemokines such as TNFα, IL6, and CCL5 being directly proportional to plasma mtDNA levels in elderly individuals [190]. Exposure of microglial (BV2) cells to mitochondrial lysates decreases triggering receptor expressed on myeloid cells (TREM)2 mRNA and increases p38 MAPK phosphorylation and NFκB translocation to the nucleus, leading to increased mRNA levels of TNFα, IL8, and matrix metalloproteinase (MMP)8. Similarly, mitochondrial lysates decreased IκBα protein levels and increased NFκB protein, TNFα mRNA, and amyloid precursor protein (APP) and mRNA levels in SH-SY5Y cells. Mitochondrial lysates lacking mtDNA fail to induce these pathological changes, confirming that mtDNA is a major driver of neuroinflammation and neurodegeneration in AD [220]. Some evidence suggests that, in addition to mtDNA, other mtDAMPs increase the mRNA and protein expression of APP and Aβ in the brain. Stereotactic injection of mitochondrial lysates or mtDNA into the hippocampi of C57BL/6 mice reduces TREM2 mRNA and elevates GFAP protein and TNFα mRNA in the hippocampus, as well as increases NFκB phosphorylation in the cortex. mtDNA, but not mitochondrial lysates, increases the expression of colony-stimulating factor 1 receptor protein and Akt phosphorylation in the cortex. Conversely, mitochondrial lysates, but not mtDNA, upregulate the mRNA and protein expression of APP and Aβ in the brain. Overall, these studies indicate that damaged mitochondria drive neuroinflammation and contribute to the progression of AD pathology [221].
Recent findings by Ochoa et al. (2023) demonstrated increased expression of dsRNA and dsRNA-sensing machinery in the brains of tau transgenic mice, as well as in astrocytes isolated from postmortem AD patient brains. Tau aggregates were shown to induce heterochromatin decondensation, leading to the activation of retrotransposons that form dsRNA, which subsequently triggers dsRNA-mediated neuroinflammation and neurodegeneration. It is also hypothesized that tau aggregates may alter mitochondrial integrity, driving mtdsRNA-mediated neuroinflammation and neurodegeneration in AD [222]. Further studies have reported that dsRNA with a high molecular weight (1–6 kb), compared to dsRNA with a low molecular weight (< 0.5 kb), induces more robust sickness behavior, the secretion of proinflammatory cytokines such as IL1β, IL6, and IFN-I, and working memory deficits in both young and aged mice. These pathologies are exacerbated in aged mice compared to younger mice [223]. The acute administration of dsRNA rapidly and persistently activates microglia and astrocytes, as well as NOS and IL1β. This activation results in the deposition of APP, Aβ, and apolipoprotein E, causing hippocampal atrophy and neuron loss in several brain regions, including the hippocampus, thalamus, cortex, and septal nucleus. Administration of the anti-inflammatory drug ibuprofen has been shown to attenuate glial cell activation and neurodegeneration, indicating that dsRNA initiates and progresses AD pathology via neuroinflammation [224]. Further research is required to evaluate the specific contribution of mtdsRNAs to neuroinflammation and AD pathology. Studies should focus on elucidating the mechanisms by which tau aggregates alter mitochondrial integrity and promote mtdsRNA-mediated neuroinflammation, as well as potential therapeutic interventions targeting these pathways to mitigate AD progression.
Parkinson’s Disease
PD is the second most prevalent neurodegenerative disorder and is characterized by motor symptoms such as bradykinesia, hypokinesia, rigidity, resting tremor, and postural instability, as well as nonmotor symptoms such as autonomic dysfunction, sleep abnormalities, depression, and dementia. The principal pathological hallmark is the intraneuronal accumulation of Lewy bodies, which are primarily composed of α-synuclein (α-syn) fibrils. These fibrils contribute to Ca2+ dysregulation, mitochondrial dysfunction, oxidative stress, neuronal damage, and neuroinflammation. Consequently, there is a progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc), leading to reduced dopamine levels in the striatum [225, 226]. Ca2+ release from damaged mitochondria is a potential contributor to neuronal damage, neuroinflammation, and the progression of PD. Familial PD has been associated with increased intracellular Ca2+ in tyrosine hydroxylase (TH)-positive neurons in the SNpc and in microglia in the striatum. This leads to mitochondrial Ca2+ overload, mitochondrial dysfunction, neuronal damage, activation of microglia and astrocytes, neuroinflammation, and death of TH-positive neurons in the SNpc, ultimately impairing motor performance [226, 227]. Mitochondrial Ca2+ overload can also enhance ROS production. α-Syn aggregates have been shown to disrupt mitochondrial functions and elevate ROS levels. Increased ROS can directly damage dopaminergic neurons and activate microglia via the NFκB signaling pathway. The SNpc is rich in microglia, making it highly susceptible to inflammatory attacks. A reduction in mitophagy proteins such as PINK1 and Parkin can activate the NLRP3/Caspase 1/GSDMD axis, exacerbating neuroinflammation and neuronal death [228, 229]. Postmortem analyses of PD patient brains suggest that mitochondrial dysfunction can exacerbate inflammation and neurodegeneration [228]. Inhibition of mtROS via Nrf2 activation or antioxidants has been shown to mitigate microglial and astrocyte activation, NFκB phosphorylation, proinflammatory cytokine release, and dopaminergic neuronal death [230, 231]. Extracellular ATP also contributes to the progression of PD pathology. Overexpression of ATP receptors such as P2X4 and P2X7 in microglia and astrocytes derived from PD rodent models has been observed. The binding of extracellular ATP to these receptors increases IL6 production, resulting in dopaminergic neuronal degeneration in the SNpc [232, 233]. Activation of the P2X4 receptor also triggers NLRP3 inflammasome-mediated neuroinflammation and dopaminergic neurodegeneration in rats [234]. Knockdown of the P2X4 receptor via small interfering (si)RNA has been shown to improve mitophagy, brain-derived neurotrophic factor (BDNF) expression, TH expression, and motor function in rats [235]. Furthermore, extracellular heme, along with ROS, can induce ferroptosis in dopaminergic neurons, promoting PD pathology [236]. The release of cardiolipin into the extracellular space has also been shown to trigger immune activation, driving neuroinflammation, dopaminergic neuronal loss, and motor deficits [237, 238]. PD pathology is linked to RAGE-mediated gliosis, which induces neuroinflammation and α-syn deposition in the SNpc and reduces motor and cognitive functions in Wistar rats. Extracellular TFAM can bind to RAGE and may contribute to the initiation and progression of PD [63, 239].
In PD patients, elevated levels of ccf-mtDNA have been reported in the CSF and serum [240, 241]. Postmortem analyses have revealed elevated STING protein levels in the neuronal and endothelial cells of PD brains [217]. Another study revealed that increased STING protein expression in the SNpc of PD patients was associated with α-syn deposition. These findings indicate that α-syn aggregates cause mtDNA damage through increased nitro-oxidative stress, leading to cGAS-STING-mediated neuroinflammation and dopaminergic neurodegeneration. Similarly, α-syn aggregates have been shown to break dsDNA in microglia and astrocytes, activating TBK1 and STING to produce IFN-I in the striatum, resulting in neuroinflammation, dopaminergic neurodegeneration, α-syn accumulation, and motor deficits in mice [242]. A reduction in mitophagy exacerbates mtDNA dysregulation and neuroinflammation in PD [241, 243]. Compared to idiopathic PD patients, those with PINK1/Parkin mutations exhibit higher serum levels of ccf-mtDNA, C-reactive protein, and IL6. Thus, ccf-mtDNA could serve as a predictive biomarker to distinguish between idiopathic PD and PINK1/Parkin-mutated PD. Additionally, IL6 levels increase with disease progression in PINK1/Parkin-mutated PD patients but not in idiopathic PD patients [241]. Sliter et al. (2018) demonstrated that Parkin−/− mice accumulate mutated mtDNA, and following exhaustive exercise, both Parkin−/− and PINK1−/− mice exhibit a strong inflammatory phenotype, which is mitigated by STING inhibition. Suppression of STING also alleviated dopaminergic neuronal loss in the SNpc and motor defects in aged Parkin−/− mice [167]. However, even with the deletion of STING or IFN-I signaling, damaged mtDNA can still induce and propagate PD-like pathology. For instance, oxidized and mutated mtDNA released from neurons induces neuropsychiatric, motor, and cognitive impairments in IFNβ−/−/IFNAR−/− mice. Neurodegeneration occurs in brain regions distant from the site of mtDNA release, indicating that damaged mtDNA not only induces but also spreads PD-like pathology. The underlying mechanism involves the activation of TLR4/9 and subsequent upregulation of oxidative stress [244]. In addition to mtDNA, dsRNA released from damaged mitochondria can drive PD-like pathology. Stereotaxic injection of polyinosinic:polycytidylic acid (poly I:C), a synthetic analog of dsRNA, into the SNpc of wild-type mice induces microglial activation, resulting in increased α-syn expression and dopaminergic neurodegeneration. Poly I:C is recognized by the Mac-1 receptor, which activates microglia and NOX, leading to the loss of dopaminergic neurons and the emergence of PD-like symptoms in mice [245]. Investigating the role of mitophagy in dsRNA release subsequent to mitochondrial damage in PD models would be a valuable addition to current research.
Amyotrophic Lateral Sclerosis
ALS is another motor neurodegenerative disorder characterized by a progressive loss of voluntary muscle control (paresis), affecting the bulbar, limb, and respiratory muscles while generally sparing sensory and cognitive functions. This disease is associated with mutations in genes such as C9orf72, SOD1, TAR DNA-binding protein (TDP)43, and fused in sarcoma (FUS), which leads to dysregulation of mRNA processing, protein misfolding, mitochondrial dysfunction, and oxidative stress. These pathological mechanisms result in axonal damage, synaptic dysfunction, microgliosis, astrogliosis, and neuroinflammation, ultimately causing the progressive loss of upper motor neurons in the cortex and lower motor neurons in the brainstem and spinal cord [246, 247]. Motor neurons in ALS patients exhibit mitochondrial abnormalities, including defective mitochondrial morphology, Ca2+ overload, reduced Cytc oxidase activity, and decreased mtDNA concentrations, with high mutation rates [247, 248]. The accumulated Ca2+ released from damaged mitochondria can initiate and exacerbate ALS pathology. Maintaining cytosolic Ca2+ levels has been shown to ameliorate microglial and astrocyte activation, reduce misfolded SOD1 accumulation, and mitigate motor neurodegeneration in the spinal cord, leading to improved motor symptoms and prolonged lifespan in ALS mice [249]. Similarly, mtROS released from damaged mitochondria can drive neuroinflammation and neurodegeneration in individuals with ALS. Reducing ROS levels through antioxidant enhancement has been shown to alleviate mitochondrial defects, neuroinflammation, and neuronal loss in the brain and to restore grip strength, locomotion, and memory in ALS rats [250]. The level of extracellular ATP, another mtDAMP, is elevated in the CSF of ALS patients and has been shown to predict disease severity [251]. Upon binding to P2X7 receptors, extracellular ATP triggers the release of mutated SOD1 (SOD1G93A) from motor neurons, which subsequently induces ER stress in healthy motor neurons and activates microglia to secrete proinflammatory cytokines such as TNFα [252]. Extracellular ATP and SOD1G93A also activate astrocytes via P2X7 receptors, causing motor neurodegeneration [253]. Although the intracellular mechanisms underlying ATP-P2X7 receptor-mediated motor neurodegeneration remain unclear, recent studies have implicated the activation of caspases, nNOS, p38 MAPK, and FAS signaling as possible contributors [254]. Pharmacological inhibition of P2X7 receptors has been shown to attenuate motor neurodegeneration, microgliosis, NFκB, NOX2, and IL1β while promoting IL10 and BDNF in the lumbar spinal cord, thereby improving motor performance in SOD1G93A male and female mice [255]. Interestingly, another independent study revealed that pharmacological inhibition of P2X7 receptors is beneficial for female SOD1G93A mice but not for male SOD1G93A mice [256]. Additionally, increased RAGE expression has been reported in astrocytes, microglia, and motor neurons in both rodent and human ALS models. RAGE activation has been shown to exacerbate ALS pathology in SOD1G93A mice [257]. Moreover, TFAM released from damaged mitochondria activates RAGE, further inducing and facilitating ALS pathology.
Recent studies have revealed elevated levels of STING protein in the brain endothelial and neuronal cells of ALS patients [217]. Increased expression of the cGAMP protein has also been observed in the spinal cord of ALS patients [258]. Both proteins are associated with the aggregation of misfolded proteins in neurons [217]. Cytoplasmic accumulation of TDP43 has been reported to trigger the opening of the mPTP, causing mtDNA to escape from the mitochondrial matrix into the cytosol. This escape leads to the upregulation of the cGAS-STING pathway, NFκB, and IFN-I in motor neurons as well as in TDP43 transgenic ALS mice. Pharmacological inhibition or genetic ablation of cGAS reverses these pathological alterations, indicating that the mtDNA-cGAS-STING cascade drives motor neuroinflammation and neurodegeneration in ALS [258]. Furthermore, cytoplasmic aggregated SOD1G93A has been shown to directly damage mitochondria, facilitating mPTP-independent release of mtDNA and RNA:DNA hybrids into the cytosol. Both mtDNA and RNA:DNA intermediates activate the cGAS and/or DDX41 (another nucleic acid sensor and STING activator)-STING-IRF3 pathway, promoting IFN-I mediated antiviral responses and neuroinflammation in in vitro and in vivo SOD1G93A ALS models. Evidence also suggests that components of the cGAS/DDX41-STING-IRF3 pathway can transfer from affected cells to nearby healthy cells via inter-neuronal gap junctions, resulting in amplification and spread of signaling pathways, worsening ALS pathology [73]. Additionally, the release of mtdsRNA from damaged mitochondria may also trigger neuroinflammation and neuronal death in individuals with ALS. Cytoplasmic TDP43 inclusions have been shown to elevate dsRNA levels in cultured human neuronal cells, triggering IFN-I-mediated cell death. This dsRNA-induced IFN-I response also propagates the death of connected neurons in the brains of C9orf72 ALS mice [259]. The accumulation of dsRNA induces microgliosis and astrogliosis, possibly via activation of protein kinase R, leading to upregulation of proinflammatory proteins and degeneration of spinal cord motor neurons in ALS mice [260, 261]. Further studies are warranted to elucidate the precise role of mtdsRNAs in ALS pathology and to explore potential therapeutic targets within these pathways.
Multiple Sclerosis
MS is an autoimmune neurodegenerative disorder characterized by neuronal demyelination, axonal loss, failure of neuronal transmission, oligodendrocyte damage, inflammatory focal lesions, microgliosis, astrogliosis, a compromised BBB, and infiltration of peripheral immune cells such as autoreactive T cells, B cells, monocytes, dendritic cells, and natural killer T cells into the CNS. Symptoms of MS include impairments in motor functions, sensory functions, and cognition, as well as visual disturbances, fatigue, and pain [262]. Damaged mitochondria play a significant role in the initiation and progression of neuroinflammation and demyelination in MS. Ca2+ released from damaged mitochondria can exacerbate MS pathology. Recent studies have shown that pharmacological inhibition or genetic suppression of the voltage-gated Ca2+ channel 1.2 attenuates microgliosis, astrogliosis, lymphocyte infiltration, and neuronal demyelination in the lumbar spinal cord and improves motor performance in MS mouse models [263]. Moreover, ROS released from dysfunctional mitochondria can damage myelinated neurons and trigger immune activation. Dysfunctional mitochondria with low ΔΨm promote ROS accumulation, which induces chemokine and cytokine production and causes axonal degeneration in the brains of rats [264]. Reducing ROS levels ameliorates NLRP3 inflammasome activation, microgliosis, T helper (Th)1 and Th17 cell infiltration, and the production of proinflammatory cytokines such as IL6, IL8, IL17, TNFα, and IFNγ while promoting the production of anti-inflammatory cytokines such as IL10 in the CNS of mice [265]. Damaged mitochondria also release stored ATP into the cytosol, which subsequently leaks into the extracellular space after cell death. Extracellular ATP activates astrocytes and oligodendrocytes and recruits T cells by binding to the P2X7 receptor. This leads to increased production of chemokines, cytokines, ROS, glutamate, and ATP, ultimately resulting in neuroinflammation and neurodegeneration [266]. Inhibition of ATP secretion into the extracellular space or blockage of the P2X7 receptor ameliorates T cell infiltration, neuroinflammation, oligodendrocyte degeneration, and clinical symptoms of MS in mice and extends their survival [267, 268]. Furthermore, clinical studies have highlighted that polymorphisms and reduced activity of the P2X7 receptor provide protection against neuroinflammation in MS [269]. Additionally, anticardiolipin antibodies have been detected in the serum of MS patients [270]. This indicates that cardiolipin released from damaged mitochondria can facilitate neurological manifestations in MS.
Furthermore, several studies have reported elevated concentrations of ccf-mtDNA in the CSF and plasma of progressive MS patients. Compared to those in primary progressive MS patients, higher plasma ccf-mtDNA levels were detected in secondary progressive MS patients. Patients with relapsing–remitting MS also reported increased levels of CSF ccf-mtDNA. Concomitantly, these patients exhibited increased levels of proinflammatory cytokines, including TNFα, neuroinflammation, neurodegeneration, and brain atrophy, suggesting that mtDNA might be an important driver of MS pathology. Suppressing MS pathology in patients through drug interventions proportionally decreased mtDNA copy numbers in the CSF of progressive MS patients, indicating that mtDNA might serve as a good prognostic marker for MS stages [56, 59, 271, 272]. Postmortem analyses of MS patients have revealed upregulation of STING protein in acute demyelinating lesions, confirming that mtDNA-mediated cGAS-STING signaling drives neuroinflammation and exacerbates MS pathology [217]. Interestingly, a study showed that activation of cGAS-STING signaling and IFN-I secretion attenuate microglial reactivity and neuroinflammation in MS mouse models [273]. Thus, further studies are required to evaluate mitochondrial damage, mtDNA release, and subsequent STING signaling-mediated neuroinflammatory status in MS models. Recent reports have shown that extracellular dsRNA triggers protein kinase R and p38 MAPK, subsequently activating NFκB and CAAT/enhancer-binding protein β, respectively, resulting in increased expression of iNOS in astrocytes [274]. Additionally, extracellular dsRNA activates oligodendrocytes by binding to TLR3, suppressing myelin gene expression and increasing the production of cytokines and chemokines, including IL1β, IFNβ, CCL2, and CXCL10, ultimately leading to neuroinflammation and neurodegeneration in the brain white matter of mice [275]. The release of mtdsRNA from damaged mitochondria might initiate and promote neuroinflammation and neuronal loss in MS.
Stroke
Stroke is the second leading cause of death worldwide, occurring due to a sudden interruption of blood supply to the brain, leading to hypoxia-induced cellular injury. This initiates a cascade of events, including microglia and astrocyte activation, peripheral immune cell infiltration, neuroinflammation, neuronal degeneration, compromised BBB integrity, and brain edema. Symptoms may include sudden numbness or weakness on one side of the body, confusion or difficulty speaking, trouble seeing in one or both eyes, loss of balance or coordination, and sudden severe headache [276]. Mitochondrial damage plays a crucial role in the development of neuroinflammation following stroke. Damaged mitochondria are unable to buffer cytosolic Ca2+, leading to Ca2+ dysregulation in affected neurons. Suppression of Ca2+ overload attenuated NLRP1 expression, ROS production, and neuronal damage, as well as promoted cerebral blood flow, limb coordination, and locomotion in cerebral ischemia–reperfusion injury (CIRI) [276, 277]. Additionally, damaged mitochondria are responsible for ROS accumulation in neuronal cells. CIRI induces mitochondrial damage and promotes ROS-RIP1/RIP3-mediated necroptosis. Moreover, the activated RIP1/RIP3 pathway was shown to attenuate autophagolysosomal degradation of damaged mitochondrial components and promote their transfer in the vicinity through exosomes. This leads to further increases in immune activation, inflammatory cytokine production, mitochondrial damage, ROS accumulation, and neurodegeneration [278]. Similarly, increased ROS activate the NLRP3 inflammasome, inhibit angiogenesis, and increase infarct size and mortality in middle cerebral artery occlusion (MCAO) model mice [101, 279]. In contrast, inhibition of ROS following mitochondrial damage alleviated NLRP3 inflammasome activation in microglia, infarct volume, and neurological deficits in CIRI rats [280]. Treatment with antioxidants prevented mitochondrial dysfunction and subsequently downregulated MMP9, COX2, iNOS, and MAPK pathways, NFκB activation, and proinflammatory cytokine production, leading to ameliorated disability and histological damage in CIRI rats[281]. Following neuronal death, the release of ATP from damaged mitochondria into the extracellular space can trigger immune activation and drive neuroinflammation and neurodegeneration in stroke. Deletion of P2Y1 receptors attenuated the glial neuroinflammatory response in the hippocampus and prevented hippocampal tissue damage as well as cognitive decline in MCAO mice [282]. Further studies also showed that inhibition of P2X7 and P2Y1 receptors attenuated microglia and astrocyte proliferation and neuroinflammation in the peri-infarct hippocampus and sensorimotor cortex, resulting in amelioration of neurological deficits, memory decline, and motor impairment in CIRI rats [283]. Moreover, blocking the P2X7 receptor inhibited Ca2+ influx and subsequently reduced microglial activation, proinflammatory cytokine secretion, neuroinflammation, brain infarction size, memory decline, as well as post-stroke depressive symptoms such as anhedonia in stroke rodents [284, 285].
Furthermore, damaged mitochondria release heme, which can exacerbate stroke pathogenesis. Intracerebral heme injection has been shown to induce lipid peroxidation and NLRP3 inflammasome activation, leading to astrogliosis, increased production of proinflammatory cytokines, brain damage, neuroinflammation, and sensorimotor deficits in mice [286]. Moreover, RAGE senses necrotic cell death and promotes microgliosis, neuroinflammation, and increased brain infarct size in MCAO mice [287]. The release of TFAM from damaged mitochondria activates RAGE, thereby driving stroke pathogenesis. Recent studies have reported elevated levels of mitochondrial-derived NFPs and increased expression of microglial FPR1 in perihematomal tissue, which correlates with extended brain edema in intracerebral hemorrhage (ICH) patients. Extracellular NFPs interact with FPR1, activating microglia, recruiting neutrophils, and promoting neuroinflammation and neurological deficits in ICH mice. Pharmacological blockade of FPR1 mitigated brain edema and neurological deficits in ICH mice, indicating that mitochondrial NFPs play a crucial role in stroke progression [151]. Additionally, damaged mitochondria release mtDNA in peri-infarct regions, which can be detected in biofluids and serve as a predictive biomarker for stroke diagnosis [280, 288, 289]. mtDNA triggers the cGAS-STING signaling pathway, promoting IRF3- and NFκB-mediated polarization of microglia toward the M1 phenotype. This exacerbates neuroinflammation, neuronal injury, brain infarction, and edema. Inhibition of STING has been shown to attenuate synapse engulfment by microglia and mitigate motor impairment in mice after cerebral venous sinus thrombosis [290, 291]. Similarly, another study demonstrated that suppression of STING ameliorates neurodegeneration in the hippocampus and improves cognitive function in MCAO mice [289]. Following cerebral venous sinus thrombosis, cGAS-STING signaling also induces NLRP3 inflammasome-mediated microglial activation and proinflammatory cytokine secretion, resulting in neutrophil recruitment, neuroinflammation, neurodegeneration, and neurological deficits in mice [292]. Another study found that oxygen–glucose deprivation and reperfusion induce mitochondrial dysfunction and promote the cytosolic accumulation of Ox-mtDNA, leading to NLRP3 inflammasome activation in microglia. This augments neuroinflammation, infarct volume, and neurological deficits in CIRI rats [280]. Recent evidence shows that histone deacetylase (HDAC)3 promotes cGAS transcription by regulating the acetylation and nuclear transfer of the NFκB p65 subunit in microglia. This potentiates mtDNA-mediated cGAS-STING signaling, microglial activation, neuroinflammation, and neuronal death. Deletion of HDAC3 ameliorated the mtDNA-cGAS-STING pathway in microglia, as well as neuroinflammation and brain injury in CIRI mice [293]. Furthermore, studies have shown that ischemic neurons are deficient in mitophagy, leading to the accumulation of damaged mitochondria, which exacerbates neuroinflammation and neurodegeneration. In contrast, promoting mitophagy has been demonstrated to alleviate neuroinflammation and stroke pathogenesis [294, 295].
Future Perspectives
Despite some controversy, substantial evidence indicates that dysfunctional mitochondria release mtDAMPs, which significantly contribute to the initiation and progression of neuroinflammation and neuronal death in neurodegenerative disorders [3]. However, several outstanding questions remain. The mechanisms involved in the release of mtDAMPs from dysfunctional mitochondria and their intercellular transfer are poorly characterized [9, 10]. Similarly, the detailed mechanisms by which mtDAMPs mediate the activation of neuroinflammatory cascades are not well understood. The threshold quantity of mtDAMPs necessary to elicit an immunogenic response also needs to be established [3]. Additionally, the potential for mtDAMPs released from the brain into the plasma to induce systemic inflammation and autoimmune diseases has yet to be investigated [296]. Furthermore, it is unknown whether dysfunctional mitochondria or mtDAMPs could serve as biomarkers for early indicators of neurodegenerative conditions [60, 190, 215, 241]. It also remains to be clarified whether targeting mitochondrial dysfunction and mtDAMPs could be viable strategies to attenuate, prevent, or cure neuroinflammation and neuronal death in neurodegenerative disorders [119, 124, 167, 297].
Several hypothetical therapeutic strategies for mitigating mtDAMP-mediated neuroinflammation in neurodegenerative disorders need to be validated (Table 2). Although a limited amount of mtDAMPs can initiate tissue repair and prevent inflammation following injury, a large quantity of mtDAMPs triggers the activation of inflammatory immune cells, leading to neuroinflammation [298]. Inhibiting mtDAMP release by reducing mitochondrial Ca2+ overload, mtROS, and the Bax:Bcl2 ratio might alleviate neuroinflammatory cascades [200,201,202]. Neutralizing circulating mtDAMPs with antibodies might be another therapeutic approach for preventing inflammatory immune activation. However, a major drawback of this strategy is the potential reduction in antibody affinities due to conformational changes in the epitopes of mtDAMPs [299]. Moreover, blocking mtDAMP-specific receptors such as TLRs, RAGE, inflammasomes, cGAS, RIG-I, MDA5, FPRs, RIPK1, and HIF1α can prevent the activation of neuroinflammatory signaling. Inhibiting downstream signaling mediators such as MyD88, p38 MAPK, ERK1/2, NFκB, caspases, and TBK1 can attenuate cytokine and chemokine production. Cytokine receptor antagonists, such as those targeting IL1β, TNFα, and IFN-I, can also ameliorate mtDAMP-induced neuroinflammation. Moreover, enhancing mtDAMP clearance and dysfunctional mitochondria via mitophagy might be the most effective way to prevent mtDAMP-mediated neuroglia and microglia activation and subsequent neuroinflammation. Studies have shown that age-related neurodegenerative disorders are associated with impaired mitophagy and lysosomal degradation [297]. Sirtuin 3, a histone deacetylase preferentially located in mitochondria, controls every aspect of mitochondrial functionality, including mitophagy. Recent studies have demonstrated that sirtuin 3 activators alleviate neuroinflammation and neuronal death in neurodegenerative disorders by improving mitophagy [295]. Thus, mitophagy might be a potential therapeutic target for ameliorating mtDAMP-driven neuroinflammation in neurodegenerative disorders.
Data Availability
No datasets were generated or analysed during the current study.
References
Casanova A, Wevers A, Navarro-Ledesma S, Pruimboom L (2023) Mitochondria: It is all about energy. Front Physiol 14. https://doi.org/10.3389/fphys.2023.1114231
Giorgi C, Marchi S, Simoes IC, Ren Z, Morciano G, Perrone M, Patalas-Krawczyk P, Borchard S et al (2018) Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int Rev Cell Mol Biol 340:209–344. https://doi.org/10.1016/bs.ircmb.2018.05.006
Lin MM, Liu N, Qin ZH, Wang Y (2022) Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol Sin 43:2439–2447. https://doi.org/10.1038/s41401-022-00879-6
Gong T, Liu L, Jiang W, Zhou R (2020) DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 20:95–112. https://doi.org/10.1038/s41577-019-0215-7
Rodríguez-Nuevo A, Zorzano A (2019) The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 3:195–207. https://doi.org/10.15698/cst2019.06.190
Wilkins HM, Weidling IW, Ji Y, Swerdlow RH (2017) Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front Immunol 8:. https://doi.org/10.3389/fimmu.2017.00508
Passaro AP, Lebos AL, Yao Y, Stice SL (2021) Immune Response in Neurological Pathology: Emerging Role of Central and Peripheral Immune Crosstalk. Front Immunol 12:. https://doi.org/10.3389/fimmu.2021.676621
Joshi AU, Minhas PS, Liddelow SA et al (2019) Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci 22:1635–1648. https://doi.org/10.1038/s41593-019-0486-0
Bock FJ, Tait SWG (2020) Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21:85–100. https://doi.org/10.1038/s41580-019-0173-8
Torralba D, Baixauli F, Sánchez-Madrid F (2016) Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front Cell Dev Biol 4:. https://doi.org/10.3389/fcell.2016.00107
Todt F, Cakir Z, Reichenbach F et al (2015) Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J 34:67–80. https://doi.org/10.15252/embj.201488806
Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, Dillon CP, Green DR (2011) A Unified Model of Mammalian BCL-2 Protein Family Interactions at the Mitochondria. Mol Cell 44:517–531. https://doi.org/10.1016/j.molcel.2011.10.001
Flores-Romero H, Hohorst L, John M, Albert MC, King LE, Beckmann L, Szabo T, Hertlein V et al (2022) BCL-2-family protein tBID can act as a BAX-like effector of apoptosis. EMBO J 41(2):e108690. https://doi.org/10.15252/embj.2021108690
O’Neill KL, Huang K, Zhang J, Chen Y, Luo X (2016) Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev 30:973–988. https://doi.org/10.1101/gad.276725.115
Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD (2010) BH3-Triggered Structural Reorganization Drives the Activation of Proapoptotic BAX. Mol Cell 40:481–492. https://doi.org/10.1016/j.molcel.2010.10.019
Subburaj Y, Cosentino K, Axmann M, Pedrueza-Villalmanzo E, Hermann E, Bleicken S, Spatz J, García-Sáez AJ (2015) Bax monomers form dimer units in the membrane that further self-assemble into multiple oligomeric species. Nat Commun 6(1):8042. https://doi.org/10.1038/ncomms9042
Dengler MA, Robin AY, Gibson L, Li MX, Sandow JJ, Iyer S, Webb AI, Westphal D et al (2019) BAX Activation: Mutations Near Its Proposed Non-canonical BH3 Binding Site Reveal Allosteric Changes Controlling Mitochondrial Association. Cell Rep 27:359-373.e6. https://doi.org/10.1016/j.celrep.2019.03.040
Gillies LA, Du H, Peters B, Knudson CM, Newmeyer DD, Kuwana T (2015) Visual and functional demonstration of growing Bax-induced pores in mitochondrial outer membranes. Mol Biol Cell 26:339–349. https://doi.org/10.1091/mbc.E13-11-0638
Keinan N, Tyomkin D, Shoshan-Barmatz V (2010) Oligomerization of the Mitochondrial Protein Voltage-Dependent Anion Channel Is Coupled to the Induction of Apoptosis. Mol Cell Biol 30:5698–5709. https://doi.org/10.1128/mcb.00165-10
Jiang X, Jiang H, Shen Z, Wang X (2014) Activation of mitochondrial protease OMA1 by Bax and Bak promotes cytochrome c release during apoptosis. Proc Natl Acad Sci U S A 111:14782–14787. https://doi.org/10.1073/pnas.1417253111
Korwitz A, Merkwirth C, Richter-Dennerlein R, Tröder SE, Sprenger HG, Quirós PM, López-Otín C, Rugarli EI et al (2016) Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J Cell Biol 212:157–166. https://doi.org/10.1083/jcb.201507022
Ader NR, Hoffmann PC, Ganeva I, Borgeaud AC, Wang C, Youle RJ, Kukulski W (2019) Molecular and topological reorganizations in mitochondrial architecture interplay during bax-mediated steps of apoptosis. Elife 8:e40712. https://doi.org/10.7554/eLife.40712
Einsele-Scholz S, Malmsheimer S, Bertram K, Stehle D, Johänning J, Manz M, Daniel PT, Gillissen BF et al (2016) Bok is a genuine multi-BH-domain protein that triggers apoptosis in the absence of Bax and Bak. J Cell Sci 129:3054. https://doi.org/10.1242/jcs.193946
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES (2019) Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10(1):1689. https://doi.org/10.1038/s41467-019-09397-2
Tait SWG, Green DR (2010) Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol 11:621–632. https://doi.org/10.1038/nrm2952
Yuanming H, Benedict MA, Ding L, Núñez G (1999) Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J 18:3586–3595. https://doi.org/10.1093/emboj/18.13.3586
Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ et al (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102:43–53. https://doi.org/10.1016/S0092-8674(00)00009-X
Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102:33–42. https://doi.org/10.1016/S0092-8674(00)00008-8
Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B, Newmeyer DD (2009) Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell 20:4871–4884. https://doi.org/10.1091/mbc.E09-07-0649
Chipuk JE, Bouchier-Hayes L, Green DR (2006) Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ 13:1396–1402. https://doi.org/10.1038/sj.cdd.4401963
Giampazolias E, Zunino B, Dhayade S, Bock F, Cloix C, Cao K, Roca A, Lopez J et al (2017) Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat Cell Biol 19:1116–1129. https://doi.org/10.1038/ncb3596
Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat Cell Biol 2:156–162. https://doi.org/10.1038/35004029
Rehm M, Düßgmann H, Prehn JHM (2003) Real-time single cell analysis of Smac/DIABLO release during apoptosis. J Cell Biol 162:1031–1043. https://doi.org/10.1083/jcb.200303123
N Dragicevic M Mamcarz Y Zhu R Buzzeo J Tan GW Arendash PC Bradshaw 2010 Mitochondrial amyloid-β levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice J Alzheimer’s Dis 20 https://doi.org/10.3233/JAD-2010-100342
Fiskum G, Starkov A, Polster BM, Chinopoulos C (2003) Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann N Y Acad Sci 991:111–119. https://doi.org/10.1111/j.1749-6632.2003.tb07469.x
Paradies G, Paradies V, Ruggiero FM, Petrosillo G (2019) Role of cardiolipin in mitochondrial function and dynamics in health and disease: Molecular and pharmacological aspects. Cells 8(7):728. https://doi.org/10.3390/cells8070728
McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S et al (2018) BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359(6378):eaao6047. https://doi.org/10.1126/science.aao6047
Riley JS, Quarato G, Cloix C, Lopez J, O’Prey J, Pearson M, Chapman J, Sesaki H (2018) Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J 37(17):e99238. https://doi.org/10.15252/embj.201899238
Cosentino K, Hertlein V, Jenner A, Dellmann T, Gojkovic M, Peña-Blanco A, Dadsena S, Wajngarten N et al (2022) The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation. Mol Cell 82:933-949.e9. https://doi.org/10.1016/j.molcel.2022.01.008
Quarato G, Llambi F, Guy CS, Min J, Actis M, Sun H, Narina S, Pruett-Miller SM et al (2022) Ca2+-mediated mitochondrial inner membrane permeabilization induces cell death independently of Bax and Bak. Cell Death Differ 29:1318–1334. https://doi.org/10.1038/s41418-022-01025-9
Brustovetsky N, Brustovetsky T, Purl KJ, Capano M, Crompton M, Dubinsky JM (2003) Increased susceptibility of striatal mitochondria to calcium-induced permeability transition. J Neurosci 23:4858–4867. https://doi.org/10.1523/jneurosci.23-12-04858.2003
Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS (2004) Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am J Physiol - Cell Physiol 287:.https://doi.org/10.1152/ajpcell.00139.2004
Odagiri K, Katoh H, Kawashima H, Tanaka T, Ohtani H, Saotome M, Urushida T, Satoh H et al (2009) Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J Mol Cell Cardiol 46:989–997. https://doi.org/10.1016/j.yjmcc.2008.12.022
Paradies G, Petrosillo G, Paradies V, Ruggiero FM (2009) Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 45:643–650. https://doi.org/10.1016/j.ceca.2009.03.012
Hansson MJ, Månsson R, Morota S, Uchino H, Kallur T, Sumi T, Ishii N, Shimazu M (2008) Calcium-induced generation of reactive oxygen species in brain mitochondria is mediated by permeability transition. Free Radic Biol Med 45:284–294. https://doi.org/10.1016/j.freeradbiomed.2008.04.021
Bauer TM, Murphy E (2020) Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ Res 126:280–293. https://doi.org/10.1161/CIRCRESAHA.119.316306
Mendoza A, Patel P, Robichaux D, Ramirez D, Karch J (2024) Inhibition of the mPTP and Lipid Peroxidation Is Additively Protective Against I/R Injury. Circ Res 134:1292–1305. https://doi.org/10.1161/CIRCRESAHA.123.323882
Patrushev M, Kasymov V, Patrusheva V, Ushakova T, Gogvadze V, Gaziev A (2004) Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell Mol Life Sci 61:3100–3103. https://doi.org/10.1007/s00018-004-4424-1
García N, García JJ, Correa F, Chávez E (2005) The permeability transition pore as a pathway for the release of mitochondrial DNA. Life Sci 76:2873–2880. https://doi.org/10.1016/j.lfs.2004.12.012
Bonora M, Giorgi C, Pinton P (2022) Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol 23:266–285. https://doi.org/10.1038/s41580-021-00433-y
Nakahira K, Hisata S, Choi AMK (2015) The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxidants Redox Signal 23:1329–1350. https://doi.org/10.1089/ars.2015.6407
Rasola A, Bernardi P (2011) Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 50:222–233. https://doi.org/10.1016/j.ceca.2011.04.007
Baev AY, Vinokurov AY, Potapova EV, Dunaev AV, Angelova PR, Abramov AY (2024) Mitochondrial Permeability Transition. Cell Death and Neurodegeneration. Cells 7:648. https://doi.org/10.3390/cells13070648
Zorov DB, Juhaszova M, Sollott SJ (2014) Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94:909–950. https://doi.org/10.1152/physrev.00026.2013
Au AK, Aneja RK, Bell MJ, Bayir H, Feldman K, Adelson PD, Fink EL, Kochanek PM et al (2012) Cerebrospinal fluid levels of high-mobility group box 1 and cytochrome C predict outcome after pediatric traumatic brain injury. J Neurotrauma 29:2013–2021. https://doi.org/10.1089/neu.2011.2171
M Nasi E Bianchini S Biasi De L Gibellini A Neroni M Mattioli M Pinti A Iannone et al 2020 Increased plasma levels of mitochondrial DNA and pro-inflammatory cytokines in patients with progressive multiple sclerosis J Neuroimmunol 338 https://doi.org/10.1016/j.jneuroim.2019.577107
Murray TE, Wenzel TJ, Simtchouk S, Greuel BK, Gibon J (2022) Klegeris A (2022) Extracellular Cardiolipin Modulates Select Immune Functions of Astrocytes in Toll-Like Receptor (TLR) 4-Dependent Manner. Mediators Inflamm 1:9946439. https://doi.org/10.1155/2022/9946439
Wojtkowska M, Karczewska N, Pacewicz K, Pacak A, Kopeć P, Florczak-Wyspiańska J, Popławska-Domaszewicz K, Małkiewicz T et al (2024) Quantification of Circulating Cell-Free DNA in Idiopathic Parkinson’s Disease Patients. Int J Mol Sci 25(5):2818. https://doi.org/10.3390/ijms25052818
Leurs CE, Podlesniy P, Trullas R, Balk L, Steenwijk MD, Malekzadeh A, Piehl F, Uitdehaag BM et al (2018) Cerebrospinal fluid mtDNA concentration is elevated in multiple sclerosis disease and responds to treatment. Mult Scler J 24:472–480. https://doi.org/10.1177/1352458517699874
BX Song E Beroncal AC Andreazza D Gallagher S Marzolini MJ Rapoport J Charles N Herrmann et al 2023 Circulating cell-free mitochondrial DNA (ccf-mtDNA) and memory in mild cognitive impairment (MCI) and Alzheimer’s disease (AD) Alzheimer’s Dement 19 https://doi.org/10.1002/alz.071912
Guescini M, Genedani S, Stocchi V, Agnati LF (2010) Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J Neural Transm 117:1–4. https://doi.org/10.1007/s00702-009-0288-8
Di Mambro T, Pellielo G, Agyapong ED, Carinci M, Chianese D, Giorgi C, Morciano G, Patergnani S et al (2023) The Tricky Connection between Extracellular Vesicles and Mitochondria in Inflammatory-Related Diseases. Int J Mol Sci 24(9):8181. https://doi.org/10.3390/ijms24098181
Deus CM, Tavares H, Beatriz M, Mota S, Lopes C et al (2022) Mitochondrial Damage-Associated Molecular Patterns Content in Extracellular Vesicles Promotes Early Inflammation in Neurodegenerative Disorders. Cells 11(15):2364. https://doi.org/10.3390/cells11152364
Wang X, Weidling I, Koppel S, Menta B, Ortiz JP, Kalani A, Wilkins HM, Swerdlow RH (2020) Detection of mitochondria-pertinent components in exosomes. Mitochondrion 55:100–110. https://doi.org/10.1016/j.mito.2020.09.006
Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, Cullen PJ, Liu J et al (2016) Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med 22:54–63. https://doi.org/10.1038/nm.3983
Muraoka S, DeLeo AM, Sethi MK, Yukawa-Takamatsu K, Yang Z, Ko J, Hogan JD, Ruan Z et al (2020) Proteomic and biological profiling of extracellular vesicles from Alzheimer’s disease human brain tissues. Alzheimer’s Dement 16:896–907. https://doi.org/10.1002/alz.12089
Yao PJ, Eren E, Goetzl EJ, Kapogiannis D (2021) Mitochondrial electron transport chain protein abnormalities detected in plasma extracellular vesicles in alzheimer’s disease. Biomedicines 9(11):1587. https://doi.org/10.3390/biomedicines9111587
Vincent AE, Turnbull DM, Eisner V, Hajnóczky G, Picard M (2017) Mitochondrial Nanotunnels. Trends Cell Biol 27:787–799. https://doi.org/10.1016/j.tcb.2017.08.009
Picca A, Calvani R, Coelho-Junior HJ, Landi F, Bernabei R, Marzetti E (2020) Inter-organelle membrane contact sites and mitochondrial quality control during aging: A geroscience view. Cells 9(3):598. https://doi.org/10.3390/cells9030598
D’Souza A, Burch A, Dave KM, Sreeram A, Reynolds MJ, Dobbins DX, Kamte YS, Zhao W et al (2021) Microvesicles transfer mitochondria and increase mitochondrial function in brain endothelial cells. J Control Release 338:505–526. https://doi.org/10.1016/j.jconrel.2021.08.038
Chakraborty R, Nonaka T, Hasegawa M, Zurzolo C (2023) Tunnelling nanotubes between neuronal and microglial cells allow bi-directional transfer of α-Synuclein and mitochondria. Cell Death Dis 14(5):329. https://doi.org/10.1038/s41419-023-05835-8
Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK et al (2012) Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18:759–765. https://doi.org/10.1038/nm.2736
Tan HY, Yong YK, Xue YC, Liu H, Furihata T, Shankar EM, Ng CS (2022) cGAS and DDX41-STING mediated intrinsic immunity spreads intercellularly to promote neuroinflammation in SOD1 ALS model. iScience 25:. https://doi.org/10.1016/j.isci.2022.104404
Grazioli S, Pugin J (2018) Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front Immunol 9:. https://doi.org/10.3389/fimmu.2018.00832
Marchi S, Guilbaud E, Tait SW, Yamazaki T, Galluzzi L (2023) Mitochondrial control of inflammation. Nat Rev Immunol 23:159–173. https://doi.org/10.1038/s41577-022-00760-x
Dabi YT, Ajagbe AO, Degechisa ST (2023) Toll-like receptors in pathogenesis of neurodegenerative diseases and their therapeutic potential. Immunity, Inflamm Dis 11(4):e839. https://doi.org/10.1002/iid3.839
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES (2012) Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287:36617–36622. https://doi.org/10.1074/jbc.M112.407130
Checa J, Aran JM (2020) Reactive oxygen species: Drivers of physiological and pathological processes. J Inflamm Res 13:1057–1073. https://doi.org/10.2147/JIR.S275595
Gouveia A, Bajwa E, Klegeris A (2017) Extracellular cytochrome c as an intercellular signaling molecule regulating microglial functions. Biochim Biophys Acta - Gen Subj 1861:2274–2281. https://doi.org/10.1016/j.bbagen.2017.06.017
Wenzel TJ, Bajwa E (1863) Klegeris A (2019) Cytochrome c can be released into extracellular space and modulate functions of human astrocytes in a toll-like receptor 4-dependent manner. Biochim Biophys Acta - Gen Subj 11:129400. https://doi.org/10.1016/j.bbagen.2019.07.009
Pullerits R, Bokarewa M, Jonsson IM, Verdrengh M, Tarkowski A (2005) Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatology 44:32–39. https://doi.org/10.1093/rheumatology/keh406
Gaikwad S, Patel D, Agrawal-Rajput R (2017) CD40 Negatively Regulates ATP-TLR4-Activated Inflammasome in Microglia. Cell Mol Neurobiol 37:351–359. https://doi.org/10.1007/s10571-016-0358-z
Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, Wang JZ, Su BY et al (2012) Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation 9:1–4. https://doi.org/10.1186/1742-2094-9-46
Yoshizaki S, Kijima K, Hara M, Saito T, Tamaru T, Tanaka M, Konno DJ, Nakashima Y et al (2019) Tranexamic acid reduces heme cytotoxicity via the TLR4/TNF axis and ameliorates functional recovery after spinal cord injury. J Neuroinflammation 16:1–5. https://doi.org/10.1186/s12974-019-1536-y
Pointer CB, Wenzel TJ, Klegeris A (2019) Extracellular cardiolipin regulates select immune functions of microglia and microglia-like cells. Brain Res Bull 146:153–163. https://doi.org/10.1016/j.brainresbull.2019.01.002
Wenzel TJ, Ranger AL, McRae SA, Klegeris A (2021) Extracellular cardiolipin modulates microglial phagocytosis and cytokine secretion in a toll-like receptor (TLR) 4-dependent manner. J Neuroimmunol 353:577496. https://doi.org/10.1016/j.jneuroim.2021.577496
Pillai VB, Bindu S, Sharp W, Fang YH, Kim G, Gupta M, Samant S, Gupta MP (2016) Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am J Physiol Heart Circ Physiol 310:H962-72. https://doi.org/10.1152/ajpheart.00832.2015
Riley JS, Tait SW (2020) Mitochondrial DNA in inflammation and immunity. EMBO Rep 21(4):e49799. https://doi.org/10.15252/embr.201949799
Julian MW, Shao G, Bao S, Knoell DL, Papenfuss TL, VanGundy ZC, Crouser ED (2012) Mitochondrial Transcription Factor A Serves as a Danger Signal by Augmenting Plasmacytoid Dendritic Cell Responses to DNA. J Immunol 189:433–443. https://doi.org/10.4049/jimmunol.1101375
Gu X, Wu G, Yao Y, Zeng J, Shi D, Lv T, Luo L, Song Y (2015) Intratracheal administration of mitochondrial DNA directly provokes lung inflammation through the TLR9-p38 MAPK pathway. Free Radic Biol Med 83:149–158. https://doi.org/10.1016/j.freeradbiomed.2015.02.034
Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M et al (2004) Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 5:1061–1068. https://doi.org/10.1038/ni1118
Zhang Q, Itagaki K, Hauser CJ (2010) Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 34:55–59. https://doi.org/10.1097/SHK.0b013e3181cd8c08
Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, Yager EJ, Ruggiero EA, Crawford DR (2012) Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J Alzheimer’s Dis 30:617–627. https://doi.org/10.3233/JAD-2012-120145
Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S et al (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560:198–203. https://doi.org/10.1038/s41586-018-0372-z
Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A (2004) Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol 75:995–1000. https://doi.org/10.1189/jlb.0703328
Salmina AB, Komleva YK, Lopatina OL, Kuvacheva NV, Gorina YV, Panina YA, Uspenskaya YA, Petrova MM et al (2015) Astroglial control of neuroinflammation: TLR3-mediated dsRNA-sensing pathways are in the focus. Rev Neurosci 26:143–159. https://doi.org/10.1515/revneuro-2014-0052
May O, Yatime L, Merle NS, Delguste F, Howsam M, Daugan MV, Paul-Constant C, Billamboz M et al (2021) The receptor for advanced glycation end products is a sensor for cell-free heme. FEBS J 288:3448–3464. https://doi.org/10.1111/febs.15667
Little JP, Simtchouk S, Schindler SM, Villanueva EB, Gill NE, Walker DG, Wolthers KR, Klegeris A (2014) Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol Cell Neurosci 60:88–96. https://doi.org/10.1016/j.mcn.2014.04.003
Schindler SM, Frank MG, Annis JL, Maier SF, Klegeris A (2018) Pattern recognition receptors mediate pro-inflammatory effects of extracellular mitochondrial transcription factor A (TFAM). Mol Cell Neurosci 89:71–79. https://doi.org/10.1016/j.mcn.2018.04.005
Xu J, Núñez G (2023) The NLRP3 inflammasome: activation and regulation. Trends Biochem Sci 48:331–344. https://doi.org/10.1016/j.tibs.2022.10.002
Panbhare K, Pandey R, Chauhan C, Sinha A, Shukla R, Kaundal RK (2024) Role of NLRP3 Inflammasome in Stroke Pathobiology: Current Therapeutic Avenues and Future Perspective. ACS Chem Neurosci 15:31–55. https://doi.org/10.1021/acschemneuro.3c0053
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–226. https://doi.org/10.1038/nature09663
Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, Yang M, Yang S et al (2018) Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol 16:32–46. https://doi.org/10.1016/j.redox.2018.02.013
Choi EH, Park SJ (2023) TXNIP: A key protein in the cellular stress response pathway and a potential therapeutic target. Exp Mol Med 55:1348–1356. https://doi.org/10.1038/s12276-023-01019-8
Muri J, Thut H, Feng Q, Kopf M (2020) Thioredoxin-1 distinctly promotes NF-κB target DNA binding and NLRP3 inflammasome activation independently of Txnip. Elife 9:e53627. https://doi.org/10.7554/eLife.53627
Harijith A, Ebenezer DL, Natarajan V (2014) Reactive oxygen species at the crossroads of inflammasome and inflammation. Front Physiol 5:352. https://doi.org/10.3389/fphys.2014.00352
Garg C, Seo JH, Ramachandran J, Loh JM, Calderon F, Contreras JE (2018) Trovafloxacin attenuates neuroinflammation and improves outcome after traumatic brain injury in mice. J Neuroinflammation 15:1–5. https://doi.org/10.1186/s12974-018-1069-9
Illes P (2020) P2X7 Receptors Amplify CNS Damage in Neurodegenerative Diseases. Int J Mol Sci 21:1–31. https://doi.org/10.3390/ijms21175996
Tapia-Abellán A, Angosto-Bazarra D, Alarcón-Vila C, Baños MC, Hafner-Bratkovič I, Oliva B, Pelegrín P (2021) Sensing low intracellular potassium by NLRP3 results in a stable open structure that promotes inflammasome activation. Sci Adv 7:eabf4468. https://doi.org/10.1126/sciadv.abf4468
Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 109:11282–11287. https://doi.org/10.1073/pnas.1117765109
Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, Tardivel A, Heuschling P et al (2015) NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 10(6):e0130624. https://doi.org/10.1371/journal.pone.0130624
Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN (2013) The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153:348–361. https://doi.org/10.1016/j.cell.2013.02.054
Yu Y, Feng S, Wei S, Zhong Y, Yi G, Chen H, Liang L, Chen H et al (2019) Extracellular ATP activates P2X7R-NF-κB (p65) pathway to promote the maturation of bone marrow-derived dendritic cells of mice. Cytokine 119:175–181. https://doi.org/10.1016/j.cyto.2019.03.019
Cao L, Cao X, Zhou Y, Nagpure BV, Wu ZY, Hu LF, Yang Y, Sethi G et al (2018) Hydrogen sulfide inhibits ATP-induced neuroinflammation and Aβ 1–42 synthesis by suppressing the activation of STAT3 and cathepsin S. Brain Behav Immun 73:603–614. https://doi.org/10.1016/j.bbi.2018.07.005
Bolívar BE, Brown-Suedel AN, Rohrman BA, Charendoff CI, Yazdani V, Belcher JD, Vercellotti GM, Flanagan JM et al (2021) Noncanonical Roles of Caspase-4 and Caspase-5 in Heme-Driven IL-1β Release and Cell Death. J Immunol 206:1878–1889. https://doi.org/10.4049/jimmunol.2000226
Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V et al (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39:311–323. https://doi.org/10.1016/j.immuni.2013.08.001
Qiu Y, Huang Y, Chen M, Yang Y, Li X, Zhang W et al (2022) Mitochondrial DNA in NLRP3 inflammasome activation. Int Immunopharmacol 108:108719. https://doi.org/10.1016/j.intimp.2022.108719
Ross C, Chan AH, von Pein JB, Maddugoda MP, Boucher D, Schroder K (2022) Inflammatory Caspases: Toward a Unified Model for Caspase Activation by Inflammasomes. Annu Rev Immunol 40:249–269. https://doi.org/10.1146/annurev-immunol-101220-030653
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–230. https://doi.org/10.1038/ni.1980
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ et al (2012) Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 36:401–414. https://doi.org/10.1016/j.immuni.2012.01.009
Zhong Z, Sanchez-Lopez E, Karin M (2016) Autophagy, NLRP3 inflammasome and auto-inflammatory/immune diseases. Clin Exp Rheumatol 34:12–16
Jabir MS, Hopkins L, Ritchie ND, Ullah I, Bayes HK, Li D, Tourlomousis P, Lupton A et al (2015) Mitochondrial damage contributes to pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11:166–182. https://doi.org/10.4161/15548627.2014.981915
Dang EV, McDonald JG, Russell DW, Cyster JG (2017) Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 171:1057-1071.e11. https://doi.org/10.1016/j.cell.2017.09.029
Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, Horng T (2014) Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A 111:15514–15519. https://doi.org/10.1073/pnas.1414859111
Ruan J, Xia S, Liu X, Lieberman J, Wu H (2018) Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557:62–67. https://doi.org/10.1038/s41586-018-0058-6
de Torre-Minguela C, Gómez AI, Couillin I, Pelegrín P (2021) Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J 35:. https://doi.org/10.1096/fj.202100085R
Liu H, Zhang H, Wu X, Ma D, Wu J, Wang L, Jiang Y, Fei Y et al (2018) Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563:131–136. https://doi.org/10.1038/s41586-018-0629-6
Barnett KC, Coronas-Serna JM, Zhou W, Ernandes MJ, Cao A, Kranzusch PJ, Kagan JC (2019) Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell 176:1432-1446.e11. https://doi.org/10.1016/j.cell.2019.01.049
Kim J, Kim HS, Chung JH (2023) Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med 55:510–519. https://doi.org/10.1038/s12276-023-00965-7
Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner KP (2013) Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498:332–337. https://doi.org/10.1038/nature12305
Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC et al (2013) The Innate Immune DNA Sensor cGAS Produces a Noncanonical Cyclic Dinucleotide that Activates Human STING. Cell Rep 3:1355–1361. https://doi.org/10.1016/j.celrep.2013.05.009
Du M, Chen ZJ (2018) DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361:704–709. https://doi.org/10.1126/science.aat1022
Andreeva L, Hiller B, Kostrewa D, Lässig C, de Oliveira Mann CC, Jan Drexler D, Maiser A, Gaidt M et al (2017) cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549:394–398. https://doi.org/10.1038/nature23890
Luecke S, Holleufer A, Christensen MH, et al (2017) cGAS is activated by DNA in a length‐dependent manner . EMBO Rep 18:1707–1715. https://doi.org/10.15252/embr.201744017
Cai X, Chiu YH, Chen ZJ (2014) The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54:289–296. https://doi.org/10.1016/j.molcel.2014.03.040
Zhao B, Du F, Xu P, Shu C, Sankaran B, Bell SL, Liu M, Lei Y et al (2019) A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 569:718–722. https://doi.org/10.1038/s41586-019-1228-x
Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ (2019) Structural basis of STING binding with and phosphorylation by TBK1. Nature 567:394–398. https://doi.org/10.1038/s41586-019-1000-2
Tanaka Y, Chen ZJ (2012) STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5:ra20. https://doi.org/10.1126/scisignal.2002521
Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM et al (2003) IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4:491–496. https://doi.org/10.1038/ni921
Harrington JS, Huh JW, Schenck EJ, Nakahira K, Siempos II, Choi AM (2019) Circulating Mitochondrial DNA as Predictor of Mortality in Critically Ill Patients: A Systematic Review of Clinical Studies. Chest 156:1120–1136. https://doi.org/10.1016/j.chest.2019.07.014
West AP, Shadel GS (2017) Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17:363–375. https://doi.org/10.1038/nri.2017.21
Zhang X, Wu J, Liu Q, Li X, Li S, Chen J, Hong Z, Wu X et al (2020) mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis 11:1050. https://doi.org/10.1038/s41419-020-03239-6
Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, Ghosh A, Vella AT et al (2018) Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 49:413-426.e5. https://doi.org/10.1016/j.immuni.2018.07.006
Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, Zhou X, Gao J et al (2017) Inflammasome Activation Triggers Caspase-1-Mediated Cleavage of cGAS to Regulate Responses to DNA Virus Infection. Immunity 46:393–404. https://doi.org/10.1016/j.immuni.2017.02.011
Swanson KV, Junkins RD, Kurkjian CJ, Holley-Guthrie E, Pendse AA, El Morabiti R, Petrucelli A, Barber GN et al (2017) A noncanonical function of cGAMP in inflammasome priming and activation. J Exp Med 214:3611–3262. https://doi.org/10.1084/jem.20171749
Wu B, Hur S (2015) How RIG-I like receptors activate MAVS. Curr Opin Virol 12:91–98. https://doi.org/10.1016/j.coviro.2015.04.004
Rehwinkel J, Gack MU (2020) RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 20:537–551. https://doi.org/10.1038/s41577-020-0288-3
Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, Sfeir A (2021) Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591:477–481. https://doi.org/10.1038/s41586-021-03269-w
Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, Crow YJ, Rice GI et al (2018) Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 560:238–242. https://doi.org/10.1038/s41586-018-0363-0
Agod Z, Fekete T, Budai MM, Varga A, Szabo A, Moon H, Boldogh I, Biro T et al (2017) Regulation of type I interferon responses by mitochondria-derived reactive oxygen species in plasmacytoid dendritic cells. Redox Biol 13:633–645. https://doi.org/10.1016/j.redox.2017.07.016
Li Z, Li Y, Han J, Zhu Z, Li M, Liu Q, Wang Y, Shi FD (2021) Formyl peptide receptor 1 signaling potentiates inflammatory brain injury. Sci Transl Med 13:eabe9890. https://doi.org/10.1126/scitranslmed.abe9890
Carp H (1982) Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med 155:264–275. https://doi.org/10.1084/jem.155.1.264
Lubkin DT, Bishawi M, Barbas AS, Brennan TV, Kirk AD (2018) Extracellular mitochondrial DNA and N-formyl peptides in trauma and critical illness: A systematic review. Crit Care Med 46:2018–2028. https://doi.org/10.1097/CCM.0000000000003381
Hazeldine J, Hampson P, Opoku FA, Foster M, Lord JM (2015) N-Formyl peptides drive mitochondrial damage associated molecular pattern induced neutrophil activation through ERK1/2 and P38 MAP kinase signalling pathways. Injury 46:975–984. https://doi.org/10.1016/j.injury.2015.03.028
Korimová A, Dubový P (2020) N-Formylated Peptide Induces Increased Expression of Both Formyl Peptide Receptor 2 (Fpr2) and Toll-Like Receptor 9 (TLR9) in Schwannoma Cells-An In Vitro Model for Early Inflammatory Profiling of Schwann Cells. Cells 9:1–14. https://doi.org/10.3390/cells9122661
Cussell PJG, Escalada MG, Milton NGN, Paterson AWJ (2020) The N-formyl peptide receptors: Contemporary roles in neuronal function and dysfunction. Neural Regen Res 15:1191–1198. https://doi.org/10.4103/1673-5374.272566
Yu Z, Jiang N, Su W, Zhuo Y (2021) Necroptosis: A Novel Pathway in Neuroinflammation. Front Pharmacol 12:701564. https://doi.org/10.3389/fphar.2021.701564
Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, Cai Q, Yang ZH et al (2017) RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 8:14329. https://doi.org/10.1038/ncomms14329
D’alessandro A, Moore HB, Moore EE, Reisz JA, Wither MJ, Ghasasbyan A, Chandler J, Silliman CC et al (2017) Plasma succinate is a predictor of mortality in critically injured patients. J Trauma Acute Care Surg 83:491–495. https://doi.org/10.1097/TA.0000000000001565
Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, Schwärzler C, Junt T et al (2008) Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol 9:1261–1269. https://doi.org/10.1038/ni.1657
Murphy MP, O’Neill LAJ (2018) Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 174:780–784. https://doi.org/10.1016/j.cell.2018.07.030
Bernardini JP, Brouwer JM, Tan IK, Sandow JJ, Huang S, Stafford CA, Bankovacki A, Riffkin CD et al (2019) Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J 38:e99916. https://doi.org/10.15252/embj.201899916
Lindqvist LM, Frank D, McArthur K, Dite TA, Lazarou M, Oakhill JS, Kile BT, Vaux DL (2018) Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differ 25:782–794. https://doi.org/10.1038/s41418-017-0017-z
Todkar K, Chikhi L, Desjardins V, El-Mortada F, Pépin G, Germain M (2021) Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat Commun 12:1971. https://doi.org/10.1038/s41467-021-21984-w
Yamazaki T, Kirchmair A, Sato AI, Buqué A, Rybstein M, Petroni G, Bloy N, Finotello F et al (2020) Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat Immunol 21:1160–1171. https://doi.org/10.1038/s41590-020-0751-0
Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D et al (2016) NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 164:896–910. https://doi.org/10.1016/j.cell.2015.12.057
Sliter DA, Martinez J, Hao L, Chen XI, Sun N, Fischer TD, Burman JL, Li Y (2018) Parkin and PINK1 mitigate STING-induced inflammation. Nature 561:258–262. https://doi.org/10.1038/s41586-018-0448-9
Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333:1109–1112. https://doi.org/10.1126/science.1201940
Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P et al (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A 113:4039–4044. https://doi.org/10.1073/pnas.1523926113
Di Rita A, Peschiaroli A, D′Acunzo P, Strobbe D, Hu Z, Gruber J, Nygaard M, Lambrughi M et al (2018) HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat Commun 9:3755. https://doi.org/10.1038/s41467-018-05722-3
Buqué A, Rodriguez-Ruiz ME, Fucikova J, Galluzzi L (2019) Apoptotic caspases cut down the immunogenicity of radiation. Oncoimmunology 8:e1655364. https://doi.org/10.1080/2162402X.2019.1655364
Arandjelovic S, Ravichandran KS (2015) Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 16:907–917. https://doi.org/10.1038/ni.3253
Wickman GR, Julian L, Mardilovich K, Schumacher S, Munro J, Rath N, Zander SA, Mleczak A et al (2013) Blebs produced by actin-myosin contraction during apoptosis release damage-associated molecular pattern proteins before secondary necrosis occurs. Cell Death Differ 20:1293–1305. https://doi.org/10.1038/cdd.2013.69
Szondy Z, Sarang Z, Kiss B, Garabuczi É, Köröskényi K (2017) Anti-inflammatory mechanisms triggered by apoptotic cells during their clearance. Front Immunol 8:909. https://doi.org/10.3389/fimmu.2017.00909
Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, Zhang R, Huang X et al (2019) Apoptotic Caspases Suppress Type I Interferon Production via the Cleavage of cGAS, MAVS, and IRF3. Mol Cell 74:19-31.e7. https://doi.org/10.1016/j.molcel.2019.02.013
Frelin C, Imbert V, Bottero V, Gonthier N, Samraj AK, Schulze-Osthoff K, Auberger P, Courtois G et al (2008) Inhibition of the NF-κB survival pathway via caspase-dependent cleavage of the IKK complex scaffold protein and NF-κB essential modulator NEMO. Cell Death Differ 15:152–160. https://doi.org/10.1038/sj.cdd.4402240
Lüthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, Brumatti G, Taylor RC et al (2009) Suppression of Interleukin-33 Bioactivity through Proteolysis by Apoptotic Caspases. Immunity 31:84–98. https://doi.org/10.1016/j.immuni.2009.05.007
Clemens MJ, Bushell M, Jeffrey IW, Pain VM, Morley SJ (2000) Translation initiation factor modifications and the regulation of protein synthesis in apoptotic cells. Cell Death Differ 7:603–615. https://doi.org/10.1038/sj.cdd.4400695
White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, Van Delft MF, Bedoui S et al (2014) Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159:1549–1562. https://doi.org/10.1016/j.cell.2014.11.036
Leonard JR, Klocke BJ, D’sa C, Flavell RA, Roth KA (2002) Strain-dependent neurodevelopmental abnormalities in caspase-3-deficient mice. J Neuropathol Exp Neurol 61:673–677. https://doi.org/10.1093/jnen/61.8.673
Honarpour N, Du C, Richardson JA, Hammer RE, Wang X, Herz J (2000) Adult Apaf-1-deficient mice exhibit male infertility. Dev Biol 218:248–258. https://doi.org/10.1006/dbio.1999.9585
Liu X, Fu R, Pan Y, Meza-Sosa KF, Zhang Z, Lieberman J (2018) PNPT1 Release from Mitochondria during Apoptosis Triggers Decay of Poly(A) RNAs. Cell 174:187-201.e12. https://doi.org/10.1016/j.cell.2018.04.017
Brokatzky D, Dörflinger B, Haimovici A, Weber A, Kirschnek S, Vier J, Metz A, Henschel J et al (2019) A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J 38:e100907. https://doi.org/10.15252/embj.2018100907
Dörflinger B, Badr MT, Haimovici A, Fischer L, Vier J, Metz A, Eisele B, Bronsert P et al (2022) Mitochondria supply sub-lethal signals for cytokine secretion and DNA-damage in H. pylori infection. Cell Death Differ 29:2218–2232. https://doi.org/10.1038/s41418-022-01009-9
Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, Haller M, Riley JS et al (2015) Limited Mitochondrial Permeabilization Causes DNA Damage and Genomic Instability in the Absence of Cell Death. Mol Cell 57:860–872. https://doi.org/10.1016/j.molcel.2015.01.018
Gama V, Swahari V, Schafer J, Kole AJ, Evans A, Huang Y, Cliffe A, Golitz B et al (2014) The E3 ligase PARC mediates the degradation of cytosolic cytochrome c to promote survival in neurons and cancer cells. Sci Signal. https://doi.org/10.1126/scisignal.2005309
Codina R, Vanasse A, Kelekar A, Vezys V, Jemmerson R (2010) Cytochrome c-induced lymphocyte death from the outside in: Inhibition by serum leucine-rich alpha-2-glycoprotein-1. Apoptosis 15:139–152. https://doi.org/10.1007/s10495-009-0412-0
Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, Pennemann FL, Schnepf D et al (2018) Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol 19:130–140. https://doi.org/10.1038/s41590-017-0013-y
Park W, Wei S, Kim BS, Kim B, Bae SJ, Chae YC, Ryu D, Ha KT (2023) Diversity and complexity of cell death: a historical review. Exp Mol Med 55:1573–1594. https://doi.org/10.1038/s12276-023-01078-x
Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, Benatti S, Gibellini L et al (2014) Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm-aging.” Eur J Immunol 44:1552–1562. https://doi.org/10.1002/eji.201343921
Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y (2020) Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener 15:1–37. https://doi.org/10.1186/s13024-020-00391-7
Verma A, Shteinfer-Kuzmine A, Kamenetsky N, Pittala S, Paul A, Nahon Crystal E, Ouro A, Chalifa-Caspi V et al (2022) Targeting the overexpressed mitochondrial protein VDAC1 in a mouse model of Alzheimer’s disease protects against mitochondrial dysfunction and mitigates brain pathology. Transl Neurodegener 11:58. https://doi.org/10.1186/s40035-022-00329-7
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. https://doi.org/10.1038/nature21029
Sharma C, Kim S, Nam Y, Jung UJ, Kim SR (2021) Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int J Mol Sci 22:4850. https://doi.org/10.3390/ijms22094850
Cai H, Qiao J, Chen S, Yang J, Hölscher C, Wang Z, Qi J, Wu M (2022) MCU knockdown in hippocampal neurons improves memory performance of an Alzheimer’s disease mouse model. Acta Biochim Biophys Sin (Shanghai) 54:1528–1539. https://doi.org/10.3724/abbs.2022138
Casaril AM, Katsalifis A, Schmidt RM, Bas-Orth C (2022) Activated glia cells cause bioenergetic impairment of neurons that can be rescued by knock-down of the mitochondrial calcium uniporter. Biochem Biophys Res Commun 608:45–51. https://doi.org/10.1016/j.bbrc.2022.03.120
Galizzi G, Di Carlo M (2023) Mitochondrial DNA and Inflammation in Alzheimer’s Disease. Curr Issues Mol Biol 45:8586–8606. https://doi.org/10.3390/cimb45110540
Aminzadeh M, Roghani M, Sarfallah A, Riazi GH (2018) TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer’s disease. Int Immunopharmacol 54:78–85. https://doi.org/10.1016/j.intimp.2017.10.024
Zhang M, Ding ZX, Huang W, Luo J, Ye S, Hu SL, Zhou P, Cai B (2023) Chrysophanol exerts a protective effect against Aβ25-35-induced Alzheimer’s disease model through regulating the ROS/TXNIP/NLRP3 pathway. Inflammopharmacology 31:1511–1527. https://doi.org/10.1007/s10787-023-01201-4
Majdi F, Taheri F, Salehi P, Motaghinejad M, Safari S (2019) Cannabinoids Δ9-tetrahydrocannabinol and cannabidiol may be effective against methamphetamine induced mitochondrial dysfunction and inflammation by modulation of Toll-like type-4(Toll-like 4) receptors and NF-κB signaling. Med Hypotheses 133:109371. https://doi.org/10.1016/j.mehy.2019.109371
Lei S, Wu S, Wang G, Li B, Liu B, Lei X (2021) Pinoresinol diglucoside attenuates neuroinflammation, apoptosis and oxidative stress in a mice model with Alzheimer’s disease. Neuroreport 32:259–267. https://doi.org/10.1097/WNR.0000000000001583
Jiang J, Pan H, Shen F, Tan Y, Chen S (2023) Ketogenic diet alleviates cognitive dysfunction and neuroinflammation in APP/PS1 mice via the Nrf2/HO-1 and NF-κB signaling pathways. Neural Regen Res 18:2767–2772. https://doi.org/10.4103/1673-5374.373715
Facci L, Barbierato M, Zusso M, Skaper SD, Giusti P (2018) Serum amyloid A primes microglia for ATP-dependent interleukin-1β release. J Neuroinflammation 15:1. https://doi.org/10.1186/s12974-018-1205-6
Martínez-Frailes C, Di Lauro C, Bianchi C, de Diego-García L, Sebastián-Serrano Á, Boscá L, Díaz-Hernández M (2019) Amyloid peptide induced neuroinflammation increases the P2X7 receptor expression in microglial cells, impacting on its functionality. Front Cell Neurosci 13:143. https://doi.org/10.3389/fncel.2019.00143
Islam J, Cho JA, Kim JY, Park KS, Koh YJ, Chung CY, Lee EJ, Nam SJ (2022) GPCR19 Regulates P2X7R-Mediated NLRP3 Inflammasomal Activation of Microglia by Amyloid β in a Mouse Model of Alzheimer’s Disease. Front Immunol 13:766919. https://doi.org/10.3389/fimmu.2022.766919
Di Lauro C, Bianchi C, Sebastián-Serrano Á, Soria-Tobar L, Alvarez-Castelao B, Nicke A, Díaz-Hernández M (2022) P2X7 receptor blockade reduces tau induced toxicity, therapeutic implications in tauopathies. Prog Neurobiol 208:102173. https://doi.org/10.1016/j.pneurobio.2021.102173
Silva DF, Candeias E, Esteves AR, Magalhães JD, Ferreira IL, Nunes-Costa D, Rego AC, Empadinhas N (2020) Microbial BMAA elicits mitochondrial dysfunction, innate immunity activation, and Alzheimer’s disease features in cortical neurons. J Neuroinflammation 17:1–8. https://doi.org/10.1186/s12974-020-02004-y
Wenzel TJ, Murray TE, Noyovitz B, Narayana K, Gray TE, Le J, He J, Simtchouk S (2023) Cardiolipin released by microglia can act on neighboring glial cells to facilitate the uptake of amyloid-β (1–42). Mol Cell Neurosci 124:103804. https://doi.org/10.1016/j.mcn.2022.103804
O Sbai M Djelloul A Auletta A Ieraci C Vascotto L Perrone 2022 RAGE-TXNIP axis drives inflammation in Alzheimer’s by targeting Aβ to mitochondria in microglia Cell Death Dis 13 https://doi.org/10.1038/s41419-022-04758-0
Zhang H, Wang D, Gong P, Lin A, Zhang Y, Ye RD, Yu Y (2019) Formyl Peptide Receptor 2 Deficiency Improves Cognition and Attenuates Tau Hyperphosphorylation and Astrogliosis in a Mouse Model of Alzheimer’s Disease. J Alzheimer’s Dis 67:169–179. https://doi.org/10.3233/JAD-180823
Slowik A, Merres J, Elfgen A, Jansen S, Mohr F, Wruck CJ, Pufe T, Brandenburg LO (2012) Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE) - and amyloid beta 1–42-induced signal transduction in glial cells. Mol Neurodegener 7:1–8. https://doi.org/10.1186/1750-1326-7-55
Xie X, Ma G, Li X, Zhao J, Zhao Z, Zeng J (2023) Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5×FAD mice. Nat Aging 3:202–212. https://doi.org/10.1038/s43587-022-00337-2
Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, Propson NE, Xu Y et al (2020) Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest 130:1912–1930. https://doi.org/10.1172/JCI133737
He S, Li X, Mittra N, Bhattacharjee A, Wang H, Zhao S, Liu F, Han X (2023) Microglial cGAS deletion protects against amyloid-β induced Alzheimer’s disease pathogenesis. bioRxiv. https://doi.org/10.1101/2023.08.07.552300
Podlesniy P, Figueiro-Silva J, Llado A, Antonell A, Sanchez-Valle R, Alcoleaet D, al, (2013) Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann Neurol 74:655–668. https://doi.org/10.1002/ana.23955
Hussain M, Chu X, Sahbaz BD, Gray S, Pekhale K, Park JH, Croteau DL, Bohr VA (2023) Mitochondrial OGG1 expression reduces age-associated neuroinflammation by regulating cytosolic mitochondrial DNA. Free Radic Biol Med 203:34–44. https://doi.org/10.1016/j.freeradbiomed.2023.03.262
Ferecskó AS, Smallwood MJ, Moore A, Liddle C, Newcombe J, Holley J, Whatmore J, Gutowski NJ et al (2023) STING-Triggered CNS Inflammation in Human Neurodegenerative Diseases. Biomedicines 11:1375. https://doi.org/10.3390/biomedicines11051375
Xia X, He X, Zhao T, Yang J, Bi Z, Fu Q, Liu J, Ao D et al (2023) Inhibiting mtDNA-STING-NLRP3/IL-1β axis-mediated neutrophil infiltration protects neurons in Alzheimer’s disease. Cell Prolif. https://doi.org/10.1111/cpr.13529
Nidadavolu LS, Feger D, Chen D, Wu Y, Grodstein F, Gross AL, Bennett DA, Walston JD et al (2023) Associations between circulating cell-free mitochondrial DNA, inflammatory markers, and cognitive and physical outcomes in community dwelling older adults. Immun Ageing 20:24. https://doi.org/10.1186/s12979-023-00342-y
Wilkins HM, Carl SM, Weber SG, Ramanujan SA, Festoff BW, Linseman DA, Swerdlow RH et al (2015) Mitochondrial lysates induce inflammation and alzheimer’s disease-relevant changes in microglial and neuronal cells. J Alzheimer’s Dis 45:305–318. https://doi.org/10.3233/JAD-142334
Wilkins HM, Koppel SJ, Weidling IW, Roy N, Ryan LN, Stanford JA, Swerdlow RH (2016) Extracellular Mitochondria and Mitochondrial Components Act as Damage-Associated Molecular Pattern Molecules in the Mouse Brain. J Neuroimmune Pharmacol 11:622–628. https://doi.org/10.1007/s11481-016-9704-7
Ochoa E, Ramirez P, Gonzalez E, De Mange J, Ray WJ, Bieniek KF, Frost B (2023) Pathogenic tau–induced transposable element–derived dsRNA drives neuroinflammation. Sci Adv 9:eabq5423. https://doi.org/10.1126/sciadv.abq5423
McGarry N, Murray CL, Garvey S, Wilkinson A, Tortorelli L, Ryan L, Hayden L, Healy D et al (2021) Double stranded RNA drives anti-viral innate immune responses, sickness behavior and cognitive dysfunction dependent on dsRNA length, IFNAR1 expression and age. Brain Behav Immun 95:413–428. https://doi.org/10.1016/j.bbi.2021.04.016
Melton LM, Keith AB, Davis S, Oakley AE, Edwardson JA, Morris CM (2003) Chronic glial activation, neurodegeneration, and APP immunoreactive deposits following acute administration of double-stranded RNA. Glia 44:1–12. https://doi.org/10.1002/glia.10276
Reichmann H, Schneider C, Löhle M (2009) Non-motor features of Parkinson’s disease: Depression and dementia. Park Relat Disord 15:S87-92. https://doi.org/10.1016/S1353-8020(09)70789-8
Scorziello A, Borzacchiello D, Sisalli MJ, Di Martino R, Morelli M, Feliciello A (2020) Mitochondrial Homeostasis and Signaling in Parkinson’s Disease. Front Aging Neurosci 12:100. https://doi.org/10.3389/fnagi.2020.00100
Sirabella R, Sisalli MJ, Costa G, Omura K, Ianniello G, Pinna A, Morelli M, Di Renzo GM et al (2018) NCX1 and NCX3 as potential factors contributing to neurodegeneration and neuroinflammation in the A53T transgenic mouse model of Parkinson’s Disease article. Cell Death Dis 9:725. https://doi.org/10.1038/s41419-018-0775-7
Soraci L, Gambuzza ME, Biscetti L, Laganà P, Lo Russo C, Buda A, Barresi G, Corsonello A et al (2023) Toll-like receptors and NLRP3 inflammasome-dependent pathways in Parkinson’s disease: mechanisms and therapeutic implications. J Neurol 270:1346–1360. https://doi.org/10.1007/s00415-022-11491-3
Zhou Q, Zhang Y, Lu L, Zhang H, Zhao C, Pu Y, Yin L (2022) Copper induces microglia-mediated neuroinflammation through ROS/NF-κB pathway and mitophagy disorder. Food Chem Toxicol 168:113369. https://doi.org/10.1016/j.fct.2022.113369
Atiq A, Lee HJ, Khan A, Kang MH, Rehman IU, Ahmad R, Tahir M, Ali J et al (2023) Vitamin E Analog Trolox Attenuates MPTP-Induced Parkinson’s Disease in Mice, Mitigating Oxidative Stress, Neuroinflammation, and Motor Impairment. Int J Mol Sci 24:9942. https://doi.org/10.3390/ijms24129942
Javed H, Meeran MN, Azimullah S, Bader Eddin L, Dwivedi VD, Jha NK, Ojha S (2020) α-Bisabolol, a Dietary Bioactive Phytochemical Attenuates Dopaminergic Neurodegeneration through Modulation of Oxidative Stress, Neuroinflammation and Apoptosis in Rotenone- Induced Rat Model of Parkinson’s disease. Biomolecules 10:1–22. https://doi.org/10.3390/biom10101421
Gao XF, Wang W, Yu Q, Burnstock G, Xiang ZH, He C (2011) Astroglial P2X7 receptor current density increased following long-term exposure to rotenone. Purinergic Signal 7:65–72. https://doi.org/10.1007/s11302-011-9218-y
Ma J, Gao J, Niu M, Zhang X, Wang J, Xie A (2020) P2X4R Overexpression Upregulates Interleukin-6 and Exacerbates 6-OHDA-Induced Dopaminergic Degeneration in a Rat Model of PD. Front Aging Neurosci 12:580068. https://doi.org/10.3389/fnagi.2020.580068
Wang J, Zhang XN, Fang JN, Hua FF, Han JY, Yuan ZQ, Xie AM (2022) The mechanism behind activation of the Nod-like receptor family protein 3 inflammasome in Parkinson’s disease. Neural Regen Res 17:898–904. https://doi.org/10.4103/1673-5374.323077
Zhang X, Wang J, Gao JZ, Zhang XN, Dou KX, Shi WD, Xie AM (2021) P2X4 receptor participates in autophagy regulation in Parkinson’s disease. Neural Regen Res 16:2505–2511. https://doi.org/10.4103/1673-5374.313053
Sun Y, He L, Wang T, Hua W, Qin H, Wang J, Wang L, Gu W et al (2020) Activation of p62-Keap1-Nrf2 Pathway Protects 6-Hydroxydopamine-Induced Ferroptosis in Dopaminergic Cells. Mol Neurobiol 57:4628–4641. https://doi.org/10.1007/s12035-020-02049-3
Petrosillo G, Matera M, Casanova G, Ruggiero FM, Paradies G (2008) Mitochondrial dysfunction in rat brain with aging. Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem Int 53:126–131. https://doi.org/10.1016/j.neuint.2008.07.001
Pope S, Land JM, Heales SJR (2008) Oxidative stress and mitochondrial dysfunction in neurodegeneration; cardiolipin a critical target? Biochim Biophys Acta - Bioenerg 1777:794–799. https://doi.org/10.1016/j.bbabio.2008.03.011
Peixoto DO, Bittencourt RR, Gasparotto J, Kessler FG, Brum PO, Somensi N, Girardi CS, dos Santos da Silva L, et al (2023) Increased alpha-synuclein and neuroinflammation in the substantia nigra triggered by systemic inflammation are reversed by targeted inhibition of the receptor for advanced glycation end products (RAGE). J Neurochem.https://doi.org/10.1111/jnc.15956
Lowes H, Pyle A, Santibanez-Koref M, Hudson G (2020) Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment. Mol Neurodegener 15:1–8. https://doi.org/10.1186/s13024-020-00362-y
Borsche M, König IR, Delcambre S, Petrucci S, Balck A, Brüggemann N, Zimprich A, Wasner K et al (2020) Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 143:3041–3051. https://doi.org/10.1093/brain/awaa246
Hinkle JT, Patel J, Panicker N, Karuppagounder SS, Biswas D, Belingon B, Chen R, Brahmachari S et al (2022) STING mediates neurodegeneration and neuroinflammation in nigrostriatal α-synucleinopathy. Proc Natl Acad Sci U S A 119:e2118819119. https://doi.org/10.1073/pnas.2118819119
Wasner K, Smajic S, Ghelfi J, Delcambre S, Prada-Medina CA, Knappe E, Arena G, Mulica P et al (2022) Parkin Deficiency Impairs Mitochondrial DNA Dynamics and Propagates Inflammation. Mov Disord 37:1405–1415. https://doi.org/10.1002/mds.29025
Tresse E, Marturia-Navarro J, Sew WQ, Cisquella-Serra M, Jaberi E, Riera-Ponsati L, Fauerby N, Hu E et al (2023) Mitochondrial DNA damage triggers spread of Parkinson’s disease-like pathology. Mol Psychiatry 28(11):4902–14. https://doi.org/10.1038/s41380-023-02251-4
Xu W, Wang Y, Quan H, Liu D, Zhang H, Qi Y, Li Q, Liao J et al (2020) Double-stranded RNA-induced dopaminergic neuronal loss in the substantia nigra in the presence of Mac1 receptor. Biochem Biophys Res Commun 533:1148–1154. https://doi.org/10.1016/j.bbrc.2020.09.101
Saberi S, Stauffer JE, Schulte DJ, Ravits J (2015) Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol Clin 33:855–876. https://doi.org/10.1016/j.ncl.2015.07.012
Zhao J, Wang X, Huo Z, Chen Y, Liu J, Zhao Z, Meng F, Su Q et al (2022) The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 11:2049. https://doi.org/10.3390/cells11132049
Sasaki S, Iwata M (2007) Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 66:10–16. https://doi.org/10.1097/nen.0b013e31802c396b
Anzilotti S, Valsecchi V, Brancaccio P, et al (2021) Prolonged NCX activation prevents SOD1 accumulation, reduces neuroinflammation, ameliorates motor behavior and prolongs survival in a ALS mouse model. Neurobiol Dis 159:. https://doi.org/10.1016/j.nbd.2021.105480
Minj E, Upadhayay S, Mehan S (2021) Nrf2/HO-1 Signaling Activator Acetyl-11-keto-beta Boswellic Acid (AKBA)-Mediated Neuroprotection in Methyl Mercury-Induced Experimental Model of ALS. Neurochem Res 46:2867–2884. https://doi.org/10.1007/s11064-021-03366-2
Nukui T, Matsui A, Niimi H, Sugimoto T, Hayashi T, Dougu N, Konishi H, Yamamoto M et al (2021) Increased cerebrospinal fluid adenosine 5’-triphosphate in patients with amyotrophic lateral sclerosis. BMC Neurol 21:255. https://doi.org/10.1186/s12883-021-02288-4
Bartlett R, Ly D, Cashman NR, Sluyter R, Yerbury JJ (2022) P2X7 receptor activation mediates superoxide dismutase 1 (SOD1) release from murine NSC-34 motor neurons. Purinergic Signal 18:451–467. https://doi.org/10.1007/s11302-022-09863-5
Gandelman M, Peluffo H, Beckman JS, Cassina P, Barbeito L (2010) Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: Implications for amyotrophic lateral sclerosis. J Neuroinflammation 7:1–9. https://doi.org/10.1186/1742-2094-7-33
Gandelman M, Levy M, Cassina P, Barbeito L, Beckman JS (2013) P2X7 receptor-induced death of motor neurons by a peroxynitrite/FAS- dependent pathway. J Neurochem 126:382–388. https://doi.org/10.1111/jnc.12286
Apolloni S, Amadio S, Parisi C, Matteucci A, Potenza RL, Armida M, Popoli P, D’Ambrosi N et al (2014) Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. DMM Dis Model Mech 7:1101–1109. https://doi.org/10.1242/dmm.017038
Ruiz-Ruiz C, García-Magro N, Negredo P, Avendaño C, Bhattacharya A, Ceusters M, García AG (2020) Chronic administration of P2X7 receptor antagonist JNJ-47965567 delays disease onset and progression, and improves motor performance in ALS SOD1G93A female mice. DMM Dis Model Mech 13:dmm045732. https://doi.org/10.1242/dmm.045732
Nowicka N, Szymańska K, Juranek J, Zglejc-Waszak K, Korytko A, Załęcki M, Chmielewska-Krzesińska M, Wąsowicz KJ et al (2022) The Involvement of RAGE and Its Ligands during Progression of ALS in SOD1 G93A Transgenic Mice. Int J Mol Sci 23:2184. https://doi.org/10.3390/ijms23042184
Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, Louis C, Low RR et al (2020) TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 183:636-649.e18. https://doi.org/10.1016/j.cell.2020.09.020
Rodriguez S, Sahin A, Schrank BR, Al-Lawati H, Costantino I, Benz E, Fard D, Albers AD et al (2021) Genome-encoded cytoplasmic double-stranded RNAs, found in C9ORF72 ALS-FTD brain, propagate neuronal loss. Sci Transl Med 13:eaaz4699. https://doi.org/10.1126/scitranslmed.aaz4699
Milstead RA, Link CD, Xu Z, Hoeffer CA (2023) TDP-43 knockdown in mouse model of ALS leads to dsRNA deposition, gliosis, and neurodegeneration in the spinal cord. Cereb Cortex 33:5808–5816. https://doi.org/10.1093/cercor/bhac461
LaRocca TJ, Mariani A, Watkins LR, Link CD (2019) TDP-43 knockdown causes innate immune activation via protein kinase R in astrocytes. Neurobiol Dis 132:104514. https://doi.org/10.1016/j.nbd.2019.104514
Haki M, Al-Biati HA, Al-Tameemi ZS, Ali IS, Al-Hussaniy HA (2024) Review of multiple sclerosis: Epidemiology, etiology, pathophysiology, and treatment. Med (United States) 103:E37297. https://doi.org/10.1097/MD.0000000000037297
Denaroso GE, Smith Z, Angeliu CG, Cheli VT, Wang C, Paez PM (2023) Deletion of voltage-gated calcium channels in astrocytes decreases neuroinflammation and demyelination in a murine model of multiple sclerosis. J Neuroinflammation 20:263. https://doi.org/10.1186/s12974-023-02948-x
Hasseldam H, Rasmussen RS, Johansen FF (2016) Oxidative damage and chemokine production dominate days before immune cell infiltration and EAE disease debut. J Neuroinflammation 13:1. https://doi.org/10.1186/s12974-016-0707-3
Yu Y, Wu DM, Li J, Deng SH, Liu T, Zhang T, He M, Zhao YY et al (2020) Bixin Attenuates Experimental Autoimmune Encephalomyelitis by Suppressing TXNIP/NLRP3 Inflammasome Activity and Activating NRF2 Signaling. Front Immunol 11:593368. https://doi.org/10.3389/fimmu.2020.593368
Zhao YF, Tang Y, Illes P (2021) Astrocytic and Oligodendrocytic P2X7 Receptors Determine Neuronal Functions in the CNS. Front Mol Neurosci 14:641570. https://doi.org/10.3389/fnmol.2021.641570
Hainz N, Wolf S, Tschernig T, Meier C (2016) Probenecid Application Prevents Clinical Symptoms and Inflammation in Experimental Autoimmune Encephalomyelitis. Inflammation 39:123–128. https://doi.org/10.1007/s10753-015-0230-1
Hainz N, Wolf S, Beck A, Wagenpfeil S, Tschernig T, Meier C (2017) Probenecid arrests the progression of pronounced clinical symptoms in a mouse model of multiple sclerosis. Sci Rep 7:17214. https://doi.org/10.1038/s41598-017-17517-5
Gu BJ, Field J, Dutertre S, Ou A, Kilpatrick TJ, Lechner-Scott J, Scott R, Lea R et al (2015) A rare P2X7 variant Arg307Gln with absent pore formation function protects against neuroinflammation in multiple sclerosis. Hum Mol Genet 24:5644–5654. https://doi.org/10.1093/hmg/ddv278
Chaleomchan W, Hemachudha T, Sakulramrung R, Deesomchok U (1990) Anticardiolipin antibodies in patients with rabies vaccination induced neurological complications and other neurological diseases. J Neurol Sci 96:143–151. https://doi.org/10.1016/0022-510X(90)90127-9
Lowes H, Pyle A, Duddy M, Hudson G (2019) Cell-free mitochondrial DNA in progressive multiple sclerosis. Mitochondrion 46:307–312. https://doi.org/10.1016/j.mito.2018.07.008
Varhaug KN, Vedeler CA, Myhr KM, Aarseth JH, Tzoulis C, Bindoff LA (2017) Increased levels of cell-free mitochondrial DNA in the cerebrospinal fluid of patients with multiple sclerosis. Mitochondrion 34:32–35. https://doi.org/10.1016/j.mito.2016.12.003
Mathur V, Burai R, Vest RT, Bonanno LN, Lehallier B, Zardeneta ME, Mistry KN, Do D et al (2017) Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron 96:1290-1302.e6. https://doi.org/10.1016/j.neuron.2017.11.032
Auch CJ, Saha RN, Sheikh FG, Liu X, Jacobs BL, Pahan K (2004) Role of protein kinase R in double-stranded RNA-induced expression of nitric oxide synthase in human astroglia. FEBS Lett 563:223–228. https://doi.org/10.1016/S0014-5793(04)00302-3
Boccazzi M, Van Steenwinckel J, Schang AL, Faivre V, Le Charpentier T, Bokobza C, Csaba Z, Verderio C et al (2021) The immune-inflammatory response of oligodendrocytes in a murine model of preterm white matter injury: the role of TLR3 activation. Cell Death Dis 12:166. https://doi.org/10.1038/s41419-021-03446-9
Qin C, Yang S, Chu YH, Zhang H, Pang XW, Chen L, Zhou LQ, Chen M et al (2022) Signaling pathways involved in ischemic stroke: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther 7:215. https://doi.org/10.1038/s41392-022-01064-1
Han Y, Li X, Yang L, Zhang D, Li L, Dong X, Li Y, Qun S et al (2022) Ginsenoside Rg1 attenuates cerebral ischemia-reperfusion injury due to inhibition of NOX2-mediated calcium homeostasis dysregulation in mice. J Ginseng Res 46:515–525. https://doi.org/10.1016/j.jgr.2021.08.001
Zeng X, Zhang YD, Ma RY, Chen YJ, Xiang XM, Hou DY, Li XH, Huang H et al (2022) Activated Drp1 regulates p62-mediated autophagic flux and aggravates inflammation in cerebral ischemia-reperfusion via the ROS-RIP1/RIP3-exosome axis. Mil Med Res 9:25. https://doi.org/10.1186/s40779-022-00383-2
Yingze Y, Zhihong J, Tong J, Yina L, Zhi Z, Xu Z, Xiaoxing X, Lijuan G (2022) NOX2-mediated reactive oxygen species are double-edged swords in focal cerebral ischemia in mice. J Neuroinflammation 19:184. https://doi.org/10.1186/s12974-022-02551-6
Peng J, Wang H, Gong Z, Li X, He L, Shen Q, Pan J, Peng Y (2020) Idebenone attenuates cerebral inflammatory injury in ischemia and reperfusion via dampening NLRP3 inflammasome activity. Mol Immunol 123:74–87. https://doi.org/10.1016/j.molimm.2020.04.013
Zhang S, Qi Y, Xu Y, Han X, Peng J, Liu K, Sun CK (2013) Protective effect of flavonoid-rich extract from Rosa laevigata Michx on cerebral ischemia-reperfusion injury through suppression of apoptosis and inflammation. Neurochem Int 63:522–532. https://doi.org/10.1016/j.neuint.2013.08.008
Chin Y, Kishi M, Sekino M, Nakajo F, Abe Y, Terazono Y, Hiroyuki O, Kato F et al (2013) Involvement of glial P2Y1 receptors in cognitive deficit after focal cerebral stroke in a rodent model. J Neuroinflammation 10:1–2. https://doi.org/10.1186/1742-2094-10-95
Huang J, You X, Liu W, Song C, Lin X, Zhang X, Tao J, Chen L (2017) Electroacupuncture ameliorating post-stroke cognitive impairments via inhibition of peri-infarct astroglial and microglial/macrophage P2 purinoceptors-mediated neuroinflammation and hyperplasia. BMC Complement Altern Med 17:1–3. https://doi.org/10.1186/s12906-017-1974-y
Wilmes M, Pinto Espinoza C, Ludewig P, Stabernack J, Liesz A, Nicke A, Gelderblom M, Gerloff C et al (2022) Blocking P2X7 by intracerebroventricular injection of P2X7-specific nanobodies reduces stroke lesions. J Neuroinflammation 19:256. https://doi.org/10.1186/s12974-022-02601-z
Shen L, Wang Z, Wang R, Chen X, Cheng S (2021) Upregulation of the P2X7 receptor promotes Ca(2+) accumulation and inflammatory response in post-stroke depression. Am J Transl Res 13:10276–10287
Vasconcellos LR, Martimiano L, Dantas DP, Fonseca FM, Mata-Santos H, Travassos L, Mendez-Otero R, Bozza MT et al (2021) Intracerebral Injection of Heme Induces Lipid Peroxidation, Neuroinflammation, and Sensorimotor Deficits. Stroke 52:1788–1797. https://doi.org/10.1161/STROKEAHA.120.031911
Muhammad S, Barakat W, Stoyanov S, Murikinati S, Yang H, Tracey KJ, Bendszus M, Rossetti G et al (2008) The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci 28:12023–12031. https://doi.org/10.1523/JNEUROSCI.2435-08.2008
Mutoh T, Kikuchi H, Jitsuishi T, Kitajo K, Yamaguchi A (2023) Spatiotemporal expression patterns of ZBP1 in the brain of mouse experimental stroke model. J Chem Neuroanat 134:102362. https://doi.org/10.1016/j.jchemneu.2023.102362
Kong L, Li W, Chang E, Wang W, Shen N, Xu X, Wang X, Zhang Y et al (2022) mtDNA-STING Axis Mediates Microglial Polarization via IRF3/NF-κB Signaling After Ischemic Stroke. Front Immunol 13:860977. https://doi.org/10.3389/fimmu.2022.860977
Wu C, Zhang S, Sun H, Li A, Hou F, Qi L, Liao H (2024) STING inhibition suppresses microglia-mediated synapses engulfment and alleviates motor functional deficits after stroke. J Neuroinflammation 21:86. https://doi.org/10.1186/s12974-024-03086-8
Chauhan C, Kaundal RK (2023) Understanding the role of cGAS-STING signaling in ischemic stroke: a new avenue for drug discovery. Expert Opin Drug Discov 18:1133–1149. https://doi.org/10.1080/17460441.2023.2244409
Ding R, Li H, Liu Y, Ou W, Zhang X, Chai H, Huang X, Yang W et al (2022) Activating cGAS–STING axis contributes to neuroinflammation in CVST mouse model and induces inflammasome activation and microglia pyroptosis. J Neuroinflammation 19:137. https://doi.org/10.1186/s12974-022-02511-0
Liao Y, Cheng J, Kong X, Li S, Li X, Zhang M, Zhang H, Yang T et al (2020) HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics 10:9644–9662. https://doi.org/10.7150/thno.47651
Wu X, Zheng Y, Liu M, Li Y, Ma S, Tang W, Yan W, Cao M et al (2021) BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy 17:1934–1946. https://doi.org/10.1080/15548627.2020.1802089
Mishra Y, Kaundal RK (2023) Role of SIRT3 in mitochondrial biology and its therapeutic implications in neurodegenerative disorders. Drug Discov Today 28:103583. https://doi.org/10.1016/j.drudis.2023.103583
Staal J, Blanco LP, Perl A (2023) Editorial: Mitochondrial dysfunction in inflammation and autoimmunity. Front Immunol 14:1304315. https://doi.org/10.3389/fimmu.2023.1304315
Sukhorukov V, Voronkov D, Baranich T, Mudzhiri N, Magnaeva A, Illarioshkin S (2021) Impaired mitophagy in neurons and glial cells during aging and age-related disorders. Int J Mol Sci 22:10251. https://doi.org/10.3390/ijms221910251
Ye J, Hu X, Wang Z, Li R, Gan L, Zhang M, Wang T (2023) The role of mtDAMPs in the trauma-induced systemic inflammatory response syndrome. Front Immunol 14:1164187. https://doi.org/10.3389/fimmu.2023.1164187
Petrara MR, Bonfante F, Costenaro P, Cantarutti A, Carmona F, Ruffoni E, Di Chiara C, Zanchetta M et al (2021) Asymptomatic and Mild SARS-CoV-2 Infections Elicit Lower Immune Activation and Higher Specific Neutralizing Antibodies in Children Than in Adults. Front Immunol 12:741796. https://doi.org/10.3389/fimmu.2021.741796
Acknowledgements
The authors gratefully acknowledge the Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Government of India, for their financial support. Additionally, Ashutosh Kumar sincerely thanks the Science and Engineering Research Board, Department of Science and Technology, Government of India, for providing funding to support ongoing research in his laboratory (grant EEQ/2021/000875). The authors also appreciate the use of BioRender.com for the creation of several figures included in this review article.
Author information
Authors and Affiliations
Contributions
Yogesh Mishra performed the literature search and wrote the manuscript. Ashutosh Kumar and Ravinder Kumar Kaundal conceptualized the study and provided feedback on earlier drafts. All authors reviewed and approved the final version for publication.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• Neurons exhibit susceptibility to the accumulation of dysfunctional mitochondria.
• Dysfunctional mitochondria release mtDAMPs, which possess immunogenic properties.
• mtDAMPs trigger inflammatory cascades, elevating the levels of cytotoxic mediators that drive neuroinflammation and neuronal death.
• mtDAMPs have emerged as potential predictive biomarkers for neuropathological conditions.
• Targeting dysfunctional mitochondria and mtDAMPs holds promise for mitigating the pathogenesis of neurodegenerative disorders.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mishra, Y., Kumar, A. & Kaundal, R.K. Mitochondrial Dysfunction is a Crucial Immune Checkpoint for Neuroinflammation and Neurodegeneration: mtDAMPs in Focus. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04412-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04412-0