
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease with an unknown pathogenesis. It has been reported that mutations in the gene for Cu/Zn superoxide dismutase (SOD1) cause familial ALS. Mutant SOD1 undergoes aggregation and forms amyloid more easily, and SOD1-immunopositive inclusions have been observed in the spinal cords of ALS patients. Because of this, SOD1 aggregation is thought to be related to the pathogenesis of ALS. Some core regions of amyloid have been identified, but the issue of whether these regions form aggregates in living cells remains unclear, and the mechanism responsible for intracellular SOD1 aggregation also remains unclear. The findings reported in this study indicate that the aggregation of the ALS-linked mutant SOD1-EGFP was significantly enhanced when the BioID2 gene was fused to the N-terminus of the mutant SOD1-EGFP plasmid for cellular expression. Expression of a series of BioID2-(C-terminal deletion peptides of SOD1)-EGFP permitted us to identify 1–35 as a minimal N-terminal sequence and Ile35 as an essential amino acid residue that contributes to the intracellular aggregation of SOD1. The findings also showed that an additional substitution of Ile35 with Ser into the ALS mutant SOD1 resulted in the significant suppression of aggregate formation. The fact that no Ile35 mutations have been reported to date in ALS patients indicates that all ALS mutant SOD1s contain Ile35. Taken together, we propose that Ile35 plays a pivotal role in the aggregation of the ALS-linked SOD1 and that this study will contribute to our understanding of the mechanism responsible for SOD1 aggregation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease that specifically damages motor neurons and eventually results in generalized muscle atrophy and paralysis. Approximately 20% of familial ALS (fALS) cases are attributed to mutations in the gene encoding cytoplasmic Cu/Zn superoxide dismutase (SOD1) [1]. SOD1, a homodimer of subunits composed of 153 amino acids, is primarily a principal anti-oxidative enzyme that catalyzes the conversion of superoxide anion radicals into the less reactive hydrogen peroxide and molecular oxygen. To date, more than 200 mutations in the SOD1 gene have been identified as causing ALS (http://alsod.iop.kcl.ac.uk). Transgenic mice that overexpress the ALS-linked mutant human SOD1 gene develop ALS-like symptoms and SOD1-immunoreactive inclusion bodies in their spinal cords [2, 3]. In contrast, SOD1 knockout mice show some impairment [4, 5] but no ALS-like phenotypes [6]. Therefore, it is widely thought that pathology associated with ALS results from a gain of toxic function rather than a loss of function. However, the mechanisms responsible for the motor neuron degeneration associated with ALS caused by mutant SOD1 have not been fully elucidated.
The misfolding of a protein molecule and the formation of inclusion bodies containing protein aggregates are major pathological hallmarks in neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and ALS [7, 8]. ALS-linked mutant SOD1 proteins are significantly less stable than wild-type SOD1 [9, 10]. Although both WT and ALS mutant SOD1 can form fibrillary amyloid structures by de-metallation and the reduction of the intramolecular disulfide bond [11,12,13], ALS mutant SOD1 proteins are more prone to misfolding, undergoing aggregation, and forming amyloid [14,15,16]. In addition, immunoreactive inclusions that are specific for misfolded SOD1 antibodies have been observed in spinal cords of both fALS and sporadic ALS patients [17]. Because of this, efforts have been made to elucidate the mechanism responsible for SOD1 aggregation. To date, some core regions involved in SOD1 fibril aggregation have been studied by computational methods and by mass spectrometric analysis of non-specific protease-digested residues of amyloid fibrils formed from the full-length SOD1 protein. The results indicated that the following amino acid residues, 1–63 [18], three different regions (1–30, 90–120, 135–153) [19], three other shorter regions (14–22, 95–114, 145–153) [20], or four regions (14–21, 30–38, 101–107, and 147–153) [21] are involved in the amyloid formation of SOD1. Notably, recombinant peptides that included core regions, 1–55 and 80–153, formed insoluble amyloid, whereas the 24–79 and 56–118 peptides remained in a supernatant fraction [19]. This result is consistent with the structure of wild-type SOD1 amyloid fibers determined by cryo-electron microscopy (Cryo-EM), showing that SOD1 amyloid is composed of N-terminal segments (3–55) and C-terminal segments (86–153) [22]. However, the issue of whether these amyloid core regions form aggregates in living cells continues to be unclear.
Previous studies have reported that the Trp32 (W32) residue in human SOD1 appeared to have a role in its ability to propagate pathological aggregation and the W32S or W32F substitution in the mutant SOD1 decreased but did not completely block this aggregation [23,24,25,26]. Therefore, a knowledge of the amino acid residues that contribute to intracellular SOD1 aggregation more effectively than W32 would be highly desirable.
In this study, we report that the aggregation of the ALS-linked mutant SOD1-EGFP is significantly enhanced when the BioID2 gene was fused to the N-terminus of the mutant SOD1-EGFP plasmid for cellular expression. This cellular assay system represents a potentially important tool for observing protein aggregates in living cells and provides the impetus to search for core sequences and amino acid residues that are directly responsible for SOD1 aggregation. By expressing a series of BioID2-truncated SOD1 fragments, EGFP, we succeeded in identifying 1–35 and Ile35 as a minimal N-terminal sequence and a key amino acid residue, respectively, responsible for intracellular SOD1 aggregation. This region, therefore, represents a new target for the development of ALS therapies.
Methods
Reagents and Antibodies
All chemicals used were of analytical grade without any further purification and were obtained from Nacalai Tesque (Kyoto, Japan) or FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Anti-vimentin (Proteintech), CF®594 goat anti-rabbit IgG (H + L) (BIOTReND), CF®594 goat anti-mouse IgG (H + L) (BIOTReND), anti-GFP (Santacruz), anti-rabbit IgG (HRP-conjugated) (Cell Signaling Technology), and anti-β-actin pAb-HRP-DirecT (MBL) were used. New anti-SOD1 mAb, 19G6G, was obtained from Kitayama Labes Co., Ltd. (Nagano Japan).
Construction of Truncated BioID2-SOD1-EGFP
The coding sequence of human wild-type (WT) SOD1 in pcDNA3.1 [10] was amplified by PCR and cloned into pEGFP-N3 (Clontech). The plasmid myc‐BioID2‐pBABE-puro was a generous gift from Kyle Roux (AddGene plasmid #80900). The amplified PCR products of myc‐BioID2 were inserted into the N-terminus of SOD1-EGFP in the N3 plasmid via In-Fusion recombination (Takara Bio, Japan) using XhoI and PstI restriction enzyme site primers 1 and 2. The remaining multicloning sites in pEGFP-N3 between BioID2 and SOD1 and the start codon of SOD1 were deleted by inverse PCR using a KOD mutagenesis kit (Toyobo, Japan), using primers 3 and 4. The primers are described below. A series of truncated WT or mutant SOD1 molecules in the myc‐BioID2‐SOD1-EGFP-N3 plasmid were produced by inverse PCR using the forward primers and the backward primers according to the human SOD1 nucleotide sequence (GenBank, X02317.1). The myc‐BioID2‐SOD1-mCherry plasmid was produced by substituting the EGFP gene for the mCherry gene. The mCherry gene was a generous gift from Dr. Tomohiko Taguchi (Tohoku University). All constructs were confirmed by DNA sequencing.
-
Primer 1 (forward)
-
GGACTCAGATCTCGAGTGGCTAGCCACCATGGAA
-
Primer 2 (reverse)
-
CGCGGTACCGTCGACTGCAGCTCGAGGCTTCTTCTCAG
-
Primer 3 (forward)
-
GCGACGAAGGCCGTGTGCGTGCTG
-
Primer 4 (reverse)
-
GCTTCTTCTCAGGCTGAACTCGCC
Cell Cultures
Human embryonic kidney 293A (HEK293A) cells were a generous gift from Dr. Makoto Urushitani (Shiga University of Medical Science). The cells were maintained in Dulbecco’s Modified Eagle Medium (Sigma-Aldrich) supplemented with 10% (v/v) fetal bovine serum (Biowest) at 37 °C, 5% CO2, and a 95% humidity. The cells (1.5 × 105 cells) were then seeded in a 6-well collagen-coated cell culture plate or 1.0 × 106 cells in a 100-mm cell culture dish 24 h before transfection. Plasmid DNA was transfected using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Fluorescence Microscope Observation
At 48 h after transfection, HEK293A cells were fixed by treatment with a 4% paraformaldehyde phosphate-buffered solution, washed with PBS, and then imaged with a fluorescent cell imager (Floid Cell Imaging Solution; Thermo Fisher Scientific, USA).
Immunofluorescence
HEK293A cells were seeded at a density of 1.0 × 104 cells/dish in Ibidi (Martinsried, Germany, product #80136) coated dishes (35 mm diameter) or at a density of 1.5 × 105 cells/well in 6-well cell culture plates on poly-L-lysine coated glass coverslips (Matsunami Glass Ind., Ltd., #C1110) 24 h before transfection. Plasmid DNA was transfected using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol. At 24 h or 48 h post transfection, the cells were fixed by treatment with a 4% paraformaldehyde phosphate-buffered solution. After blocking with 5% (w/v) bovine serum albumin BSA and 0.2% (v/v) Triton X-100 in PBS for 1 h, the cells were incubated anti-vimentin (1:500, Proteintech) or anti-SOD1 (mAb 19G6G) in PBS containing 0.1% (v/v) Triton X-100 for 1 h at room temperature and then washed with PBS three times. The cells were then incubated with anti-rabbit or mouse IgG conjugated with CF®594 goat anti-rabbit IgG (H + L) (1:500, BIOTReND) in PBS containing 0.1% (v/v) Triton X-100 for 1 h at room temperature. After washing three times with PBS, DAPI (Nacalai Tesque) diluted in PBS (1.0 μg/mL) was added for 10 min to stain nuclei. The immunostained cells were imaged on LSM780 confocal laser scanning microscopy (Carl Zeiss, Jena, Germany) or a fluorescent cell imager (FLoid Cell Imaging Solution; Thermo Fisher Scientific, USA).
Structural Analysis
The starting structure for human SOD1 was obtained from a 1.8 Å crystal structure (3T5W) [27]. Induction of the I35S mutation and preparation of the molecular graphics were performed with the UCSF ChimeraX 1.5 software, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases [28].
Fractionation of RIPA-Soluble and Insoluble Proteins
At 48 h post transfection, the cultured HEK293A cells were placed on ice, the culture medium was removed, and the cells were washed twice with PBS ( −). The cells were detached by treatment with 1 mL of cold RIPA buffer (50 mM tris–HCl, pH8.0, 150 mM sodium chloride, 0.5% (w/v) sodium deoxycholate, 0.1% (w/v) sodium dodecyl sulfate, 1.0% (w/v) NP-40 substitute) (FUJIFILM Wako) containing complete protease inhibitor (Roche, Basel, Switzerland)), followed by incubation at 4 °C for 20 min. The detached cells were collected into a microtube and centrifuged at 15,000 × g for 20 min at 4 °C, and the supernatant was collected as RIPA-soluble fractions. The remaining pellet was resuspended in 1 mL of the cold RIPA buffer and centrifuged at 15,000 × g for an additional 20 min at 4 °C. After the removal of the supernatant fraction, the pellet was resolved in 200 µL of 8 M urea by sonication (Bioruptor Cosmo Bio) for 10 min and centrifuged at 15,000 × g for 20 min at 4 °C. The supernatant fraction was collected as RIPA-insoluble proteins. These samples were examined by SDS-PAGE and immunoblotting as a “soluble-insoluble assay.”
Immunoblotting
Protein concentrations of the soluble and insoluble protein fractions were measured by the BCA assay (Nacalai Tesque). The protein samples were separated by SDS-PAGE on a 10% gel (ATTO) and transferred onto a PVDF membrane (Millipore, USA) by semi-dry blotting (Bio-Rad). The membrane was incubated for 1 h at room temperature in TBS-T containing 5% (w/v) skim milk (Difco, Detroit, MI, USA). The blocked membrane was reacted with the appropriate antibodies. The target proteins were detected by a chemiluminescence method using Luminata Western HRP substrate (Merck Millipore) using an LAS3000 luminescent image analyzer (FUJIFILM, Tokyo, Japan).
Statistical Analyses
Numerical values are expressed as the mean ± SEM. The statistical analyses were performed using a one-way ANOVA test with the Tukey post hoc test.
Results
BioID2 Conjugation at N-Terminus Facilitates the Aggregation of SOD1-EGFP
The expression plasmids in which the BioID2 sequence was fused to the N-terminus of each mutant SOD1-EGFP and the original SOD1-EGFP plasmids were transiently transfected into HEK293A cells. As shown in Fig. 1A, we observed that the cells expressing the BioID2-ALS mutant (A4V, H46R, G93A) SOD1-EGFP formed more abundant larger aggregates/inclusions compared with cells expressing the original ALS mutant SOD1 (A4V, H46R, G93A)-EGFP. The frequency of aggregate-positive cells showed that the BioID2 conjugation significantly enhanced the aggregation of these ALS mutant SOD1-EGFPs by approximately tenfold (Fig. 1B). Even cells that were expressing BioID2-SOD1 (WT)-EGFP formed some aggregates (approximately 15%), while the frequency of aggregate-positive cells of all the BioID2 conjugated ALS mutant EGFP was significantly higher than that of BioID2-WT-EGFP (p < 0.001). In the case of the empty control, BioID2-EGFP, the EGFP fluorescence was distributed diffusely throughout the cells without any aggregates being detected (Fig. 1). Moreover, the other typical ALS-linked mutant SOD1, G37R, G85R, C111Y, and I113T (http://alsod.iop.kcl.ac.uk) also formed intracellular aggregates (Fig. S1A and S1C). In contrast, the non-ALS mutant SOD1, cytoprotective T2D [29] and W32S [23], murine SOD1 sequence L42Q [30], and the oxidative stress stable C111S [31, 32] only formed aggregates at a level similar to that for BioID2-SOD1 (WT)-EGFP (Fig. S1B and S1C). These results indicate that the fusion of the BioID2 protein at the N-terminus significantly promotes the aggregation of ALS-associated mutant SOD1 in the absence of any stresses. In addition, HEK293A cells expressing BioID2-G93A-EGFP were immunostained with an anti-SOD1 monoclonal antibody (mAb 19G6G). As shown in Fig. S2, the aggregates were stained with the mAb 19G6G (SOD1 in red). These data eliminated the concern that the green fluorescence in the aggregates could be due to the presence of free EGFP or truncated SOD1-EGFP fusion proteins.

BioID2 conjugation at N-terminus facilitates the formation of intracellular aggregates of SOD1-EGFP. A HEK293A cells were transiently transfected with expression plasmids for SOD1-EGFP or BioID2-conjugated SOD1-EGFP. At 48 h post transfection, cells were fixed with 4% paraformaldehyde, and then, images were then captured. B The aggregate-positive cells and GFP-positive cells in eight images (approximately 60–90 cells/image) were counted to quantify the frequency of aggregate-positive cells. Data are presented as the mean ± SEM. The statistical analyses were performed using a one-way ANOVA test with the Tukey post hoc test. The frequency of aggregate-positive cells of BioID2-SOD1-EGFP was significantly higher than that of SOD1-EGFP including WT (***p < 0.001). In addition, the frequency of aggregate-positive cells of all BioID2-conjugated ALS mutant EGFP was significantly higher than that of BioID2-WT-EGFP (p < 0.001)
To further investigate the role of BioID2 in promoting aggregate formation in SOD1, we constructed several plasmids expressing BioID2-SOD1-mCherry, in which EGFP was replaced by the red fluorescent protein, mCherry. As shown in Fig. S3A, both BioID2-A4V-mCherry and BioID2-G93A-mCherry rarely formed aggregates, as did BioID2-WT SOD1-mCherry. Their aggregate-positive cell frequencies were significantly lower than that of BioID2-WT SOD1-EGFP (Fig. S3B). These data indicate that not only BioID2 fusion but also EGFP fusion promotes the aggregation of ALS mutant SOD1. The significant difference observed between EGFP (aggregation promotion) and mCherry (no aggregation promotion) is particularly intriguing. The crystal structures of EGFP (PDB 4EUL) and mCherry (PDB 2H5Q) both display a similar β-barrel structure comprised of 11 β-strands and a core helix containing the chromophore. Despite these structural similarities, it remains unclear why EGFP and mCherry exhibit different effects on aggregation promotion when fused with BioID2-SOD1. Further studies will be needed to elucidate this phenomenon. Nevertheless, the sandwich expression system between BioID2 and EGFP would be an excellent tool for observing intracellular aggregates of ALS-associated mutant SOD1s or other polypeptides that are prone to misfolding. In addition, HEK293A cells were used in this study because their large cell size makes it easier to observe intracellular aggregates, although they are not neuronal cells.
The Amyloidogenic Region and the Intracellular Aggregation Region Do Not Necessarily Coincide
To date, several amyloidogenic core regions of SOD1 have been identified by MS–MS analysis after the digestion of the amyloid with a non-specific protease. Furukawa et al. reported that recombinant SOD1 peptides, 1–55 and 80–153, that include the amyloidogenic core region, became insoluble amyloid, whereas peptides 24–79 and 56–118 remained in the supernatant fraction [19]. However, the issue of whether these amyloidogenic regions form intracellular aggregates remains unclear. We therefore examined the issue of whether or not these regions form aggregates in living cells using BioID2-fused SOD1 peptide-EGFP expression constructs. As shown in Fig. 2A, the 1–55 and 56–118 peptides formed intracellular aggregates, whereas 24–79 and 80–153 formed only minimal levels of aggregates. The frequency of aggregate formation for the 24–79 and 80–153 peptides was significantly lower than that of WT (Fig. 2B). These results indicate that the N-terminal side, 1–55 (aggregate and amyloid) and 24–79 (no aggregates and soluble), were consistent between this intracellular aggregation experiment and the previous in vitro amyloidogenesis study [19]. However, the C-terminal sides, 56–118 (aggregate vs. soluble) and 80–153 (no aggregates vs. amyloid), were not in agreement with these studies. Since the conditions for amyloid formation and intracellular aggregation could be different, it is possible that both regions do not necessarily coincide.

The intracellular aggregate-forming region is consistent with the amyloid formation only on the N-terminal side. A HEK293A cells were transiently transfected with expression plasmids for 1–55, 24–79, 56–118, and 80–153 of wild-type SOD1 conjugated between BioID2 and EGFP. At 48 h post transfection, the cells were fixed with 4% paraformaldehyde, and images were then captured. B The aggregate-positive cells and GFP-positive cells in eight images (approximately 60–90 cells/image) were counted to quantify the frequency of aggregate-positive cells. Data are presented as the mean ± SEM. The statistical analyses were performed using a one-way ANOVA test with the Tukey post hoc test. The frequency of aggregate-positive cells of all peptides was significantly different compared to WT (***p < 0.001)
1–35 Is Required for Intracellular SOD1 Aggregation
Nonsense or frameshift mutations, such as L126X, G127X, G141X, or C146X, have been reported in fALS patients [33,34,35,36], which indicates that the C-terminal sequences of SOD1 may not be involved in the pathology of ALS and the formation of inclusion bodies in the lesion sites. Consistent with previous in vitro amyloidogenesis studies [18, 19], 1–55 formed intracellular aggregates as shown in Fig. 2. We therefore focused on a search for the N-terminal sequence responsible for SOD1 aggregation using our BioID2-SOD1-EGFP system, which makes it much easier to observe the intracellular aggregates. As shown in Fig S4A, plasmids containing a series of C-terminal truncated SOD1 molecules (1–30, 1–60, 1–90, 1–120, and 1–140) between the BioID2 and EGFP were constructed, and the plasmids were transiently transfected into HEK293A cells. The levels of expression of the series of BioID2‐trancated SOD1-EGFP were confirmed by the immunoblotting of the cell lysates (Fig. S4B).
As shown in Fig. 3A, BioID2-SOD1 peptide-EGFP containing amino acid residues, 1–60, 1–90, 1–120, and 1–140 (ALS mutant, G141X) of SOD1, formed more abundant aggregates compared to the full-length (WT-SOD1) although the 1–30 peptide formed no aggregates. Since both 1–40 and 1–50 formed the aggregates (Fig. 3B), the amino acid sequence was further shortened. As a result, residues 1–36 and 1–38 formed aggregates, but 1–32 and 1–34 did not (Fig. 3C). We also demonstrated that 1–35 and 1–36, but not 1–34, formed aggregates (Fig. 3D). Significant differences were observed in frequency of aggregate-positive cells for BioID2-1–35-EGFP versus BioID2-1–34-EGFP (Fig. 3E). Therefore, we finally concluded that a peptide containing amino acid residues “1–35” of SOD1 is the minimal N-terminal sequence that contributes to intracellular aggregation.

Determination of the minimum N-terminal sequence required for SOD1 aggregation. C-terminal deletion constructs of SOD1 fused between BioID2 and EGFP were transiently transfected in HEK293A cells. After 48 h, the cells were fixed with 4% paraformaldehyde, and then, images were then captured. A Expression plasmids for 1–30, 1–60, 1–90, 1–120, 1–140, and 1–153 (full length, WT) of wild-type SOD1 conjugated between BioID2 and EGFP were transfected. B Expression plasmids for 1–30, 1–40, and 1–50 of wild-type SOD1 conjugated between BioID2 and EGFP were transfected. C Expression plasmids for 1–32, 1–34, 1–36, and 1–38 of wild-type SOD1 conjugated between BioID2 and EGFP were transfected. D Expression plasmids for 1–34, 1–35, and 1–36 of wild-type SOD1 conjugated between BioID2 and EGFP were transfected. E The aggregate-positive cells and GFP-positive cells in eight images (approximately 55–150 cells/image) were counted to quantify the frequency of aggregate-positive cells. Data are presented as the mean ± SEM. The statistical analyses involved the use of the one-way ANOVA test with the Tukey post hoc test. The frequency of aggregate-positive cells of all peptides was significantly different compared to BioID2-1–30-EGFP (left graph) or BioID2-1–32-EGFP (right graph), respectively (***p < 0.001)
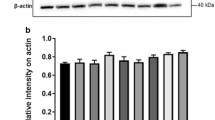
To evaluate the relationship between hydrophobicity and aggregation, we compared the solubility of 1–32, 1–34, 1–35, and 1–36 of wild-type SOD1 expressed in the BioID2-EGFP sandwich system in a soluble-insoluble assay. Proteins that were insoluble in the RIPA buffer were solubilized by treatment with 8 M urea. As shown in Fig. 4A, B, the levels of the soluble fractions of 1–35 and 1–36 were significantly lower than those of 1–32 and 1–34, while the levels of insoluble fractions of 1–35 and 1–36 were significantly higher. These data indicate that there is a significant difference in solubility between 1–34 and 1–35 expressed in the BioID2-EGFP sandwich system.

The N-terminal SOD1 peptides containing I35 are more hydrophobic. A Expression plasmids for 1–32, 1–34, 1–35, and 1–36 of wild-type SOD1 conjugated between BioID2 and EGFP were transfected in HEK293A cells. After 48 h, the cells were harvested, and then, soluble proteins and insoluble proteins were fractionated. A total of 15 μg of protein per lane for soluble fractions and 1 μg of protein per lane for insoluble fractions were applied on SDS-PAGE and analyzed by immunoblotting. Anti-GFP antibody (1:3000, Santacruz) was used as the primary antibody, and anti-rabbit IgG (1:3000, Cell Signaling Technology) was used as the secondary antibody. The same membrane was stained with anti-β-actin pAb-HRP-DirecT (1:3000, MBL) as an internal control. Representative immunoblot for soluble and insoluble fractions of cells expressing 1–32, 1–34, 1–35, and 1–36 conjugated between BioID2 and EGFP is shown. B Quantification of the immunoblotting data shown is the mean ± SEM. (n = 3). The statistical analyses were performed using a one-way ANOVA test with the Tukey post hoc test. The immunoblot levels of BioID2-peptide-EGFP/β-actin were compared to that of BioID2-(1–34)-EGFP/β-actin (**p < 0.005, *p < 0.05)
Substitution of I35 by Hydrophilic Amino Acids Blocked Aggregation of the 1–35 Peptide
Figure 5A shows the N-terminal sequences of various species of SOD1. The 35th amino acid residue of SOD1 is conserved as Ile in mammals and fish and Leu or Val in Drosophila and plants (Fig. 5A). Moreover, there is no report of a mutation linked to fALS at I35 (http://alsod.iop.kcl.ac.uk), which indicates that all ALS mutant SOD1s harbor I35. We therefore hypothesized that a branched-chain amino acid conserved at the 35th amino acid residue would be needed for SOD1 aggregation. I35 in BioID2‐(1–35)-EGFP was then substituted with other branched-chain amino acids, i.e., leucine (I35L) or valine (I35V), and hydrophilic amino acids, serine (I35S), threonine (I35T), or lysine (I35K). As shown in Fig. 5B, C, I35L and I35V clearly formed aggregates, while I35S, I35T, and I35K did not form any aggregates. These results indicate that the presence of a branched-chain amino acid at the 35th amino acid residue is required for aggregation and that the replacement of I35 with a hydrophilic amino acid could block this type of aggregation. In addition, Zhong et al. proposed that the normally buried nuclear export signal (NES)-like sequence of SOD1 encompasses residues 33–51 (Fig. 5A) [30]. Indeed, SOD1 peptides shorter than 1–40, lacking the NES-like sequence, were therefore found to be localized in both the nucleus and cytoplasm (Fig. 3), but 1–55 and 24–79 harboring the NES-like sequence were observed to be localized in the cytoplasm (Fig. 2).

Substitution of I35 conserved in mammalian SOD1s to hydrophilic amino acids blocked aggregation of the 1–35 peptide. A Comparison of N-terminal sequences of SOD1 proteins between different species. Amino acids highlighted in yellow in human SOD1 are reported to be ALS-associated mutations. The 35th amino acid is highlighted in blue, and the 32nd amino acid is boxed in red. B Expression plasmids for 1–35, 1–35 (I35L), 1–35 (I35V), 1–35 (I35S), 1–35 (I35T), and 1–35 (I35K) of wild-type SOD1 conjugated between BioID2 and EGFP were transfected in HEK293A cells. After 48 h, the cells were fixed with 4% paraformaldehyde, and then, images were captured. C The aggregate-positive cells and GFP-positive cells in eight images (approximately 75–110 cells/image) were counted to quantify the frequency of aggregate-positive cells. Data are presented as the mean ± SEM. The statistical analyses were performed using a one-way ANOVA test with the Tukey post hoc test. The frequency of aggregate-positive cells of all peptides was significantly different compared to BioID2-1–35 (I35)-EGFP (***p < 0.001)
Substituting I35 with Serine (I35S) Suppressed the Aggregation of the ALS Mutant SOD1 to a Greater Extent than Substituting Trp32 with Serine (W32S)
Based on previous studies, it is possible that the tryptophan residue at position 32 (W32) of human SOD1 is important for the pathology of ALS because the oxidation of W32 results in the formation of covalently crosslinked non-native SOD1 dimers which could be a cytotoxic seed for aggregation [24]. In addition, substituting W32 with serine (W32S) reduced the seeding aggregation of the mutant SOD1 [23, 37]. However, our results showed that 1–32 and 1–34 formed no aggregates even though they contained W32 (Fig. 3). Therefore, to determine which residue between W32 and I35 is more important for SOD1 aggregation, the effects of substituting I35 and W32 with serine on the aggregation of the ALS mutant SOD1 were compared. This experiment also allowed us to confirm whether our assay system using the artificial fusion protein, BioID2-SOD1-EGFP, reflects the previous results showing that W32S inhibited the aggregation of the ALS mutant SOD1 [23, 37]. As shown in Fig. 6A, the presence of either W32S or I35S decreased the extent of aggregation of the A4V, H46R, and G93A mutations including WT SOD1 when using the BioID2-EGFP sandwich expression system. Measurement of the frequency of aggregate-positive cells clearly demonstrated that both the W32S and I35S substitutions statistically reduced aggregate formation compared to the original SOD1s. It is noteworthy that the I35S substitution significantly reduced the extent of aggregation in all ALS mutant SOD1s more than the W32S substitution (Fig. 6B). These results indicate that the contribution of I35 is much greater than that of W32 in the aggregation of SOD1 in living cells and that our BioID2-EGFP sandwich system accurately reproduced findings in previous reports [23, 37]. The soluble-insoluble assay also showed that the levels of the insoluble fractions of A4V or G93A were significantly higher than those for A4V/I35S or G93A/I35S, respectively (Fig. 7A, B). These data indicate that substituting I35 with serine (I35S) suppressed the hydrophobicity of the ALS mutant SOD1 when expressed in the BioID2-EGFP sandwich system.

Substituting Ile35 with serine (I35S) suppressed the aggregation of the ALS mutant SOD1 to a greater extent than substituting Trp32 with serine (W32S). A Expression plasmids for BioID2 conjugated SOD1 (wild-type (WT), A4V, H46R, and G93A)-EGFP with additional mutations of W32S or I35S were transfected in HEK293A cells. After 48 h, the cells were fixed with 4% paraformaldehyde, and images were then captured. B The aggregate-positive cells and GFP-positive cells in eight images (approximately 60–90 cells/image) were counted to quantify the frequency of aggregate-positive cells. Data are presented as the mean ± SEM. Statistical analyses were performed using a one-way ANOVA test with the Tukey post hoc test (*p < 0.05; **p < 0.01, ***p < 0.001)

Addition of the I35S mutation increased the hydrophilicity of BioID2-G93A-EGFP and BioID2-A4V-EGFP. A Expression plasmids for A4V, A4V/I35S, G93A, and G93A/I35S conjugated between BioID2 and EGFP were transfected in HEK293A cells. After 48 h, the cells were harvested, and then, soluble proteins and insoluble proteins were fractionated. A total of 15 μg of protein per lane for soluble fractions and 1 μg of protein per lane for insoluble fractions were applied on SDS-PAGE and analyzed by immunoblotting. Anti-GFP antibody (1:3000, Santacruz) was used as the primary antibody, and anti-rabbit IgG (1:3000, Cell Signaling Technology) was used as the secondary antibody. The same membrane was stained with anti-β-actin pAb-HRP-DirecT (1:3000, MBL) as an internal control. Representative immunoblot for soluble and insoluble fractions of cells expressing A4V, A4V/I35S, G93A, and G93A/I35S conjugated between BioID2 and EGFP is shown. B Quantification of the immunoblotting data shown are the mean ± SEM. (n = 3). The statistical analyses were performed using a one-way ANOVA test with the Tukey post hoc test. The immunoblot levels of BioID2-A4V/I35S-EGFP/β-actin and BioID2-G93A/I35S-EGFP/β-actin were compared to BioID2-A4V-EGFP/β-actin and BioID2-G93A-EGFP/β-actin, respectively (**p < 0.005, *p < 0.05)
Additional G33V Mutation Promotes the Aggregation of the ALS Mutant SOD1
Sangwan et al. reported that corkscrew-like antiparallel beta-sheet oligomers, not amyloid, consisting of a segment 28–38 of SOD1 are toxic and that the substitution of G33 to Val or Trp destroys the corkscrew-like antiparallel beta-sheet oligomers [38]. The authors also demonstrated that the additional G33V mutation reduced neuronal cytotoxicity when these 28–38 oligomers were supplemented with medium and that in a zebrafish ALS model expressing G93A or A4V, the addition of G33V mutation alleviated the axonal damage and mitochondrial defects associated with ALS [38]. Therefore, we constructed new plasmids expressing BioID2-28–38-EGFP, BioID2-WT SOD1/G33V-EGFP, and BioID2-A4V/G33V-EGFP to determine whether 28–38 is an aggregation-prone peptide and whether G33V substitution suppresses aggregate formation. Unexpectedly, the findings indicated that BioID2-28–38-EGFP did not form aggregates even though it includes I35 (Fig. 8A, B). More surprisingly, the addition of a G33V mutation significantly promoted the aggregate formation of BioID2-WT-EGFP and BioID2-A4V-EGFP (Fig. 8C, D).

BioID2-28–38-EGFP formed no aggregates, and the addition of G33V promoted the aggregation of BioID2-SOD1-EGFP. A Expression plasmids for 1–34, 1–38, and 28–38 of wild-type SOD1 conjugated between BioID2 and EGFP were transfected in HEK293A cells. After 48 h, the cells were fixed with 4% paraformaldehyde, and images were then captured. B The aggregate-positive cells and GFP-positive cells in eight images (approximately 90–130 cells/image) were counted to quantify the frequency of aggregate-positive cells. Data are presented as the mean ± SEM. The statistical analyses were performed using a one-way ANOVA test with the Tukey post hoc test. The frequency of aggregate-positive cells of 1–38 but not 28–38 is significantly different compared to 1–34 (***p < 0.001). C Expression plasmids for BioID2-conjugated SOD1 (WT and A4V)-EGFP with or without additional mutation of G33V were transfected in HEK293A cells. After 48 h, the cells were fixed with 4% paraformaldehyde, and images were then captured. D The aggregate-positive cells and GFP-positive cells in eight images (approximately 90–160 cells/image) were counted to quantify the frequency of aggregate-positive cells. Data are presented as the mean ± SEM. The statistical analyses were performed using a one-way ANOVA test with the Tukey post hoc test (***p < 0.001)
Intracellular BioID2-SOD1-EGFP Aggregates Resemble Aggresomes
Our BioID2-EGFP sandwich system generated perinuclear aggregates of mutant SOD1 in the absence of inhibitors of proteasome or autophagy. The large perinuclear aggregates of the mutant SOD1 with a strong fluorescence intensity resembled structures that are commonly referred to as aggresomes [39,40,41], which are trafficked to the centrosomes and surrounded by a vimentin cage type of structure. Therefore, we immunostained these aggregates with anti-vimentin as a marker for aggresomes. As shown in Fig. 9A and S5, vimentin formed ring-like structures (white arrows) around the perinuclear aggregates of BioID2-G93A-EGFP ((d) and (d’) (enlarged view of enclosure part of (d))), similar to previous reports regarding aggresomes [39,40,41]. The Z-stacking images and the 3D image (Fig. S5) indicate that the aggregate appears to be surrounded by vimentin. The tiny aggregates of BioID2-(1–35)-EGFP were also present in close proximity to the vimentin, as shown in Fig. 9B, (h), (h’) (enlarged view of the enclosure part of (h)) although a clear relationship with vimentin was not observed. Further research will be needed to clarify how BioID2-ALS mutant SOD1-EGFP or BioID2-SOD1 peptide-EGFP forms such structure within cells.

BioID2-G93A-EGFP aggregates are surrounded by vimentin cages. Expression plasmids for BioID2-G93A-EGFP (A) and BioID2-(1–35)-EGFP (B) were transfected in HEK293A cells. After 24 h, the cells were fixed with 4% paraformaldehyde and processed for immunofluorescence of vimentin. The cells were observed with a LSM780 confocal laser scanning microscope
Discussion
BioID2 is a smaller promiscuous biotin ligase and an improved version of the “Biotin IDentification (BioID) method” for biotinylating and identifying neighboring proteins that interact with proteins in living cells [42]. Naturally, the objective of our ongoing study was to identify neighboring proteins that interact with the ALS mutant SOD1. On the other hand, our findings indicated that an N-terminal fusion of the BioID2 protein unexpectedly promoted the aggregation of the ALS-associated mutant SOD1-EGFP (Fig. 1 and Fig. S1). However, replacing EGFP with mCherry failed to promote aggregate formation in BioID2-SOD1 (Fig. S3). Therefore, we hypothesized that the BioID2-EGFP sandwich expression system could be a good tool for identifying aggregation-prone sequences of SOD1. By expressing a series of BioID2-(a C-terminal deletion peptide of SOD1)-EGFP, we succeeded in identifying 1–35 and Ile35 (I35) as the minimal N-terminal sequences and amino acid residues required for intracellular aggregation. Indeed, the substitution of I35 with serine (I35S) blocked the aggregation of residues 1–35 (Fig. 5) and the ALS-linked mutant SOD1 (Fig. 6). The positions of residues 1–35 and I35 in the three-dimensional structure of SOD1 are indicated in Fig. 10A. I35 is present in beta-strand III, and its hydrophobic side chain points to the interior of the SOD1 molecule. As shown in Fig. 10B-a, I35 is surrounded by 9 hydrophobic amino acid residues within a distance of 3 Å. In contrast, there are only 4 hydrophobic amino acid residues within a distance of 3 Å around S35 when the computational mutation of I35S is induced (Fig. 10B(b)). Indeed, the substitution of I35 in BioID2‐(1–35)-EGFP with other branched-chain amino acids also resulted in the formation of aggregates, while no aggregation was observed when I35 was substituted with hydrophilic amino acids formed (Fig. 5B, C). The soluble-insoluble assay also showed that the levels of the insoluble fractions of the I35-containing segments, 1–35 and 1–36, were significantly higher than 1–32 and 1–34 when expressed in the BioID2-EGFP sandwich system (Fig. 4). Furthermore, the levels of the insoluble fractions of A4V or G93A were significantly higher than those for A4V/I35S or G93A/I35S, respectively, when expressed in the BioID2-EGFP sandwich system (Fig. 7). We therefore conclude that the hydrophobicity of I35 contributes to the aggregation of SOD1 in living cells and that the substitution of I35 with Ser (I35S) reduces the hydrophobic effect and prevents aggregation. Additionally, our atomistic molecular dynamics simulations of the SOD1 structure suggest that I35 is involved in the large fluctuations in the electrostatic loop (loop VII) due to the G93A mutation (Fukushima K and Fujiwara N, unpublished work). Since conformational change in the electrostatic loop is considered to be closely related to the aggregation process of ALS mutant SOD1, I35 is thought to be an important residue for conformational change and the aggregation of SOD1.

Illustration of Ile35 in three-dimensional structure of SOD1. A Schematic diagram of the three-dimensional structure of SOD1 (PDB:3T5W). Residues 1–35 and Ile35 of the SOD1 dimer are colored pink and red in chain A and light blue and blue in chain B, respectively. B I35S mutation was induced by using the UCSF ChimeraX 1.5. The C atoms of Ile35 are colored orange-red (a) and the C atoms and the O atom of Ser35 are colored blue and red, respectively. Hydrophobic amino acids contacted with Ile35 or Ser35 are colored green, and other amino acids are colored pale orange. The molecular graphics were prepared with UCSF ChimeraX 1.5
Prior studies have shown that W32 potentiates SOD1 aggregation because the substitution of W32 with Ser or Phe suppressed aggregation [23,24,25,26]. We also observed that W32S partially blocked the aggregation of the ALS mutant SOD1 in the BioID2 fusion system, although the suppression efficiency was lower than that of I35S (Fig. 6). In fact, W32 is not conserved among mammalian SOD1, and indeed, the mouse and horse SOD1 have S32 and K32, respectively (Fig. 5A). Transgenic mice that express mouse SOD1 with a G86R mutation, which corresponds to a human G85R mutation, developed an ALS-like disease that features motor neuron loss and SOD1-immunopositive inclusions [43, 44]. These reports indicate that W32 is not required for the development of ALS. Furthermore, 1–32 and 1–34, despite the fact that they include W32, exhibited no aggregation in our assay system (Fig. 3C–E). In contrast, I35 is conserved in mammalian SOD1 (Fig. 5A), and no mutations at I35 have been reported in fALS patients to date (http://alsod.iop.kcl.ac.uk), indicating that all ALS mutant SOD1s harbor the I35 that is responsible for SOD1 aggregation. The inhibitory effect of I35S on ALS mutant SOD1 aggregation was also stronger than that of W32S when using our BioID2-EGFP sandwich system (Fig. 6). These data indicate that the contribution of I35 to the SOD1 aggregation linked to the pathogenesis associated with ALS is greater than that of W32. A comparative study of symptoms between traditional ALS model organisms (such as G93A) and organisms expressing the ALS mutation with I35S (such as G93A/I35S) will be needed to answer the question of whether the additional mutation, I35S, suppresses ALS development. In contrast, the addition of a G33V mutation significantly enhanced BioID2-WT-EGFP and BioID2-A4V-EGFP aggregate formation (Fig. 8C, D). Gly33 is also present in beta-strand III near the I35 forming the hydrophobic region of SOD1 (Fig. 10). It is thought that the substitution of G33 to Val would further increase the hydrophobicity of this hydrophobic region, thus making aggregate formation easier. However, it was reported that the addition of the G33V mutation to G93A or A4V alleviated the axonal damage and mitochondrial defects associated with ALS in a zebrafish ALS model [38]. These data suggest that aggregate formation might not be directly involved in the pathogenesis of ALS although the BioID2-ALS mutant SOD1-EGFP does indeed form aggregates (Fig. 1 and S1).
Aggresomes have been observed mostly under conditions of proteasome or autophagy inhibition [41, 45, 46]. However, BioID2-conjugated ALS mutant SOD1 aggregates were formed, even in the absence of proteasome or autophagy inhibitors. Notably, large aggregates of BioID2‐G93A-EGFP closely resemble aggresomes since they appear to be surrounded by a vimentin cage (Fig. 9A and S5). On the other hand, these aggregates did not co-localize with the Golgi marker, GM-130 (data not shown), which is consistent with previous studies of cystic fibrosis transmembrane conductance regulator aggresomes [41]. To date, it has been proposed that “aggresomes” composed of misfolded SOD1 proteins result in cell death [47]. However, Zhu et al. reported that large SOD1 aggregates were not responsible for cytotoxicity and that large aggregates might rather be protective [48]. In the case of polyglutamine-containing proteins, it was proposed that aggresomes sequester misfolded proteins to protect cells from their toxicity [49]. We also observed that large aggregates of BioID2-ALS mutant SOD1-EGFP remained in surviving cells even at 72 h after transfection. We therefore hypothesize that encapsulating toxic soluble ALS mutant SOD1 proteins (oligomers) into aggregates may protect cells from their toxicity, while non-toxic proteins do not need to be sequestered and therefore do not form aggregates. Further studies will be needed to determine whether the aggregate formation of SOD1 is directly involved in the pathogenesis of ALS including neuronal cytotoxicity.
Additionally, it is important to elucidate the mechanism by which BioID2 accelerates the propensity of mutant SOD1-EGFP to undergo aggregation. The BioID2 used in this study is the biotin protein ligase from Aquifex aeolicus, a 26 kDa protein composed of two domains with 11 β-strands and five α-helices [50]. BioID2 may influence the stability and/or solubility of SOD1-EGFP. Furthermore, its relatively large size as a tag protein results in an artificial fusion protein. However, given that the BioID2-EGFP system can detect the impact of single amino acid changes on aggregate formation within a protein or peptide, we conclude that it has potential as a useful tool for studying aggregate formation in proteins associated with various neurodegenerative diseases. On the other hand, the BioID2 fusion system was originally established as a tool for biotin labeling and identifying proximate proteins that interact with proteins in cells [42]. However, in this study, we focused on the identification of amino acid residues or sequences that are involved in the aggregate formation of SOD1 using this BioID2-EGFP sandwich system. We recently initiated a new study to identify proteins that bind to ALS mutant SOD1 but not to its I35 serine mutant SOD1 (and vice versa). This research is expected to shed light on the mechanism responsible for the aggregation of SOD1 and lead to the discovery of therapeutic drugs for ALS.
Conclusions
In the present study, we report that BioID2 fusion greatly enhanced the formation of intracellular aggregates of ALS-linked mutant SOD1-EGFP. In addition, this expression plasmid made it possible to distinguish between aggregate-forming and non-aggregate-forming sequences by the presence or absence of only one amino acid residue. Using a BioID2-EGFP sandwich expression system, we successfully identified that Ile35 plays a pivotal role in the intracellular aggregation of SOD1. We propose that the hydrophobicity of Ile35 contributes to SOD1 aggregation and that making this site hydrophilic would suppress aggregation. The current study and future research will contribute to the elucidation of the mechanism responsible for the aggregation of ALS-linked mutant SOD1 and the development of therapeutics for ALS.
Data Availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- fALS:
-
Familial ALS
- SOD1:
-
Cu/Zn superoxide dismutase
- EGFP:
-
Enhanced green fluorescent protein
- PBS:
-
Phosphate-buffered saline
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- RIPA:
-
Radio-immunoprecipitation assay
- BCA:
-
Bicinchoninic acid
- ANOVA:
-
Analysis of variance
- MS:
-
Mass spectrometry
- SEM:
-
Standard error of the mean
References
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM (1996) Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci U S A 93:3155–3160
Bruijn LI, Miller TM, Cleveland DW (2004) Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 27:723–749
Yoshihara D, Fujiwara N, Kitanaka N, Kitanaka J, Sakiyama H, Eguchi H, Takemura M, Suzuki K (2016) The absence of the SOD1 gene causes abnormal monoaminergic neurotransmission and motivational impairment-like behavior in mice. Free Radic Res 50:1245–1256
Sakiyama H, Fujiwara N, Yoneoka Y, Yoshihara D, Eguchi H, Suzuki K (2016) Cu, Zn-SOD deficiency induces the accumulation of hepatic collagen. Free Radic Res 50:667–677
Reaume A, Elliott J, Hoffman E et al (2016) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13:43–47
Cicardi ME, Marrone L, Azzouz M, Trotti D (2021) Proteostatic imbalance and protein spreading in amyotrophic lateral sclerosis. EMBO J 40:e106389
Davis AA, Leyns CEG, Holtzman DM (2018) Intercellular spread of protein aggregates in neurodegenerative disease. Annu Rev Cell Dev Biol 34:545–568
Perry JJ, Shin DS, Getzoff ED, Tainer JA (2010) The structural biochemistry of the superoxide dismutases. Biochim Biophys Acta 1804(2):245–262
Fujiwara N, Miyamoto Y, Ogasahara K, Takahashi M, Ikegami T, Takamiya R, Suzuki K, Taniguchi N (2005) Different immunoreactivity against monoclonal antibodies between wild-type and mutant copper/zinc superoxide dismutase linked to amyotrophic lateral sclerosis. J Biol Chem 280(6):5061–5070
Fujiwara N, Wagatsuma M, Oba N, Yoshihara D, Tokuda E, Sakiyama H, Eguchi H, Ichihashi M, Furukawa Y, Inoue T, Suzuki K (2018) Cu/Zn-superoxide dismutase forms fibrillar hydrogels in a pH-dependent manner via a water-rich extended intermediate state. PLoS ONE 13(10):e0205090
Vassall KA, Stubbs HR, Primmer HA, Tong MS, Sullivan SM, Sobering R, Srinivasan S, Briere LA, Dunn SD, Colón W, Meiering EM (2011) Decreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALS. Proc Natl Acad Sci U S A 108(6):2210–2215
Münch C, Bertolotti A (2012) Exposure of hydrophobic surfaces initiates aggregation of diverse ALS-causing superoxide dismutase-1 mutants. J Mol Biol 421(4–5):491–498
DiDonato M, Craig L, Huff ME, Thayer MM, Cardoso RM, Kassmann CJ, Lo TP, Bruns CK, Powers ET, Kelly JW, Getzoff ED, Tainer JA (2003) ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J Mol Biol 332(3):601–615
Valentine JS, Doucette PA, Zittin PS (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem 74:563–593
Lindberg MJ, Tibell L, Oliveberg M (2002) Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc Natl Acad Sci U S A 99(26):16607–16612
Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH Jr (2010) Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci 13(11):1396–1403
Chan PK, Chattopadhyay M, Sharma S, Souda P, Gralla EB, Borchelt DR, Whitelegge JP, Valentine JS (2013) Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc Natl Acad Sci U S A 110(27):10934–10939
Furukawa Y, Kaneko K, Yamanaka K, Nukina N (2010) Mutation-dependent polymorphism of Cu, Zn-superoxide dismutase aggregates in the familial form of amyotrophic lateral sclerosis. J Biol Chem 285(29):22221–22231
Ida M, Ando M, Adachi M, Tanaka A, Machida K, Hongo K, Mizobata T, Yamakawa MY, Watanabe Y, Nakashima K, Kawata Y (2016) Structural basis of Cu, Zn-superoxide dismutase amyloid fibril formation involves interaction of multiple peptide core regions. J Biochem 159(2):247–260
Ivanova MI, Sievers SA, Guenther EL, Johnson LM, Winkler DD, Galaleldeen A, Sawaya MR, Hart PJ, Eisenberg DS (2014) Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc Natl Acad Sci U S A 111(1):197–201
Wang LQ, Ma Y, Yuan HY, Zhao K, Zhang MY, Wang Q, Huang X, Xu WC, Dai B, Chen J, Li D, Zhang D, Wang Z, Zou L, Yin P, Liu C, Liang Y (2022) Cryo-EM structure of an amyloid fibril formed by full-length human SOD1 reveals its conformational conversion. Nat Commun 13(1):3491
Pokrishevsky E, McAlary L, Farrawell NE, Zhao B, Sher M, Yerbury JJ, Cashman NR (2018) Tryptophan 32-mediated SOD1 aggregation is attenuated by pyrimidine-like compounds in living cells. Sci Rep 8(1):15590
Taylor DM, Gibbs BF, Kabashi E, Minotti S, Durham HD, Agar JN (2007) Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J Biol Chem 282(22):16329–16335
DuVal MG, Hinge VK, Snyder N, Kanyo R, Bratvold J, Pokrishevsky E, Cashman NR, Blinov N, Kovalenko A, Allison WT (2019) Tryptophan 32 mediates SOD1 toxicity in a in vivo motor neuron model of ALS and is a promising target for small molecule therapeutics. Neurobiol Dis 124:297–310
Crown A, McAlary L, Fagerli E, Brown H, Yerbury JJ, Galaleldeen A, Cashman NR, Borchelt DR, Ayers JI (2020) Tryptophan residue 32 in human Cu-Zn superoxide dismutase modulates prion-like propagation and strain selection. PLoS ONE 15(1):e0227655
Ihara K, Fujiwara N, Yamaguchi Y, Torigoe H, Wakatsuki S, Taniguchi N, Suzuki K (2012) Structural switching of Cu, Zn-superoxide dismutases at loop VI: insights from the crystal structure of 2-mercaptoethanol-modified enzyme. Biosci Rep 32(6):539–548
Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE (2021) UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci 30:70–82
Fay JM, Zhu C, Proctor EA, Tao Y, Cui W, Ke H, Dokholyan NV (2016) A phosphomimetic mutation stabilizes SOD1 and rescues cell viability in the context of an ALS-associated mutation. Structure 24(11):1898–1906
Zhong Y, Wang J, Henderson MJ, Yang P, Hagen BM, Siddique T, Vogel BE, Deng HX, Fang S (2017) Nuclear export of misfolded SOD1 mediated by a normally buried NES-like sequence reduces proteotoxicity in the nucleus. Elife 2(6):e23759
Fujiwara N, Nakano M, Kato S, Yoshihara D, Ookawara T, Eguchi H, Taniguchi N, Suzuki K (2007) Oxidative modification to cysteine sulfonic acid of Cys111 in human copper-zinc superoxide dismutase. J Biol Chem 282(49):35933–35944
Cozzolino M, Amori I, Pesaresi MG, Ferri A, Nencini M, Carrì MT (2008) Cysteine 111 affects aggregation and cytotoxicity of mutant Cu, Zn-superoxide dismutase associated with familial amyotrophic lateral sclerosis. J Biol Chem 283(2):866–874
Guissart C, Mouzat K, Kantar J, Louveau B, Vilquin P, Polge A, Raoul C, Lumbroso S (2020) Premature termination codons in SOD1 causing amyotrophic lateral sclerosis are predicted to escape the nonsense-mediated mRNA decay. Sci Rep 10(1):20738
Wu J, Shen E, Shi D, Sun Z, Cai T (2012) Identification of a novel Cys146X mutation of SOD1 in familial amyotrophic lateral sclerosis by whole-exome sequencing. Genet Med 14(9):823–826
Alvarez S, Calin A, Graffmo KS, Moldovan M, Krarup C (2013) Peripheral motor axons of SOD1(G127X) mutant mice are susceptible to activity-dependent degeneration. Neuroscience 25(241):239–249
Maglemose R, Hedegaard A, Lehnhoff J, Dimintiyanova KP, Moldovan M, Grøndahl L, Meehan CF (2017) Potassium channel abnormalities are consistent with early axon degeneration of motor axons in the G127X SOD1 mouse model of amyotrophic lateral sclerosis. Exp Neurol 292:154–167
Grad LI, Guest WC, Yanai A, Pokrishevsky E, O’Neill MA, Gibbs E, Semenchenko V, Yousefi M, Wishart DS, Plotkin SS, Cashman NR (2011) Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci U S A 108(39):16398–16403
Sangwan S, Zhao A, Adams KL, Jayson CK, Sawaya MR, Guenther EL, Pan AC, Ngo J, Moore DM, Soriaga AB, Do TD, Goldschmidt L, Nelson R, Bowers MT, Koehler CM, Shaw DE, Novitch BG, Eisenberg DS (2017) Atomic structure of a toxic, oligomeric segment of SOD1 linked to amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 114(33):8770–8775
Ogrodnik M, Salmonowicz H, Brown R, Turkowska J, Średniawa W, Pattabiraman S, Amen T, Abraham AC, Eichler N, Lyakhovetsky R, Kaganovich D (2014) Dynamic JUNQ inclusion bodies are asymmetrically inherited in mammalian cell lines through the asymmetric partitioning of vimentin. Proc Natl Acad Sci U S A 111(22):8049–8054
Rujano MA, Bosveld F, Salomons FA, Dijk F, van Waarde MA, van der Want JJ, de Vos RA, Brunt ER, Sibon OC, Kampinga HH (2006) Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol 4(12):e417
Johnston JA, Ward CL, Kopito RR (1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143(7):1883–1898
Sears RM, May DG, Roux KJ (2019) BioID as a tool for protein-proximity labeling in living cells. Methods Mol Biol 2012:299–313
Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW (1995) Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 92(3):689–693
Morrison BM, Janssen WG, Gordon JW, Morrison JH (1998) Time course of neuropathology in the spinal cord of G86R superoxide dismutase transgenic mice. J Comp Neurol 391(1):64–77
Kaganovich D, Kopito R, Frydman J (2008) Misfolded proteins partition between two distinct quality control compartments. Nature 454(7208):1088–1095
Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115(6):727–738
Matsumoto G, Stojanovic A, Holmberg CI, Kim S, Morimoto RI (2005) Structural properties and neuronal toxicity of amyotrophic lateral sclerosis-associated Cu/Zn superoxide dismutase 1 aggregates. J Cell Biol 171(1):75–85
Zhu C, Beck MV, Griffith JD, Deshmukh M, Dokholyan NV (2018) Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 115(18):4661–4665
Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH (2003) Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet 12(7):749–757
Minde DP, Ramakrishna M, Lilley KS (2020) Biotin proximity tagging favours unfolded proteins and enables the study of intrinsically disordered regions. Commun Biol 3(1):38
Acknowledgements
We appreciate Dr. Leonard Chavas (Nagoya University) for their discussion of the BioID2-SOD1-EGFP fusion protein structure. In addition, we thank Dr. Makoto Urushitani (Shiga University of Medical Science) and Dr. Tomohiko Taguchi (Tohoku University) for the generous gift of the HEK293A cells and the mCherry gene, respectively. We also wish to thank Dr. Milton S Feather from Technical Editing Services for editing a draft of this manuscript.
Funding
This work was supported by Grants-in-Aid 22K11870 for Scientific Research (C) (to NF), a grant from the Japan Foundation for Applied Enzymology (to NF), and in part supported by “Hyogo Medical University Diversity Grant for Research Promotion” under MEXT Funds for the Development of Human Resources in Science and Technology, “Initiative for Realizing Diversity in the Research Environment (Characteristic-Compatible Type) (to NF) and Hyogo Medical University Physician-Scientist Training Program (to YA).
Author information
Authors and Affiliations
Contributions
Noriko Fujiwara directed the project and all authors contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Yoshiyuki Asai, Kyoka Yano, Tomoyuki Higashino, Daisaku Yoshihara, Haruhiko Sakiyama, Hironobu Eguchi, Keiichiro Suzuki, and Noriko Fujiwara. Structural analysis was performed by Kazuaki Fukushima. The first draft of the manuscript was written by Noriko Fujiwara and all authors commented on previous versions of the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Asai, Y., Yano, K., Higashino, T. et al. The Ile35 Residue of the ALS-Associated Mutant SOD1 Plays a Crucial Role in the Intracellular Aggregation of the Molecule. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04369-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04369-0