
Abstract
Neuroinflammation is a common pathological feature in a number of neurodegenerative diseases, which is mediated primarily by the activated glial cells. Nucleotide-binding oligomerization domain-like receptor pyrin domain-containing-3 (NLRP3) inflammasome-associated neuroinflammatory response is mostly considered. To investigate the situation of the NLRP3-related inflammation in prion disease, we assessed the levels of the main components of NLRP3 inflammasome and its downstream biomarkers in the scrapie-infected rodent brain tissues. The results showed that the transcriptional and expressional levels of NLRP3, caspase-1, and apoptosis-associated speck-like protein (ASC) in the brains of scrapie-infected rodents were significantly increased at terminal stage. The increased NLPR3 overlapped morphologically well with the proliferated GFAP-positive astrocytes, but little with microglia and neurons. Using the brain samples collected at the different time-points after infection, we found the NLRP3 signals increased in a time-dependent manner, which were coincidental with the increase of GFAP. Two main downstream cytokines, IL-1β and IL-18, were also upregulated in the brains of prion-infected mice. Moreover, the gasdermin D (GSDMD) levels, particularly the levels of GSDMD-NT, in the prion-infected brain tissues were remarkably increased, indicating activation of cell pyroptosis. The GSDMD not only co-localized well with the astrocytes but also with neurons at terminal stage, also showing a time-dependent increase after infection. Those data indicate that NLRP3 inflammasomes were remarkably activated in the infected brains, which is largely mediated by the proliferated astrocytes. Both astrocytes and neurons probably undergo a pyroptosis process, which may help the astrocytes to release inflammatory factors and contribute to neuron death during prion infection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Prion diseases, also named transmissible spongiform encephalopathies, are a kind of fatal, progressive neurodegenerative diseases which affect humans and various species of mammals. Human prion diseases can be sporadic, inherited, or acquired, with a long incubation period and a short disease course [1,2,3]. The typical neuropathological features of prion diseases include cavernous degeneration, amyloid plaque deposition of PrPSc, neuron loss, and glial cell proliferation [2, 4]. Up to now, there is lacking of specific therapeutic and prophylactic tools for efficiently relieving symptoms or delaying onset.
Glial cells mainly include microglia and astrocytes, which are generally considered to be important neuroimmune cells in the central nervous system (CNS). Neuroinflammation is usually manifested by microglial activation and astrocyte proliferation, accompanied by transcriptome changes, which is a common marker of various neurodegenerative diseases, including prion diseases. Neuroinflammatory responses are mainly induced by a variety of inflammasomes, in which NLRP3 is a key regulatory protein [5,6,7]. NLRP3 inflammasome is a protein complex composed of one NLRP3 containing a PYD domain, one pro-caspase-1 (proCas1) containing a CARD domain, and one ASC containing both domains mentioned above [5, 8, 9]. Stimulated by pathogen-associated molecular patterns (PAMP) or host-derived damage-associated molecular patterns (DAMP), neuroimmune cells rapidly recruit amounts of proCas1 and ASC that assemble with NLRP3 into inflammasomes via PYD and CARD domains. This process leads to the activation of proCas1 into active caspase-1 (Cas1) that further proteolyzes some pro-inflammatory cytokines (e.g., Il-1β, IL-18) and gasdermin-D (GSDMD) [10,11,12,13]. The proteolyzed N-terminal structure (GSDMD-NT) may bind to the acidic phospholipids in the inner lobe of the plasma membrane, punch holes in cell membrane, release the proinflammatory agents, and eventually induce the death of inflammatory cells [14, 15].
NLRP3 inflammasome are described to promote deterioration on the animal models of some neurodegeneration diseases, such as Alzheimer disease (AD), Parkinson disease (PD), and Huntington disease (HD)[5, 16, 17]. Knockout of NLRP3 in a transgenic mouse model of AD protects from spatial memory dysfunction and reduces the deposition of β-amyloid protein [9, 18]. In the PD mouse model, siRNA-mus-NLRP3 plays a significant neuroprotective role through inhibiting the injury neuron and the proliferation of glial cells [19, 20]. However, the role of NLRP3 inflammasomes in the neuroinflammatory response of prion diseases is unclear.
In the present study, the status of NLRP3 inflammasome in the brain tissues of several prion-infected rodent models was comprehensively assessed by different methodologies. The mRNA levels of NLPR3, Cas1, ASC, IL-1β, IL-18, and GSDMD in the brain of infected rodents were tested by reverse transcription quantitative PCR (qRT-PCR). The brain levels of NLPR3, Cas1, ASC, and GSDMD were assessed by Western blots and immunofluorescence assay. The morphological overlaps of NLPR3 with neurons, microglia, and astrocytes were detected by double-stained immunofluorescent assays. The brain levels of molecular complexes of NLRP3 with proCas1 and ASC were detected by immunoprecipitation (IP) assay and Western blots. Il-1β and IL-18 were also observed in the brains of scrapie-infected animals through ELISA.
Materials and Methods
Ethics Statement
Usage of animal specimens in this study was approved by the Ethical Committee of the National Institute for Viral Disease Prevention and Control, China CDC under protocol 2009ZX10004-101. Animal housing and experimental protocols were in accordance with the Chinese Regulations for the Administration of Affairs Concerning Experimental Animals.
Brain Samples of Scrapie-Infected Rodent Models
The brain specimens of the experimental rodents infected with various scrapie agents were enrolled, including 263 K-infected hamsters, 139A- and ME7-infected mice. The animal challenge of intracerebral inoculation was described previously [21,22,23] and the incubation periods of scrapie agent 263 K-infected hamsters, as well as 139A- and ME7-infected mice, were 66.7 ± 11 days, 183.9 ± 23.1 days, and 184.2 ± 11.8 days, respectively. The brain tissues of the age-matched normal hamsters (80 days old) and normal mice (180 days old) were used as the control. Additionally, the brain tissues of 139A- and ME7-infected mice collected on 80-, 120-, 150-, and 180-day post-inoculation (dpi) were recruited into this study, which represented early, middle-early, middle-late, and terminal stage, respectively. Totally, nine groups of mice (groups of control, 139A-80d, 139A-120d, 139A-150d, 139A-180d, ME7-80d, ME7-120d, ME7-150d, ME7-180d) and two groups of hamsters (groups of control, 263 K) were recruited in this study, each group containing three animals. After surgical removal of the brain of the experimental rodent, the whole brain was vertically separated, half of hemisphere for preparation of tissue homogenate and the other for formalin fixation.
Preparation of Brain Homogenates
According to the protocol described previously [2], to prepare the brain homogenates of mice and hamsters, the brain samples were homogenized in cold lysis buffer (containing 100 mM NaCl, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, 10 mM EDTA and protease inhibitor cocktail set III (Calbiochem)) after weighing (1 g/10 mL). Crude homogenates were spun shortly, and aliquots of the supernatants were immediately frozen at 80 °C for subsequent experiments.
Preparation of Paraffin Sections of Brain Tissue
The brains of normal and scrapie-infected rodents were fixed with 4% paraformaldehyde at 4 °C for 16 h, and then moved to an embedding box and rinsed under running tap water for 3 h. The fixed brain tissues were serially dehydrated and soaked in xylene for 1.5 h. The soaked tissue was embedded, and the tissue slices were prepared according to the routine neuropathological protocol.
Quantitative Reverse Transcription PCR (qRT-PCR)
Total RNAs from the brain tissues of 139A- and ME7-infected mice collected at end-stage and the age matched health mice were extracted with of the Total RNA Rapid Extraction Kit (R218, GeneBetter, China) according to the instructions of the manufacturer. The first-strand cDNA synthesis was conducted with commercial Script III RT MasterMix kit (P518, GeneBetter, China). The specific primers were designed based on the sequences issued in GenBank and summarized in Table 1. Real-time PCR was performed on a PCR instrument (ABI 7900HT, Applied Biosystems, USA) with the ChamQ Universal SYBR qPCR Master Mix (Q711-02, Vazyme, China). All PCR processes were performed in triplicate with a total of 40 cycles (30 s at 95 °C, 30 s at 60 °C, 60 s at 72 °C). The relative transcriptional level of mRNA was calculated by Ct method (2 − ΔΔCt).
Immunofluorescence Assay (IFA)
After incubated in the repair buffer (containing 2 mM citric acid and 10 mM trisodium citrate) and heated in microwave oven for 30 min, the brain sections were permeabilized with 0.3% Triton X-100 in PBS for 30 min and then blocked with normal goat serum for 1 h. The slices were incubated with different specific antibodies, including 1:100 diluted mouse anti-NLRP3, 1:200 diluted rabbit anti-GFAP (HPA056030, Millipore, USA), 1:200 diluted rabbit anti-NeuN (ab177487, Abcam, Britain), and 1:500 diluted rabbit anti-Iba1 (019–19741, WAKO) at 4 °C overnight. After washing, the sections were incubated with 1:200-diluted Alexa Fluor 488-labeled goat-derived anti-rabbit (A11034, Invitrogen, USA) and Alexa Fluor 568-labeled goat-derived anti-mouse (A11031, Invitrogen, USA) secondary antibodies at 37 °C for 1 h, and stained with DAPI (1 mg/mL) at room temperature (RT) for 15 min. The slices were sealed, and the images were viewed and analyzed using confocal laser microscope system (Leica TCS/SP8, Germany). The images of cortex and hippocampus were captured and the relevant optical values were quantitatively analyzed.
Western Blots
Ten percent of brain homogenates were separated by 12% SDS-PAGE and electronically transferred to nitrocellulose membranes with a semi-dry facility. After blocking with 5% nonfat-dried milk in TBS at 37 °C for 1 h, the membranes were incubated at 4 °C overnight with specific monoclonal or polyclone antibody, including anti-NLRP3 (1:000 dilution, AG-20B-0014-C100, AdipoGen, Switzerland), anti-ASC (1:500 dilution, Santa Cruz, USA), and anti-Caspase-1 (1:500 dilution, 06–503-I, Millipore, USA). After washing with TBST (containing 0.1% Tween-20, pH 7.6) for four times, the membranes were incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibody (1:5000 dilution, 115–035-003/111–035-003, Jackson, USA). Immunoreactive signals were developed using an enhanced ECL kit (PE Applied Biosystems, Foster City, CA, USA). Images were captured by ChemiDocTM XRSC Imager (Bio-Rad, USA). The gray values of the target blots were calculated by software ImageJ. The relative gray value of each target protein was normalized with that of the individual internal control protein β-actin. Each group contained three animals.
Immunoprecipitation (IP) Assay
The possible molecular interaction of NLRP3 with ACS or proCas1 in the brains of prion-infected rodents and their normal controls were evaluated by immunoprecipitation assays. Briefly, Dynabeads Protein G (Life Technologies, 10004D) were separately incubated with 10 μg anti-NLRP3, or same amount of normal mouse and hamster IgG on a vertical rotating mixer at 4 °C for 4 h. After washed with PBST (PBS containing 0.05% Tween 20) for five times to remove unbound antibodies, various 10% brain homogenates with the total protein content of 100 μg were added and incubated at 4 °C overnight on a vertical rotating mixer. Protein G compounds were pelleted with a magnet and washed five times with PBST. The precipitated products were eluted by 5 μl of protein loading buffer and 20 μl of PBS with vertexing and heated at 100 °C for 5 min prior to separating by 12% SDS-PAGE. The signals of ASC or proCas1 in the immunoprecipitated products were assayed by the Western blots with anti-ASC or anti-proCas1 separately. Aliquots of brain homogenates were directly loaded as the input control.
ELISA
The brain homogenates were diluted to 500 μg/μl (total protein concentration) with a bicinchoninic acid protein assay kit (71,285–3, Novagen, USA). The levels of IL-1β and IL-18 were separately measured with commercial enzyme-linked immunosorbent assay (ELISA) kits (EK0394 for IL-1β, EK0433 for IL-18, Boster, China) according to the manufacturer’s instruction.
Statistics
All experiments were repeated at least three times in this study. Quantitative analyses of immunoblots were carried out with software ImageJ. The integral optical density (IOD) values of each field-specific fluorescence staining were collected automatically by the software equipped in the confocal laser microscope. Each IOD values were averaged with the data of three different views. The relative IOD values of the specific staining were determined by normalized with that of DAPI-specific staining. Data shown were means ± SD of triplicate samples from a single experiment and were representative of three independent experiments. Statistical analyses were conducted using Student’s t test. *P < 0.05; **P < 0.01; ***P < 0.001.
Results
Increased Expression of NLRP3 in the Brains of Prion-Infected Rodent Models at Terminal Stage
To assess the status of the inflammasome during prion infection, the brain tissues of several prion-infected rodents at terminal stage were enrolled into this study. The transcriptional levels of brain NLRP3, one of the components of inflammasome, were evaluated by NLRP3-specifc qRT-PCR. Compared to were 1.5–3 folds increased (Fig. 1A). IFAs with anti-NLRP3 revealed more and larger NLRP3-stained cells in the brain sections (cortex and hippocampus regions) of 139A- and ME7-infected mice, as well as that of 263 K-infected hamsters, with significantly higher IOD values compared to the individual normal control (Fig. 1B). NLRP3-specifc Western blots identified notably stronger bands in the brain homogenates of 139A- and ME7-infected mice (Fig. 1C), and 263 K-infected hamsters (Fig. 1D), with statistical differences in the quantitative assays normalized with the data of the individual normal control. It strongly indicates that the expressions of brain NLRP3 in the prion-infected rodent models at final stage are remarkably upregulated.

Analyses of brain NLRP3 in the prion-infected rodent models at terminal stageA NLRP3-specific qRT-PCR of the brain RNAs of scrapie agents 139A- or ME7-infected mice and the age-matched mice. Relative fold changes of NLRP3 mRNAs are marked on Y-axis. Graphical data denote mean + SD (n = 3). B Representative images of NLRP3-specific IFAs of the brain sections of scrapie agents 139A- and ME7-infected mice (left) and 263 K-infected hamsters (right). Relative fluorescent intensities of NLRP3 signals (red) in the brain slices of prion-infected animals compared to the individual controls (n = 3) are showed on the bottom. C NLRP3-specific Western blots of the brain homogenates of 139A- and ME7-infected mice. D NLRP3-specific Western blots of the brain homogenates of 263 K-infected hamsters. The relative gray values of NLRP3 normalized with the individual β-actin (n = 3) are marked below each panel
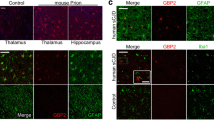
Morphological Colocalization of the Increased NLRP3 with the Proliferated Astrocytes in the Brains of Prion-Infected Rodents
The brain sections of 139A- and ME7-infected mice and 263 K-infected hamsters were subjected into double-stained IFAs with anti-NLRP3 and anti-GFAP, Iba1, or anti-NeuN separately. Confocal images showed obvious morphological overlaps of NLRP3 (red) with GFAP (green) signals in the background of amounts of large astrocytes both in the regions of cortex and hippocampus of 139A- and ME7-mice (Fig. 2A, left panel), and those of 263 K-infected hamsters (2B, left panel). NLRP3-stained asteroid cells (red) distributed around the Iba1 (middle panel) and NeuN (right panel)-stained cells (green), however, did not overlap each other in the brain sections of normal and infected animals. It implies the increased NLRP3 in the prion-infected experimental rodents mainly distributes in the proliferated astrocytes.

Double-stained IFAs for morphological colocalization of NLRP3 with various biomarkers in the brain sections of the prion-infected rodent models at terminal stage. A Representative merged images of NLRP3 (red) with GFAP (green, left), NeuN (green, middle), or Iba1(green, right) in the brain slices of 139A- and ME7-infected mice. B Representative merged images of NLRP3 (red) with GFAP (green, left), Iba1 (green, middle), or NeuN (green, right) in the brain slices of 263 K-infected hamsters. Brain regions of cortex and hippocampus are indicated on the left. The enlarged images of prion-infected animals are showed on the right each graph. Pearson’s correlation coefficient test results of NLRP3 and GFAP are marked below or on the right
Increased Expression of Cas1 and ACS in the Brains of Prion-Infected Rodent Models at Terminal Stage
The expressions of Cas1 and ACS, the other two components of inflammasome, in the brain tissues of prion-infected rodents were also evaluated. Remarkably upregulated Cas1 transcriptions in the brains of 139A- and ME7-infected mice were found by Cas-l-specific qRT-PCR (Fig. 3A). Similarly, ACS-specific qRT-PCR assays also revealed an increased transcription of ACS in the brains of both 139A- and ME7-infected mice compared to that of normal control (Fig. 3B). IFAs with anti-Cas1 showed more green signals in the brain sections of 139A- and ME7-infected mice and 263 K-infected hamsters, with statistical difference to that of normal control (Fig. 3C).

Analyses of brain caspase-1 and ASC in the prion-infected rodent models at terminal stage. A Caspase-1-specific qRT-PCR of the brain RNAs of 139A- or ME7-infected mice and the age-matched mice. B ASC-specific qRT-PCR of the brain RNAs of 139A- or ME7-infected mice and the age-matched mice. Relative fold changes of caspase-1 or ASC mRNAs are marked on Y-axis. Graphical data denote mean + SD (n = 3). C Representative images of caspase-1-specific IFAs of the brain sections of scrapie agents 139A- and ME7-infected mice (left) and 263 K-infected hamsters (right). Relative fluorescent intensities of NLRP3 signals in the brain slices of prion-infected animals compared to the individual controls (n = 3) are shown on the bottom. D Representative merged images of double-stained IFAs with anti-NLRP3 (red) and anti-caspase-1 (green) in the brain sections of the prion-infected rodent models. Brain regions of cortex and hippocampus are indicated on the left. The enlarged images of prion-infected animals are shown on the right each graph. Upper panels: 139A- and ME7-infected mice. Bottom panels: 263 K-infected hamsters. E IP assays of the brain homogenates of the prion-infected rodent models using anti-NLPR3 as capturing antibody and anti-caspase-1 as blotting antibody. F IP assays of the brain homogenates of the prion-infected rodent models using anti-NLPR3 as capturing antibody and anti-ASC as blotting antibody. IgG, isotopic mouse IgG. Input: an aliquot of brain homogenates directly loaded into SDS-PAGE as control. The relative gray values of the precipitated caspase-1 and ASC bands after normalized with the individual β-actin (n = 3) are marked below each graph
The brain sections were further employed to double-stained IFAs with anti-NLRP3 and anti-Cas1. Differing to the large asteroid-like cells stained by anti-NLRP3 (red), the Cas1-specific signals were small graininess (green) widely distributing in the brain tissues (cortex and hippocampus) of three scrapie-infected rodent models (Fig. 3D). Amounts of yellow signals were identifiable in the merged images, highlighting colocalizations between NLRP3 and Cas1 morphologically. Furthermore, the molecular interaction of NLRP3 with Cas1 and ACS in brain tissues was estimated by IP assays. Same amounts of 10% brain homogenates of 139A- and ME7-infected mice, as well as that of 263 K-infected hamsters, were separately immunoprecipitated with anti-NLRP3 controlled by the brain homogenates of individual normal control. In the blots with anti-Cas1 (Fig. 3E), specific Cas1 bands were detected in the elution fractions but not in that precipitated by isotype IgG. Quantitative assays verified significantly higher relative gray values in prion-infected mice (upper panel) and hamsters (low panel) compared to the control. Specific ACS bands were also identified in the IP assays of both mouse and hamster brain homogenates, using anti-NLRP3 as capturing antibody and anti-ACS as detecting antibody (Fig. 3F). Markedly stronger ACS bands were observed in the preparations of prion-infected mice and hamsters, showing statistical differences compared to the controls in the quantitative assays. Those date indicate that the expressions of Cas1 and ACS also increase in the brains of prion-infected rodents. NLRP3 can form protein complexes with Cas1 and ACS in brain tissues.
Time-Dependent Increases of Brain NLRP3 Levels During Prion Infection
To estimate the alteration of brain NLRP3 during prion infection, the brain sections of 139A- and ME7-infected mice collected on 80, 120, 150, and 180 dpi were immunofluorescently stained by anti-NLPR3 and anti-GFAP separately. As shown in Fig. 4, notably more GFAP-stained astrocytes (green) were detectable in the slices of 80 dpi and continually increased in the subsequent slices of both 139A- (left) and ME7 (right)-infected mice. NLRP3 signals (red) also increased gradually along with the sampling time. Further assays of the fluorescent intensities proposed that time-dependent increases of both brain GFAP and NLPR3 during the incubation of prion infection. Compared to the data of normal mice with the age of 180 days, GFAP-stained cells became significant since 80 dpi both in cortex and hippocampus regions, while NLRP3 signals were significantly upregulated since 120 dpi, particularly in the region of cortex. It highlights a time-dependent increase of brain NLPR3 during prion infection, which is coincidental with the increase of proliferative astrocytes.

NLPR3- and GFAP-specific IFAs of the brain sections of prion-infected mice collected at different time-points post-inoculation. A Representative images of 139A-infected mice. B Representative images of ME7-infected mice. The brain sections of the infected mice collected at 80, 120, 150, and 180 (terminal stage) dpi are indicated above the images. Brain regions of cortex and hippocampus are indicated on the left. Relative fluorescent intensities of NLRP3 (red) and GFAP (green) in the brain regions of the infected mice compared to the normal control are shown below the graphs.
Increased Il-1β and IL-18 Levels in the Brains of Prion-Infected Rodent Models at Terminal Stage
As one of the important downstream components of inflammasome, the brain levels of Il-1β and IL-18 in the prion-infected rodents at terminal stage were evaluated. qRT-PCR assays of the extracted brain RNAs found that the brain Il-1β and IL-18 transcriptional levels in 139A- and ME7-infected mice were significantly higher than those of normal mice (Fig. 5A). Subsequently, the Il-1β and IL-18 levels in the brains of various experimental rodents were measured by a commercial kit. Compared to the individual control, the brain Il-1β levels of two prion-infected mice models and the hamster model were significantly increased, while IL-18 levels of prion-infected mice significantly upregulated and that of prion-infected increased but without statistical difference (Fig. 5B). Il-1β-specific IFAs revealed more green particles in the regions of cortex and hippocampus of 139A- and ME7-infected mice and 263 K-infected hamsters, which distributed widely in the brains, and some of them overlapped with the NLRP3-stained asteroid-like cells (red) (Fig. 5C).

Analyses of brain IL-1β and IL-18 in the prion-infected rodent models at terminal stage. A IL-1β- and IL-18-specific qRT-PCRs of the brain RNAs of 139A- or ME7-infected mice and the age-matched mice. Relative fold changes of IL-1β (upper) and IL-18 (lower) mRNAs are marked on Y-axis. Graphical data denote mean + SD (n = 3). B ELISA for IL-1β and IL-18 in the brain homogenates. Upper panel: 139A- and ME7-infected mice controlled with normal mice. Low panel: 263 K-infected hamsters controlled with normal hamsters. C Representative merged images of double-stained IFAs with anti-NLRP3 (red) and anti-IL-1β (green). Brain regions of cortex and hippocampus are indicated on the left. The enlarged images are shown on the right each graph. Upper panels: 139A- and ME7-infected mice. Bottom panels: 263 K-infected hamsters
Increased GSDMD Levels in the Brains of Prion-Infected Rodent Models
Gasdermin D (GSDMD) is the biomarker for pyroptosis, which is hydrolyzed and activated by Cas1. To assess the status of GSDMD in the brain tissues during prion infected, the brain specimens of 139A-infected mice at terminal stage were evaluated. qRT-PCR assays identified significantly increased mRNA level of GSDMD in the brains of 139A-infected mice (Fig. 6A). Western blots revealed stronger GSDMD-NT-specific bands in the brains of 139A-infected mice with statistical significance compared to that of normal mice in the quantitative assay (Fig. 6B). IFAs illustrated remarkably more and larger GSDMD-specific signals (green) in the brain regions (cortex and hippocampus) of 139A-infected mice, showing significantly higher IOD values compared to those of normal control (Fig. 6C).

Analyses of brain GSDMD in the prion-infected rodent models. A GSDMD-specific qRT-PCR assays of the brain RNAs of 139A-infected mice at terminal stage. Relative fold changes of GSDMD mRNAs in the infected mice normalized with the data of control are marked on Y-axis. B GSDMD-NT-specific Western blot of brain homogenates of 139A-infected mice at terminal stage. The relative gray values of GSDMD-NT normalized with the individual β-actin (n = 3) are marked on the right. C Western blot representative images of GSDMD-specific IFAs of the brain sections from 139A-infected mice at terminal stage and normal control. The enlarged images of 139A-infected mice are shown on the right of the graphs. Relative fluorescent intensities of GSDMD signals in the brain slices of 139A-infected mice compared to the normal control (n = 3) are shown on the right. D Representative images of double-stained IFAs with anti-GSDMD (green) and anti-NLPR3 (red) of the brain slices from 139A-infected mice collected at different times post-inoculation. Relative fluorescent intensities of NLRP3 (red) and GFAP (green) in the brain regions of the infected mice compared to the normal control are shown below the graphs. E Representative images of double-stained IFAs by anti-GSDMD (green) together with anti-GFAP, anti-NeuN, or anti-Iba1 (red) of the brain sections from 139A-infected mice at terminal stage
Subsequently, the brain sections of 139A-infected mice collected on 120, 150, and 180 (terminal stage) dpi were subjected into double-stained IFAs with anti-GSDMD and anti-NLRP3. Accompanying with the increases of NLRP3 signals (red), the intensities of GSDMD signals (green) in the cortex and hippocampus of prion-infected mice increased gradually along with the incubation period (Fig. 6D). Analysis of the enlarged images identified that the GSDMD signals distributed not only in the asteroid-like cells that colocalized well with NLRP3 signals (yellow in the merged images), but also in the round and elliptical cells that overlapped little with NLRP3 signals. To estimate the distribution of the increased GSDMD in the different cell types in the prion-infected brains, the brain slices of 139A-infected mice at terminal stage were subjected into double-stained IFAs of anti-GSDMD together with anti-GFAP, anti-NeuN, or anti-Iba1. GSDMD signals overlapped with GFAP- and NeuN-positive cells, but not with Iba1-positive cells (Fig. 6E). Those data strongly indicate a time-dependent increase of brain GSDMD during prion infection, which distributes widely in astrocytes and neurons at the end of disease.
Discussion
Inflammasomes are a class of multiprotein complexes assembled with the participation of intracytoplasmic pattern recognition receptors (PRRs), which are important components of natural immune system. A variety of inflammasomes, such as NLRP1-, NLRP3-, NLRC4-, IPAF-, and AIM2-inflammasomes, have been described to be involved in host responses against a wide range of pathogens [10, 24, 25]. By mediating neuroinflammation, NLRP3 inflammasomes are considered to play a vital role in the progressions of some neurodegenerative diseases. For example, Aβ can activate NLRP3 inflammasomes in glial cells, which further promotes the formation of ASC spots, maturation and release of IL-1β, and accelerate memory loss and other symptoms in AD mouse models [9]. In a PD mouse model, stimulation of mitochondrial autophagy or acceleration of removing damaged mitochondria inhibits the activation of NLRP3 inflammasomes in microglia, which remits the loss of dopaminergic neurons and ameliorates the clinical symptoms [19]. Activation of NLRP3 inflammasomes has also reported in other neurodegenerative disorders, e.g., amyotrophic lateral sclerosis (ALS) and multiple sclerosis [26, 27]. Here, using the brain samples of several prion-infected rodent models, we have verified overexpression of NLRP3 inflammatory vesicle components with time-dependent manner after inoculation. Coincidentally, the brain levels of the downstream inflammatory cytokines IL-1β and IL-18, and the membrane perforating protein GSDMD-NT are significantly increased, which may promote the release of inflammatory factors during prion infection. Our data here demonstrate again the similar response of brain NLRP3 inflammasomes of prion diseases as other neurodegenerative diseases, although their etiological, neuropathological, and clinical features are different.
Astrocytes in CNS play an important physiological role in providing energy and nutrients to neurons, regulating synaptic activity, and regulating extracellular glutamate levels. On the other hand, astrocytes may produce toxic effects on neighbor neurons by releasing inflammatory factors during the pathogenic courses of neurodegenerative diseases [28, 29]. Our IFAs in this study demonstrate an overwhelming morphological overlap of NLRP3 with astrocytes but not with microglia and neurons in the prion-infected experimental rodents, which highly indicates that the over-proliferative astrocytes are likely to be responsible for the enhanced brain NLRP3 inflammasomes during prion infection. In many AD and PD mice models, overexpression of NLRP3 is identified mainly associated with the activated microglia [5, 9], and a few studies demonstrate NLRP3 inflammasomes in astrocytes [5, 29, 30]. Several in vitro studies also figure out overexpression of NLRP3 inflammasomes in microglia after stimulation of PrP106-126 peptide [31, 32]. Notably, microglia are the main source for NLRP3 inflammasomes in AD and PD mice models. The exact reason why NLRP3 expression confines in astrocytes in this study is unknown. Certainly, we could not exclude the technical reasons, e.g., the NLRP3 antibody used in this study. However, our IFAs for GSDMD also illustrate similar pattern as NLRP3 that GSDMD signals overlap well with astrocytes but little with microglia. Recent studies on AD and PD have shown that the classical astrocyte-mediated neuroinflammatory response is dependent on the induction of microglia activation [5, 33]. Further comparative analysis of NLRP3 inflammasomes activation in AD, PD, and PrD, regardless of patients’ brains or animal models, is deserved for address the potential difference among those neurodegenerative diseases.
NLRP3 inflammasome consists of three components, NLRP3, ASC, and Cas1. Upon activation, these three components undergo conformational change to assemble and form the NLRP3 inflammasomes [34, 35]. In line with the increase of brain NLRP3 in the prion-infected rodents, the overall transcriptional and expressional levels of ASC and caspase-1 are also upregulated. Importantly, our data here also demonstrate the direct molecular interaction of NLRP3 with ASC and Cas1 in the brain tissues, and the markedly more of NLRP3-ASC and NLRP3-Cas1 complexes in the prion-infected brains. Coincidentally, the elevated brain levels of IL-1β and IL-18, the main downstream targets of NLRP3 inflammasomes, are also detected in prion-infected mice. Apparently, infection of prion agents sufficiently induces increases of expression, assembly, and activity of NLRP3 inflammasomes in the brain tissues.
Another positive evidence for activation of NLRP3 inflammasomes during prion infection is the aberrant upregulation of GSDMD, particularly GSDMD-NT, in brain tissues. It is well known that the activated gasdermin proteins are closely related with cellular pyroptosis, also known as cellular inflammatory necrosis. This process relies on the activation of the caspases IL-1β and IL-18 by the inflammasomes. The active caspases further cleave GSDMD, and the cleaved GSDMD-NT translocate to the cell membrane and form pores, ultimately leading to cytoplasmic efflux, the release of inflammatory factors and cell rupture [14, 36]. The well morphological colocalizations of the increased GSDMD signals with NLRP3- and GFAP-positive cells in the brains of prion-infected mice suggest that cell pyroptosis occurs aggravatedly in the proliferated astrocytes, which is dependent on activation of inflammasomes. One may speculate that the pyrotostic process in astrocytes helps the cells to release inflammatory factors that participate in the neuroinflammation during prion infection. Unlike the distribution of NLRP3, GSDMD signals here are also detectable in the neurons of prion-infected mice, particularly in the region of hippocampus at terminal stage. Neuron loss is one of neuropathological features for prion diseases [37, 38] and our previous studies using Nissl stain have demonstrated severe neuron loss in those prion-infected rodent models at final stage [39]. As cell pyroptosis is one of the pathways for programmed cell death pathophysiologically, one may assume the possibility that the pyroptosis contributes, at least partially, to the neuron death in prion infection, although the mechanism of cross-talking of NLRP3-inflammasomes between astrocytes and neurons is unclear.
Our data here also present a time-related increase of brain GFAP-NLRP3-GSDMD in the incubation period of prion infection, in which increase of GFAP appears apparently earlier. It highlights a time-dependent correlation among the astrogliosis, NLRP3-inflammasomes, and cell pyroptosis in the brains of prion infection. Aggregations of the misfolding proteins in neurodegenerative diseases are able to trigger the NLRP3-associated neuroinflammatory responses [40, 41]. Certain cellular pathologies, e.g., dysfunction of autophagy, ROS production, mitochondrial injury, lysosomal damage, are also responsible for the activation of NLRP3 inflammasomes [42, 43]. The accumulation of PrPSc and above cellular pathologies worsens gradually during prion disease. Thereby, it is supposable that the activation of NLRP3 inflammasomes during prion infection is a combined outcome mediated by multiple pathological elements. Given the gradual enhancement and persistent presence of NLRP3 inflammasomes signature in a number of neurodegenerative diseases, including prion disease, the development of NLRP3 inhibitors has been becoming a therapeutic approach to delay the disease progression [17, 29, 44, 45]. However, considering the markedly short duration of prion disease after onset, there will be more challenges in the R&D of therapeutic tools, including NLRP3 modulator, for such rare but fatal disease.
In summary, we have verified remarkable increases of the brain levels of NLPR3, ACS, and proCas1 in various scrapie-infected rodents, with a time-dependent manner along with the incubation period. The increased NLPR3 mainly overlapped with the proliferated astrocytes. The downstream cytokines Il-1β and IL-18 were increased in the brains of scrapie-infected animals. Furthermore, the brain levels of GSDMD, particularly GSDMD-NT, in the scrapie-infected mice were upregulated progressively post-infection. Our data here suggest that prion infection induces astrocyte activation, which mediates the activation of the NLRP3-GSDMD pathway, leading to neuronal pyroptosis and a series of neuroinflammatory responses.
Data Availability
No datasets were generated or analyzed during the current study.
References
Ma Y, Shi Q, Xiao K, Wang J, Chen C, Gao LP, Gao C, Dong XP (2019) Stimulations of the culture medium of activated microglia and TNF-alpha on a scrapie-infected cell line decrease the cell viability and induce marked necroptosis that also occurs in the brains from the patients of human prion diseases. ACS Chem Neurosci 10(3):1273–1283. https://doi.org/10.1021/acschemneuro.8b00354
Zhou DH, Wang J, Xiao K, Wu YZ, Maimaitiming A, Hu C, Gao LP, Chen J, Gao C, Chen C, Shi Q, Dong XP (2020) Stilbene compounds inhibit the replications of various strains of prions in the levels of cell culture, PMCA, and RT-QuIC possibly via molecular binding. ACS Chem Neurosci 11(14):2117–2128. https://doi.org/10.1021/acschemneuro.0c00218
Mead S, Khalili-Shirazi A, Potter C, Mok T, Nihat A, Hyare H, Canning S, Schmidt C, Campbell T, Darwent L, Muirhead N, Ebsworth N, Hextall P, Wakeling M, Linehan J, Libri V, Williams B, Jaunmuktane Z, Brandner S, Rudge P, Collinge J (2022) Prion protein monoclonal antibody (PRN100) therapy for Creutzfeldt-Jakob disease: evaluation of a first-in-human treatment programme. Lancet Neurol 21(4):342–354. https://doi.org/10.1016/s1474-4422(22)00082-5
Ma Y, Shi Q, Wang J, Xiao K, Sun J, Lv Y, Guo M, Zhou W, Chen C, Gao C, Zhang BY, Dong XP (2017) Reduction of NF-κB (p65) in scrapie-infected cultured cells and in the brains of scrapie-infected rodents. ACS Chem Neurosci 8(11):2535–2548. https://doi.org/10.1021/acschemneuro.7b00273
Heneka MT, McManus RM, Latz E (2018) Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19(10):610–621. https://doi.org/10.1038/s41583-018-0055-7
Moonen S, Koper MJ, Van Schoor E, Schaeverbeke JM, Vandenberghe R, von Arnim CAF, Tousseyn T, De Strooper B, Thal DR (2023) Pyroptosis in Alzheimer’’s disease: cell type-specific activation in microglia, astrocytes and neurons. Acta Neuropathol 145(2):175–195. https://doi.org/10.1007/s00401-022-02528-y
Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, Griep A, Santarelli F, Brosseron F, Opitz S, Stunden J, Merten M, Kayed R, Golenbock DT, Blum D, Latz E, Buée L, Heneka MT (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575(7784):669–673. https://doi.org/10.1038/s41586-019-1769-z
Zhang Y, Huang R, Cheng M, Wang L, Chao J, Li J, Zheng P, Xie P, Zhang Z, Yao H (2019) Gut microbiota from NLRP3-deficient mice ameliorates depressive-like behaviors by regulating astrocyte dysfunction via circHIPK2. Microbiome 7(1):116. https://doi.org/10.1186/s40168-019-0733-3
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT (2013) NLRP3 is activated in Alzheimer’’s disease and contributes to pathology in APP/PS1 mice. Nature 493(7434):674–678. https://doi.org/10.1038/nature11729
Barnett KC, Li S, Liang K, Ting JP (2023) A 360° view of the inflammasome: mMechanisms of activation, cell death, and diseases. Cell 186(11):2288–2312. https://doi.org/10.1016/j.cell.2023.04.025
Nozaki K, Maltez VI, Rayamajhi M, Tubbs AL, Mitchell JE, Lacey CA, Harvest CK, Li L, Nash WT, Larson HN, McGlaughon BD, Moorman NJ, Brown MG, Whitmire JK, Miao EA (2022) Caspase-7 activates ASM to repair gasdermin and perforin pores. Nature 606(7916):960–967. https://doi.org/10.1038/s41586-022-04825-8
Knorr M, Hoppe J, Steuhl KP, Dartsch PC (1992) Effect of PDGF-AB heterodimer on a corneal epithelial cell line. Eur J Cell Biol 57(2):202–209
Ma X, Hao J, Wu J, Li Y, Cai X, Zheng Y (2022) Prussian blue nanozyme as a pyroptosis inhibitor alleviates neurodegeneration. Adv Mater 34(15):e2106723. https://doi.org/10.1002/adma.202106723
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535(7610):153–158. https://doi.org/10.1038/nature18629
Wang X, Li X, Liu S, Brickell AN, Zhang J, Wu Z, Zhou S, Ding Z (2020) PCSK9 regulates pyroptosis via mtDNA damage in chronic myocardial ischemia. Basic Res Cardiol 115(6):66. https://doi.org/10.1007/s00395-020-00832-w
Siew JJ, Chen HM, Chen HY, Chen HL, Chen CM, Soong BW, Wu YR, Chang CP, Chan YC, Lin CH, Liu FT, Chern Y (2019) Galectin-3 is required for the microglia-mediated brain inflammation in a model of Huntington’’s disease. Nat Commun 10(1):3473. https://doi.org/10.1038/s41467-019-11441-0
Wu AG, Zhou XG, Qiao G, Yu L, Tang Y, Yan L, Qiu WQ, Pan R, Yu CL, Law BY, Qin DL, Wu JM (2021) Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res Rev 65:101202. https://doi.org/10.1016/j.arr.2020.101202
Xu F, Wu Y, Yang Q, Cheng Y, Xu J, Zhang Y, Dai H, Wang B, Ma Q, Chen Y, Lin F, Wang C (2022) Engineered extracellular vesicles with SHP2 high expression promote mitophagy for Alzheimer’’s disease treatment. Adv Mater 34(49):e2207107. https://doi.org/10.1002/adma.202207107
Panicker N, Kam TI, Wang H, Neifert S, Chou SC, Kumar M, Brahmachari S, Jhaldiyal A, Hinkle JT, Akkentli F, Mao X, Xu E, Karuppagounder SS, Hsu ET, Kang SU, Pletnikova O, Troncoso J, Dawson VL, Dawson TM (2022) Neuronal NLRP3 is a parkin substrate that drives neurodegeneration in Parkinson’’s disease. Neuron 110(15):2422-2437.e2429. https://doi.org/10.1016/j.neuron.2022.05.009
Lawrence G, Holley CL, Schroder K (2022) Parkinson’’s disease: connecting mitochondria to inflammasomes. Trends Immunol 43(11):877–885. https://doi.org/10.1016/j.it.2022.09.010
Xiao K, Zhang BY, Zhang XM, Wang J, Chen C, Chen LN, Lv Y, Shi Q, Dong XP (2016) Re-infection of the prion from the scrapie-infected cell line SMB-S15 in three strains of mice, CD1, C57BL/6 and Balb/c. Int J Mol Med 37(3):716–726. https://doi.org/10.3892/ijmm.2016.2465
Kim YS, Carp RI, Callahan SM, Wisniewski HM (1987) Incubation periods and survival times for mice injected stereotaxically with three scrapie strains in different brain regions. J Gen Virol 68(Pt 3):695–702. https://doi.org/10.1099/0022-1317-68-3-695
Shi Q, Zhang BY, Gao C, Zhang J, Jiang HY, Chen C, Han J, Dong XP (2012) Mouse-adapted scrapie strains 139A and ME7 overcome species barrier to induce experimental scrapie in hamsters and changed their pathogenic features. Virol J 9:63. https://doi.org/10.1186/1743-422x-9-63
Chou WC, Jha S, Linhoff MW, Ting JP (2023) The NLR gene family: from discovery to present day. Nat Rev Immunol 23(10):635–654. https://doi.org/10.1038/s41577-023-00849-x
Kibby EM, Conte AN, Burroughs AM, Nagy TA, Vargas JA, Whalen LA, Aravind L, Whiteley AT (2023) Bacterial NLR-related proteins protect against phage. Cell 186(11):2410-2424.e2418. https://doi.org/10.1016/j.cell.2023.04.015
Singh J, Habean ML, Panicker N (2023) Inflammasome assembly in neurodegenerative diseases. Trends Neurosci 46(10):814–831. https://doi.org/10.1016/j.tins.2023.07.009
Shao S, Chen C, Shi G, Zhou Y, Wei Y, Fan N, Yang Y, Wu L, Zhang T (2021) Therapeutic potential of the target on NLRP3 inflammasome in multiple sclerosis. Pharmacol Ther 227:107880. https://doi.org/10.1016/j.pharmthera.2021.107880
Osso LA, Chan JR (2015) Astrocytes underlie neuroinflammatory memory impairment. Cell 163(7):1574–1576. https://doi.org/10.1016/j.cell.2015.12.001
Zhu J, Hu Z, Han X, Wang D, Jiang Q, Ding J, Xiao M, Wang C, Lu M, Hu G (2018) Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of β-arrestin2 and NLRP3. Cell Death Differ 25(11):2037–2049. https://doi.org/10.1038/s41418-018-0127-2
Giordano AMS, Luciani M, Gatto F, Abou Alezz M, Beghè C, Della Volpe L, Migliara A, Valsoni S, Genua M, Dzieciatkowska M, Frati G, Tahraoui-Bories J, Giliani SC, Orcesi S, Fazzi E, Ostuni R, D’Alessandro A, Di Micco R, Merelli I, Lombardo A, Reijns MAM, Gromak N, Gritti A, Kajaste-Rudnitski A (2022) DNA damage contributes to neurotoxic inflammation in Aicardi-Goutières syndrome astrocytes. J Exp Med 219(4):e20211121. https://doi.org/10.1084/jem.20211121
Shi F, Yang L, Kouadir M, Yang Y, Wang J, Zhou X, Yin X, Zhao D (2012) The NALP3 inflammasome is involved in neurotoxic prion peptide-induced microglial activation. J Neuroinflammation 9:73. https://doi.org/10.1186/1742-2094-9-73
Lai M, Yao H, Shah SZA, Wu W, Wang D, Zhao Y, Wang L, Zhou X, Zhao D, Yang L (2018) The NLRP3-caspase 1 inflammasome negatively regulates autophagy via TLR4-TRIF in prion peptide-infected microglia. Front Aging Neurosci 10:116. https://doi.org/10.3389/fnagi.2018.00116
Li S, Fang Y, Zhang Y, Song M, Zhang X, Ding X, Yao H, Chen M, Sun Y, Ding J, Wang Q, Lu M, Wu G, Hu G (2022) Microglial NLRP3 inflammasome activates neurotoxic astrocytes in depression-like mice. Cell Rep 41(4):111532. https://doi.org/10.1016/j.celrep.2022.111532
Niu T, De Rosny C, Chautard S, Rey A, Patoli D, Groslambert M, Cosson C, Lagrange B, Zhang Z, Visvikis O, Hacot S, Hologne M, Walker O, Wong J, Wang P, Ricci R, Henry T, Boyer L, Petrilli V, Py BF (2021) NLRP3 phosphorylation in its LRR domain critically regulates inflammasome assembly. Nat Commun 12(1):5862. https://doi.org/10.1038/s41467-021-26142-w
Hochheiser IV, Pilsl M, Hagelueken G, Moecking J, Marleaux M, Brinkschulte R, Latz E, Engel C, Geyer M (2022) Structure of the NLRP3 decamer bound to the cytokine release inhibitor CRID3. Nature 604(7904):184–189. https://doi.org/10.1038/s41586-022-04467-w
Wang C, Yang T, Xiao J, Xu C, Alippe Y, Sun K, Kanneganti TD, Monahan JB, Abu-Amer Y, Lieberman J, Mbalaviele G (2021) NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci Immunol 6(64):eabj3859. https://doi.org/10.1126/sciimmunol.abj3859
Wang J, Zhang BY, Zhang J, Xiao K, Chen LN, Wang H, Sun J, Shi Q, Dong XP (2016) Treatment of SMB-S15 cells with resveratrol efficiently removes the PrP(Sc) accumulation in vitro and prion infectivity in vivo. Mol Neurobiol 53(8):5367–5376. https://doi.org/10.1007/s12035-015-9464-z
Sigurdson CJ, Bartz JC, Glatzel M (2019) Cellular and molecular mechanisms of prion disease. Annu Rev Pathol 14:497–516. https://doi.org/10.1146/annurev-pathmechdis-012418-013109
Xu Y, Tian C, Wang SB, Xie WL, Guo Y, Zhang J, Shi Q, Chen C, Dong XP (2012) Activation of the macroautophagic system in scrapie-infected experimental animals and human genetic prion diseases. Autophagy 8(11):1604–1620. https://doi.org/10.4161/auto.21482
Trudler D, Nazor KL, Eisele YS, Grabauskas T, Dolatabadi N, Parker J, Sultan A, Zhong Z, Goodwin MS, Levites Y, Golde TE, Kelly JW, Sierks MR, Schork NJ, Karin M, Ambasudhan R, Lipton SA (2021) Soluble α-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc Natl Acad Sci U S A 118(15):e2025847118. https://doi.org/10.1073/pnas.2025847118
Yang T, Zhang L, Shang Y, Zhu Z, Jin S, Guo Z, Wang X (2022) Concurrent suppression of Aβ aggregation and NLRP3 inflammasome activation for treating Alzheimer’’s disease. Chem Sci 13(10):2971–2980. https://doi.org/10.1039/d1sc06071f
Su L, Zhang J, Gomez H, Kellum JA, Peng Z (2023) Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 19(2):401–414. https://doi.org/10.1080/15548627.2022.2084862
Zhu M, Du L, Zhao R, Wang HY, Zhao Y, Nie G, Wang RF (2020) Cell-penetrating nanoparticles activate the inflammasome to enhance antibody production by targeting microtubule-associated protein 1-light chain 3 for degradation. ACS Nano 14(3):3703–3717. https://doi.org/10.1021/acsnano.0c00962
Harrison D, Billinton A, Bock MG, Doedens JR, Gabel CA, Holloway MK, Porter RA, Reader V, Scanlon J, Schooley K, Watt AP (2023) Discovery of clinical candidate NT-0796, a brain-penetrant and highly potent NLRP3 inflammasome inhibitor for neuroinflammatory disorders. J Med Chem. https://doi.org/10.1021/acs.jmedchem.3c01398
. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O’Neill LA (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases Nat Med 21 (3):248-255 https://doi.org/10.1038/nm.3806
Funding
This work was supported by the grant (2021SKLID101, 2019SKLID501, 2019SKLID603) from the State Key Laboratory for Infectious Disease Prevention and Control, China CDC.
Author information
Authors and Affiliations
Contributions
Dong-Hua Zhou, Qi Shi, and Xiao-Ping Dong wrote the main manuscript text. Xiao-Xi Jia, Yue-Zhang Wu, Wei-Wei Zhang, Yuan Wang, Dong-Lin Liang, Li-Ping Gao, King Xiao, and Can Chen participated in analyzing the results. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Ethics Approval
Approved. Details are provided in the beginning of the “Materials and Methods” section.
Consent to Participate
Not applicable
Consent for Publication
Not applicable
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhou, DH., Jia, XX., Wu, YZ. et al. Aberrant Enhanced NLRP3 Inflammasomes and Cell Pyroptosis in the Brains of Prion-Infected Rodent Models Are Largely Associated with the Proliferative Astrocytes. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04169-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04169-6