
Abstract
Observational studies have faced challenges in identifying replicable causes for amyotrophic lateral sclerosis (ALS). To address this, we employed an unbiased and data-driven approach to discover and explore potential causal exposures using two-sample Mendelian randomization (MR) analyses. In the phenotype discovery stage, we assessed 3948 environmental exposures from the UK Biobank and utilized ALS summary statistics (Europeans, 20,806 cases, 59,804 controls) as the outcome within a phenome-wide MR pipeline. Through a range of sensitivity analyses, two medication traits were identified to be protective for ALS. In the target exploration stage, we further conducted drug target MR analyses using the latest and trans-ethnic summary data on lipid-related traits and ALS (Europeans, 27,205 cases, 110,881 controls; East Asians, 1234 cases, 2850 controls). Our aim was to explore potential causal drug targets through six lipid-modifying effects. These comprehensive analyses revealed significant findings. Specifically, “cholesterol-lowering medication” and “atorvastatin” survived predefined criteria in the phenotype discovery stage and exhibited a protective effect on ALS. Further in the target exploration stage, we demonstrated that the therapeutic effect of APOB through LDL-lowering was associated with reduced ALS liability in Europeans (OR = 0.835, P = 5.61E − 5). Additionally, the therapeutic effect of APOA1 and LDLR through TC-lowering was associated with reduced ALS liability in East Asians (APOA1, OR = 0.859, P = 5.38E − 4; LDLR, OR = 0.910, P = 2.73E − 5). Overall, we propose potential protective effects of cholesterol-lowering drugs or statins on ALS risk from thousands of exposures. Our research also suggests APOB, APOA1, and LDLR as novel therapeutic targets for ALS and supports their potential protective mechanisms may be mediated by LDL-lowering or TC-lowering effects.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive paralysis and eventual death. It is widely acknowledged that ALS arises from a complex interplay of genetic and environmental factors [1,2,3]. However, unraveling the specific role of environmental risk factors has proven to be a challenging task. Observational studies investigating potential causes for ALS have been hindered by unmeasured confounders and the issue of reverse causality.
Mendelian randomization (MR) is a novel method to investigate the potential causality between exposure and outcome by utilizing genetic variants to proxy related traits or diseases [4]. Compared to traditional observational studies, MR offers several advantages in inferring or refuting causal associations. It helps minimize confounding biases and addresses concerns related to reverse causality, providing a robust framework for exploring the causal effects of exposures of interest on outcomes. Furthermore, by using the genetic variants near or within the genes encoding drug target proteins (i.e., cis-variants), MR studies allow the effect of long-term modulation of drug targets on disease risk to be tested, a concept called drug target MR. This type of study is less susceptible to bias arising from horizontal pleiotropy and can, therefore, be designed to mimic the therapeutic effect of specific drug targets in randomized controlled trials [5].
The availability of summary-level data from genome-wide association studies (GWAS) has rapidly increased, enabling more accessible MR analyses. Large biobanks, such as the UK Biobank (UKB) and FINNGEN, have incorporated publicly available data, facilitating MR research. Additionally, the development of R packages, such as “TwoSampleMR,” has streamlined the process of obtaining data from the IEU GWAS database, supporting systematic causal inference between the human phenome and diseases of interest.
In such context, the present study aimed to discover and explore the causal factors for ALS by employing an unbiased and data-driven approach. Specifically, we conducted two-sample MR analyses using a two-step approach.
First, using a range of traits from UKB [6], we comprehensively screen the causal factors for ALS by performing phenome-wide MR analyses. As a result, two lipid-modifying medication traits were found to be protective for ALS after sensitivity analyses. Notably, while previous epidemiological studies have produced inconsistent findings, the relationship between statin use [7,8,9], blood lipids [10,11,12,13,14,15,16], and ALS has remained a topic of interest. However, research on the causal effects of lipid-modifying targets [17] on ALS is lacking, creating a significant knowledge gap in the field.
Understanding the potential impact of lipid-modifying targets on ALS risk could provide valuable insights into the underlying mechanisms of the disease and potentially identify novel therapeutic strategies. Therefore, we further conducted drug target MR analyses using the latest and trans-ethnic summary data on lipid-related traits and ALS, aiming to comprehensively explore causal drug targets through multiple lipid-modifying effects.
Materials and Methods
Phenotype Discovery Stage
In the phenotype discovery stage, openly accessible GWAS summary statistics curated and centralized by the Medical Research Council Integrative Epidemiology Unit (MRC-IEU) open GWAS database (https://gwas.mrcieu.ac.uk) were accessed via the MR-Base platform [6] (accessed on 1 April 2022).
Exposure Data
We used 3948 traits of European ancestry from UKB data released by Neale’s lab and MRC-IEU (http://www.nealelab.is/uk-biobank). Considering that the second round GWAS release from Neale’s lab has generally larger sample sizes than the first round and has a better-curated analysis pipeline (http://www.nealelab.is/uk-biobank/ukbround2announcement), we excluded the 596 traits from the first round. Furthermore, duplicated traits (n = 17), categorical/binary ones with cases < 1000 (n = 0), and continuous ones without normalization [18] (n = 31) were excluded. A total of 3304 traits were included in the primary analysis. Subsequently, 784 traits (SNPs ≥ 3) entered the analytic pipeline, which has been categorized into 14 broad categories based on the information provided by the UKB website. Detailed methodology related to phenotype categorization has been described in Method S1.
Outcome Data
Summary statistics of ALS were derived from a recent large-scale meta-analysis of GWAS confined to Europeans (20,806 cases; 59,804 controls) [19] (Table S1). A detailed description of the participants and study design was provided in the original study [19].
Mendelian Randomization
Main Analysis
MR requires meeting three core assumptions: Assumption 1, the genetic variants utilized as the instrumental variables (IV) should exhibit robust associations with the exposure of interest; Assumption 2, the genetic instruments are not associated with any major confounders; Assumption 3, the genetic instruments affect the outcome only through exposure. We ran a two-sample MR to assess the potential causal effect of each candidate trait on ALS, using the multiplicative random-effects inverse variance weighted (IVW) method as the main analysis [20]. MR analyses only included independent single nucleotide polymorphisms (SNPs) (R2 < 0.001 and window size = 10 Mb) with P < 5E − 08 in the exposure, and the exposure would be removed when its independent SNPs were less than three to enhance the stability of MR results [18]. For exposure variants not found in the outcome, GWAS proxies were used instead, R2 ≥ 0.8 (obtained using 1000 Genomes European sample). Traits were included in the subsequent analyses when the P value of the IVW method was < 0.05. Steiger analyses were performed to verify that the proposed instruments were directly associated with the outcome or effect estimate directionality [21].
Sensitivity Analyses
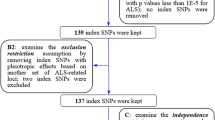
We conducted a combined method to reduce the risk of false-positive associations (Fig. 1A). For IVW results with P value < 0.05, sensitivity analyses, including the MR-Egger regression and weighted median methods, were conducted to assess the robustness of the main findings. Exposures were considered consistent with a causal effect only if the P values of both MR-Egger and weighted median methods were < 0.05.

Flow diagram of the phenotype discovery (A) and target exploration (B) of lipid-modifying drugs for amyotrophic lateral sclerosis in two-sample MR analyses
Furthermore, we used Cochran’s Q and MR-Egger intercept to test the presence of heterogeneity and directional pleiotropy, respectively. We used MR-PRESSO, which performs a test to detect horizontal pleiotropy (MR-PRESSO global test), and if detected, it removes horizontal pleiotropic outliers and then performs the IVW method using the remaining instruments [22]. Exposures with evidence of heterogeneity (Q test P < 0.05) and directional pleiotropy (MR-Egger intercept P < 0.05 or global test P < 0.05) were excluded. The leave-one-out analysis was also conducted within the IVW method to assess the influence of individual variants on the observed association.
Finally, two significant exposures passed the phenotype discovery stage, including “cholesterol-lowering medication” and “atorvastatin.” The correct direction of effect was checked using a positive control of coronary heart disease (CHD) from the CARDIoGRAM GWAS data (60,801 CHD cases and 123,504 controls) [23].
Target Exploration Stage
Then, we used drug target MR analyses to explore potential lipid-modifying targets beyond the phenotype of “cholesterol-lowering medication” and “atorvastatin.”
Data Sources
For exposures, we chose the latest and largest trans-ancestry meta-analysis of GWAS for each lipid category. Summary statistics for triglycerides (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL), and high-density lipoprotein cholesterol (HDL) were from Europeans and East Asians [24], while apoprotein B (ApoB) and apoprotein A1 (ApoA1) were only available in Europeans [25].
For outcomes, we obtained large-scale European-based ALS GWAS summary data from the most recently published study, which included 27,205 ALS patients and 110,881 controls [2]. All ALS patients were diagnosed according to the revised El Escorial criteria by specialized neurologists. To examine the causal effect of lipid targets on ALS in East Asians, we also obtained association summary statistics from a Chinese ALS GWAS that analyzed ∼6.6 million genotyped and imputed SNPs on up to 4084 individuals (1234 cases and 2850 controls) [26].
Details of the summary data are listed in Table S1, and the study design, including the collection of samples, quality control procedures, and imputation methods, have been described in the original paper. The research protocol of each GWAS was approved by the relevant institutional review boards or ethics committees.
Instrument Selection
To broadly evaluate the impact of lipid-modifying drug targets on the risk of ALS, we employed a collaborative approach to identify existing lipid-modifying drug targets available for study. Detailed methodology and related information on the identification process can be seen in Method S2.
After a comprehensive literature review [17, 27,28,29,30], we identified 23 lipid-modifying drug targets in total, including HMGCR, PCSK9, NPC1L1, CETP, LDLR, APOB, LPL, ANGPTL3, ACLY, PPARA, ABCG5/ABCG8, DGAT1, MTTP, LPA, APOA1, LIPA, LCAT, APOC3/APOA5, ANGPTL4, ANGPTL8, and ASGR1 (Fig. 2A and B, and Table S4). Considering the diverse lipid-modifying effects of selected targets and corresponding drugs, it is challenging to disentangle the specific lipid-modifying effects through which drug targets confer their causal effect. For example, although reducing LDL and TC levels is the major effect of statins (HMGCR), modifying effects on TG [31], HDL [32], ApoB [33], and ApoA1 [32] have also been suggested. As previous MR research indicated that higher LDL concentrations may increase ALS risk [34], the primary analysis focused on the LDL-lowering effect for each drug target, with sensitivity analyses considering the remaining lipid-modifying effects of TG-lowering, TC-lowering, HDL-raising, ApoB-lowering, and ApoA1-raising. Specifically, these effects refer to candidate lipid targets that may impact ALS risk by reducing circulating levels of TG (TG-lowering), TC (TC-lowering), LDL-C (LDL-lowering), or ApoB (ApoB-lowering), while increasing circulating levels of HDL-C (HDL-raising) or ApoA1 (ApoA1-raising).

Flowchart of the A database search, B systematic identification, and C population distribution of lipid-modifying drug targets. A Details on the database search can be seen in Table S2. All included studies fulfill the inclusion criteria which can be seen in Table S3. B The review of interest is the work by Hegele and Tsimikas [17] (PMID 30702996), while the basic research of interest is the work by Wang et al. [] (PMID 35922515). C NA indicates that lipid-modifying targets through ApoB-lowering and ApoA1-raising effects were not available in East Asians considering the lack of corresponding summary data on lipid fractions. Lipid-modifying targets in light green circles but out of light blue circles have available SNPs of less than 3, indicating that such targets were less likely to significantly affect the blood concentrations of this lipid fraction. The number of available lipid-modifying targets was less in East Asians than in Europeans which may be attributed to the relatively small sample size of GWAS in East Asians
Genetic instruments consisted of variants that were associated with each lipid-modifying effect at genome-wide significance (P < 5E − 08) and located near (± 100 kb) or within genes encoding regions of 23 lipid-modifying drug targets, with effect estimates for each genetic variant derived for each lipid target from the trans-ancestry lipid GWAS (Fig. 1B). To include more SNPs, we set a moderate to low LD threshold (R2 < 0.4) [35, 36] in the primary analyses. More stringent LD thresholds (R2 < 0.3 [37], R2 < 0.2 [38], and R2 < 0.1 [29]) were conducted in sensitivity analyses. The lipid target (e.g., PCSK9) through each lipid-modifying effect (e.g., TG-lowering) would be removed when its available SNPs were less than three [39], indicating that this target was less likely to significantly affect the blood concentrations of this lipid fraction. For all the selected IVs in this study, F-statistics were above 10, indicating that weak instrumental bias is minimal [40].
Positive Control Analysis
To help validate the IV selection strategy of lipid-modifying targets, positive control analyses were performed in our study. We examined the association of lipid-modifying targets through LDL-lowering effect with CHD because higher LDL is a well-established causative factor for CHD [23], for which we may expect to see the therapeutic effect of LDL-lowering targets including HMGCR [41], CETP [35], PCSK9 [42], and NPC1L1 [41] on CHD as previous evidence. For the positive control outcome, GWAS summary data for CHD was based on the CARDIoGRAMplusC4D Consortium, which conducted a meta-analysis of 60,801 CHD cases and 123,504 controls [23].
Statistical Analysis
For the IVW method, we used a random-effects model when the results were heterogeneous, and a fixed-effects model was used when there was no heterogeneity. The association is considered to be significant after Bonferroni correction for each lipid target in Europeans (P < 4.39E − 4 (0.05/19 × 6)) (19 targets multiplied by 6 lipid-modifying effects) and in East Asians (P < 1.14E − 3 (0.05/11 × 4))] (11 targets multiplied by 4 lipid-modifying effects). A P value above Bonferroni’s corrected P value but below 0.05 was considered suggestive of evidence for a potential association. Tests for heterogeneity and horizontal pleiotropy were similar to the phenotype discovery stage. Here, we considered the lipid target to be causal for ALS only if (1) IVW P < 4.39E − 4 (EUR) or 1.14E − 3 (EAS) for primary analysis (R2 < 0.4); (2) IVW P < 0.05 for all sensitivity analyses with more stringent LD threshold (R2 < 0.3, R2 < 0.2, and R2 < 0.1); and (3) no evidence of heterogeneity (Q test P < 0.05) and horizontal pleiotropy (MR-Egger intercept P < 0.05 and Global test P < 0.05). Furthermore, we use another four MR methods, namely weighted median, MR-Egger, RadialMR, and MR-RAPS, to support the causal lipid targets after the screening stage, which makes the MR result more robust. For drug targets passing the predefined criteria, we performed colocalization analysis as a sensitivity analysis (detailed methodology provided in Method S3). Associations of lipid targets with outcomes were scaled to 1-SD reduction/increase in corresponding lipid fractions to represent the therapeutic effect of lipid-modifying drugs, considering the MR analysis package preference to automatically change these into a positive direction of effect (i.e., lipid/apoprotein raising).
MR analyses were mainly performed using the packages TwoSampleMR (0.5.6), RadialMR (1.0), MR-PRESSO (1.0), mr.raps (0.4.1), and coloc (5.2.0) of the statistical software R (4.1.3).
Standard Protocol Approvals, Registrations, and Patient Consents
The GWAS summary statistics supporting this research are publicly available, and the original studies obtained ethical approval from relevant ethics review boards.
Results
Phenotype Discovery Stage
The flowchart of the phenotype discovery stage is seen in Fig. 1A. Of the initial 3304 analyzed exposures, 784 exposures have no less than three IVs in the outcome (Fig. 3A and Table S5), and 58 exposures showed associations with ALS at P < 0.05 by IVW analyses in the phenotype discovery stage (Fig. 3B and Table S6). We found five showed consistent associations with ALS through sensitivity analysis methods, including MR-Egger and Weighted median (Fig. 3C). Two of the five significant exposures showed a robust effect on ALS with non-significant heterogeneity and horizontal pleiotropy in post-analyses (Fig. 3C). Furthermore, the MR-PRESSO approach and leave-one-out analyses failed to find outliers for “cholesterol-lowering medication” or “atorvastatin,” indicating that our estimates were stable for a single SNP (Fig. 3C and Figure S1). Interestingly, both “cholesterol-lowering medication” and “atorvastatin” belong to lipid-modifying drugs.

Overview of the data composition and analytical process for UKB traits in the phenotype discovery stage. Notes: A total of 784 traits from 14 categories have no less than three IVs in the outcome (A). A total of 58 traits from 12 categories were found to be significant by the IVW method (P < 0.05) (B). Two medication traits passed the predefined criteria in the phenotype discovery stage (C). We considered that there was evidence of a causal relationship if (1) IVW P < 0.05; (2) the same direction of effect as the IVW and P < 0.05 was found for the weighted median and MR-Egger; and (3) no evidence of heterogeneity (Q test P < 0.05) and horizontal pleiotropy (MR-Egger intercept P < 0.05 and Global test P < 0.05). Exposures with odds ratios (OR) greater than 1 were considered risk exposures, while exposures with OR less than 1 were considered protective exposures. *It indicates that the direction of effect was corrected by the positive control of CHD
After the direction of effect was corrected by the positive control of CHD, “cholesterol-lowering medication” (30 IVs; OR 0.427; 95% CI 0.251–0.726; P = 1.66E − 03) and “atorvastatin” (21 IVs; OR 0.041; 95% CI 0.006–0.302; P = 1.69E − 03) confer a protective effect on ALS. As both two exposures were medication traits, we further conducted drug target MR analyses to explore potential targets beyond them using the latest and trans-ethnic independent GWAS datasets (Table S1).
Target Exploration Stage
Instrument Selection
The flowchart of the target exploration stage is seen in Fig. 1B. We identified 23 unique lipid-modifying drug targets through a literature review (Table S4), which have been classified into six categories based on different lipid-modifying pathways (TG-lowering, TC-lowering, LDL-lowering, HDL-raising, ApoB-lowering, and ApoA1-raising). The Venn diagrams illustrated in Fig. 2C not only offer evidence that the target affects one or more lipid fractions but also imply an interrelationship of lipid targets across different populations. Due to the proximity of the genes encoding ApoA5 and ApoC3 (also ABCG5 and ABCG8), variants in the vicinity of these genes were combined in MR models. DGAT1 and LIPA were excluded before the main analysis because insufficient genetic instruments (< 3) were available in the outcome GWAS. Consequently, 19 drug targets for Europeans and 11 drug targets for East Asians were ultimately analyzed (Fig. 2C).
LDL-Lowering Targets and ALS Risk
In MR analyses designed to proxy the LDL-lowering effects of lipid-modifying drugs, we identified two significant targets with protective effects on ALS. Genetically proxied LDL-lowering via APOB (OR, 0.835 per SD decrease in LDL; 95% CI 0.765–0.912; P = 5.61E − 5) for Europeans and LDLR for East Asians (OR, 0.920 (0.878–0.964), P = 4.9E − 4) was associated with decreased risk of ALS after Bonferroni correction (Fig. 4). There was no evidence of heterogeneity indicated by the Q test and horizontal pleiotropy indicated by the MR-Egger bias intercept in the causal effect estimates for both lipid targets (all P values > 0.05; Table S7 and Table S8). Furthermore, the MR-PRESSO approach and leave-one-out analysis indicated that the significant protective effect was not driven by outliers or single genetic variants for APOB and LDLR. When more stringent LD thresholds were set at R2 < 0.3, R2 < 0.2, and R2 < 0.1, the results consistently remained significant for only APOB in the secondary analyses (Fig. 4).

Causal map for the effect of lipid targets through different lipid-modifying pathways on amyotrophic lateral sclerosis (significant results). Notes: Shown are the IVW results for each causal association, with colors representing the P value. Red indicates higher risk, while green indicates lower risk. The significant targets after correcting for 104 tests (19 targets multiplied by 6 lipid-modifying effects, P < 4.39E − 4) in Europeans and for 44 tests (11 targets multiplied by 4 lipid-modifying effects, P < 1.14E − 3) in East Asians are labeled with the symbol “*.” Causal estimates bracketed in red indicate nominal significant causal effects (P < 0.05) that showed no evidence for heterogeneity or horizontal pleiotropy. The symbol “/” indicates that summary data for ApoA1 and ApoB were not available in East Asians. The symbol “-” indicates that targets with available SNPs of less than 3 were removed. Finally, lipid targets that passed the corrected P value in the primary analyses (R2 < 0.4), remained nominal significant in the secondary analyses (R.2 < 0.3, 0.2, and 0.1), and had no heterogeneity or horizontal pleiotropy were APOB (LDL-lowering effect, EUR), APOA1 (TC-lowering effect, EAS), and LDLR (TC-lowering effect, EAS). The non-significant causal map can be seen in Figure S2
Other Lipid Targets and ALS Risk
Next, we selected TC-lowering variants within genes encoding lipid targets as proxies for the effects of lipid-modifying drugs and examined their effects on ALS (see Table S7 and Table S8). All SNPs had F values > 10, suggesting they were unlikely to introduce marked weak instrument bias into the MR analyses.
The MR analysis showed an association of ApoA1 lower risk of ALS in the EAS population (OR = 0.859, 95% CI = 0.788–0.936, P = 5.38E − 4, SNPs = 7; Fig. 4). The Cochran Q statistic of the IVW method (P = 0.905) indicated no notable heterogeneity across instrument SNP effects (Table S7 and Table S8). Egger analysis did not show evidence of directional pleiotropy (P > 0.05). There was no distortion in the leave-one-out plot, suggesting that no single SNP was driving the observed effect in any analysis. In addition, reduction in TC through variants in genes encoding LDLR target was associated with a lower risk of ALS in the EAS population (OR = 0.910, 95% CI = 0.871–0.951, P = 2.73E − 5, SNPs = 17; Fig. 4). No evidence of heterogeneity and pleiotropy in the causal effect estimates was found (all P > 0.05, Table S7 and Table S8). The leave-one-out analysis did not change the overall direction. When more stringent LD thresholds were set at R2 < 0.3, R2 < 0.2, and R2 < 0.1, the results consistently remained significant for ApoA1 and LDLR (Fig. 4).
However, we did not find evidence of causal effects of other lipid targets on the risk of ALS through TG-lowering, HDL-raising, ApoB-lowering, and ApoA1-raising after Bonferroni correction.
Summary
Overall, according to the predefined screening criteria, lipid targets that passed the corrected P value in the primary analyses (R2 < 0.4), remained nominal significant in the secondary analyses (R2 < 0.3, 0.2, and 0.1), and had no heterogeneity or horizontal pleiotropy were APOB (LDL-lowering effect, EUR), APOA1 (TC-lowering effect, EAS), and LDLR (TC-lowering effect, EAS).
Furthermore, we use sensitivity analysis methods, including weighted median, MR-Egger, RadialMR, and MR-RAPS to confirm our results (Figure S3). As for APOB, all sensitivity methods supported the primary results. As for APOA1 and LDLR, sensitivity methods except MR-Egger supported the primary results. Colocalization analyses showed some evidence of a shared causal variant for APOA1 (PP.H4 = 36.48%; PP.H4/(PP.H4 + PP.H3) = 92.31%), and deemed confounding by LD unlikely (PP.H3 = 3.04%). For APOB and LDLR, we did not observe strong evidence suggesting colocalization between corresponding blood lipid fractions and ALS within the two gene regions (PP.H4 = 0.31% in APOB and PP.H4 = 2.50% in LDLR). For more details regarding the colocalization results, please see Table S9.
Positive Control Analyses
As shown in Table S10, genetic variations in the targets of ABCG5/G8, ANGPTL3, APOA1, APOA5/C3, APOB, ASGR1, CETP, HMGCR, LDLR, LPA, MTTP, NPC1L1, and PCSK9 through LDL-lowering effect were all associated with a decreased risk of CHD, except ANGPTL8 (R2 = 0.4). In summary, positive control analyses confirmed the validity of the IV selection methodology.
Discussion
In this study, we evaluated the causal relationships between a broad range of environmental exposures from UKB and ALS at first. Through this comprehensive strategy, we found consistent evidence across methods for a protective effect of the genetic liability of “cholesterol-lowering medication” and “atorvastatin” on ALS. Furthermore, we investigated the effects of genetic variation in lipid-modifying drug targets on ALS risk using independent, latest, and trans-ethnic GWAS data. We found causal evidence to indicate that genetic variation in the targets APOB (EUR) through LDL-lowering, APOA1 (EAS) through TC-lowering, and LDLR (EAS) through TC-lowering had a protective role in ALS. A conceptual framework for the phenotype discovery and target exploration of lipid-modifying drugs can be seen in Fig. 5. Our study may shed light on the lipid-modifying effects through which corresponding drugs confer a reduced risk of ALS and also indicate that repurposing the lipid-modifying drugs targeting APOB, APOA1, and LDLR in different ethnic populations for ALS prevention may be promising.

Conceptual framework for the phenotype discovery and target exploration of lipid-modifying drugs
Recently, several MR studies have been conducted to identify causal lipid-modifying targets for ALS [43,44,45]. However, these previous investigations were limited to European populations and failed to provide a comprehensive evaluation of potential targets using an unbiased methodology. In contrast, our study employed an unbiased and data-driven approach during the phenotype discovery stage, revealing two causal lipid-modifying medication traits for ALS from a vast array of UKB phenotypes. Unlike other studies that merely offer a one-stop shop of risk factors for ALS [46, 47], we further delved into the underlying mechanism or targets associated with these potential phenotypes by adopting a two-stage approach. Building upon the initial findings, we systematically identified 23 candidate lipid-modifying targets for subsequent drug target MR analysis through an extensive literature review. During the target exploration stage, it was postulated that genetic proxies for lipid targets exert their effects through multiple lipid-modifying pathways, surpassing the narrow focus on LDL/ApoB-lowering observed in previous studies [43,44,45]. Moreover, our investigation aimed to elucidate the potential differences in causal lipid targets between Europeans and East Asians for ALS. While consistent results were observed between our study and previous findings, our research also revealed novel insights. For instance, both our study and prior research failed to reveal the causal effects of common targets for ALS such as CETP, PCSK9, and NPC1L1. In contrast, our study suggests that APOA1 may impact ALS risk in East Asians through pathways related to lowering total cholesterol levels. A detailed comparison between our MR study and others on lipid-modifying targets for ALS has been summarized in Table S11.
ALS is a progressive neurodegenerative disorder characterized by an absence of well-established etiology and efficacious therapeutic interventions. The increased efficiency of MR studies and the abundance of GWAS data make it possible to broadly screen for causes and repurpose potential drugs. UKB is a large population cohort with genetics, broad phenotypes, and clinical information on half a million individuals. We conducted extensive screening for traits causally associated with ALS using UKB data and found that “cholesterol-lowering medication” and “atorvastatin” were protective against ALS. Statin use has previously been implicated in increased ALS risk through pharmaceutical surveillance [7], although this has largely been refuted by recent unbiased population-based studies which failed to identify any association between statin use and risk of ALS [8, 9]. However, our phenome-wide MR analyses indicate a protective effect of cholesterol-lowering drugs or atorvastatin against ALS, supported by a series of sensitivity analyses. Notably, during the phenotype discovery stage, we also included rosuvastatin and simvastatin. Although these two statins failed to pass all sensitivity analyses, the main analysis by IVW indicated their potential protective effects against ALS (Table S5). Therefore, it is plausible that the observed significant effect of cholesterol-lowering medication may be attributed solely to atorvastatin or a combined effect of various statins.
Previous epidemiological studies have also explored the role of blood lipids in the pathogenesis of ALS. These observational studies have yielded controversial results, with many reporting that hyperlipidemia increases disease risk and others suggesting the opposite [10,11,12,13,14,15,16]. In addition to being underpowered, much of previous research was based on blood lipid profiles obtained after diagnosis of ALS when confounding factors may influence these levels [10,11,12,13,14,15,16]. Based on the UKB datasets, a recent large prospective cohort study found that premorbid higher ApoB and LDL levels were associated with a higher risk of subsequent ALS diagnosis [48]. Observational studies cannot disentangle the causal direction of the association between lipid profile and the development of ALS. Apart from reverse causation, epidemiologic studies could have also been subject to unmeasured confounders, such as socioeconomic status, lifestyle, and drug use. A series of MR studies have found causal evidence of an association between ALS risk and specific environmental exposure. As indicated by previous MR studies, there is a strong consensus that high levels of LDL and TC in the blood are positively correlated with an increased risk of developing ALS [34, 43, 47, 49, 50].
Circulating blood cholesterol are multifunctional molecules, involved primarily in energy generation, as precursors or cofactors for signaling molecules, and in neuronal development and function [51]. Although the mechanisms by which LDL and TC might confer an increased risk of ALS have not been revealed, we further identified three protective lipid targets for ALS, including APOB by LDL-lowering effect, APOA1 by TC-lowering effect, and LDLR by TC-lowering effect. ApoB, encoded by APOB, is the main apolipoprotein of chylomicrons and LDL particles and serves as the ligand for the LDL receptor. In plasma, there are two main isoforms of ApoB: ApoB-48 and ApoB-100. ApoB-48 is synthesized exclusively in the gut, while ApoB-100 is produced in the liver. Utilizing an innovative animal model injected with cerebrospinal fluid (CSF) from ALS patients, a recent study has identified apolipoprotein B-100 in sporadic ALS CSF as the putative agent responsible for pathological translocation of TDP-43, motor neuron degeneration, and subsequent induction motor disability [52]. ApoA1, encoded by APOA1, forms the main lipoprotein constituent of HDL particles and is crucial for the process of reverse cholesterol transport from peripheral tissues to the liver [53]. HDL and ApoA1 have anti-inflammatory effects, reducing monocyte migration and dendritic cell function [54]. HDL and ApoA1 are also antioxidants and preserve mitochondrial function in models of ischemic heart disease [54]. Increased cerebrospinal fluid HDL and ApoA1 have also been observed following spinal cord injury, and exogenous HDL enhances neuronal growth via the ERK pathway [55]. LDL receptor, encoding by LDLR, is a protein expressed on the cell surface that binds and mediates the endocytosis of LDL particles. Using TDP-43 proteinopathies–related model, a recent study indicated that LDLR is involved in coaggregation with TDP-43 in the oligodendrocyte, suggesting that LDLR may have a role in cholesterol dysmetabolism associated with ALS pathogenesis [56]. Of course, regarding APOB, APOA1, and LDLR, more studies are needed to reveal the exact mechanisms by which they contribute to the disease. Although we found the protective role of statin use in ALS in the phenotype discovery stage, we failed to replicate our results using HMGCR as a drug target for statin in the target exploration stage. Considering the pleiotropic effects of statin, this may be attributed to the off-target effects of this drug on ALS.
The present study has some strengths. First, we conduct a hypothesis-free, unbiased, data-driven approach using broad environmental exposures from UKB in the phenotype discovery stage and explore the preliminary results using subsequent drug target MR analyses. Second, genetic proxies for lipid targets were hypothesized to act through the multiple lipid-modifying effects, which would help us explore the potential causal mechanism that really acts in ALS etiology. Third, positive control analyses were performed to validate the IV selection strategies and confirm that the approach was appropriate. Last, comprehensive analyses, including sensitivity analyses, were undertaken to reduce the false-positive possibility in our results.
This study has several limitations. First, our study can only predict the on-target effects of lipid drugs because only the well-documented protein targets were included in our analysis. Drug effects that are not exerted through these protein targets (off-target effects) cannot be captured in our MR models. Second, the genetically predicted drug effects may be somewhat different from therapeutic practice. An exposure instrumented by genetic variants is present from birth and lasts for a lifetime. This study should therefore be interpreted to evaluate the long-term modulation of genetic predisposition to using cholesterol-lowering drugs or statin on ALS risk in the phenotype discovery stage, as well as the long-term modulation of lipid-modifying drug targets on ALS risk in the target exploration stage. Moreover, given that genetic effects are lifelong, our estimates cannot reflect the effects of exposure to lipid drugs during a certain period of life. Fourth, the posterior probability of the shared causal variant was generally low (less than the conventional threshold of PP.H4 > 80%) in colocalization analyses. Since colocalization analyses were initially designed to identify evidence of colocalization between mRNA expression and diseases or traits, therefore, the default prior probabilities may not be ideal for the pairs of traits (e.g., lipid targets) and disease (e.g., ALS) within our study. Furthermore, it is important to note that the assumption of a single causal variant in genetic colocalization methods may not always hold, even when prior conditional analyses are performed. Moreover, considering the potential protective role of hyperlipidemia in the survival of ALS patients [12], it is important to acknowledge the potential impact of survivor bias, which may result in an overrepresentation of ALS patients with elevated blood lipid levels in our study cohort. Despite thoroughly addressing the reverse causality between blood lipids and ALS as demonstrated in the original ALS study [2], and conducting multiple sensitivity analyses to ensure the stability of our results, we recognize that the influence of survivor bias cannot be entirely eliminated. It is noteworthy that the largest ALS GWAS study conducted by van Rheenen et al. [2] was not included in the MR-Base platform (accessed on 1 April 2022). To identify potentially causal phenotypes for ALS using these available sources, we employed the accessible largest ALS GWAS at that time [19] as an outcome in the discovery stage. We acknowledge and clarify the design choice as a limitation, recognizing its potential impact on the robustness of the MR analyses in the discovery stage and the subsequent drug target results.
In conclusion, by screening thousands of environmental traits for their association with ALS in a phenome-wide MR framework, we propose potential protective effects of cholesterol-lowering drugs or statins on ALS risk. Further drug target analyses suggested that genetic variation in the targets APOB (EUR) through LDL-lowering effect, APOA1 (EAS) through TC-lowering effect, and LDLR (EAS) through TC-lowering effect had a protective role in ALS. Future fundamental research into lipid targets using established ALS models may yield novel insights into the underlying pathophysiology of the disease.
Availability of Data and Materials
The GWAS summary statistics supporting this research are available from the corresponding GWAS consortium. The main paper and supplementary materials present all data supporting our findings. The code or algorithm used to generate results in this study is available from the corresponding authors upon reasonable request.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- CHD:
-
Coronary heart disease
- UKB:
-
UK Biobank
- MRC-IEU:
-
Medical Research Council Integrative Epidemiology Unit
- GWAS:
-
Genome-wide association studies
- SNP:
-
Single nucleotide polymorphism
- MR:
-
Mendelian randomization
- IVW:
-
Inverse variance weighted
- IV:
-
Instrumental variables
- Q test:
-
Cochran’s Q test
- MR-PRESSO:
-
MR pleiotropy residual sum and outlier
- OR:
-
Odds ratio
- CI:
-
Confidence interval
- TG:
-
Triglycerides
- TC:
-
Total cholesterol
- LDL:
-
Low-density lipoprotein cholesterol
- HDL:
-
High-density lipoprotein cholesterol
- ApoB:
-
Apoprotein B
- ApoA1:
-
Apoprotein A1
- EUR:
-
Europeans
- EAS:
-
East Asians
- NA:
-
Not available
References
Chia R, Chio A, Traynor BJ (2018) Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol 17:94–102
van Rheenen W, van der Spek RAA, Bakker MK et al (2021) Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat Genet 53:1636–1648
Brown RH, Al-Chalabi A (2017) Amyotrophic lateral sclerosis. N Engl J Med 377:162–172
Skrivankova VW, Richmond RC, Woolf BAR et al (2021) Strengthening the reporting of observational studies in epidemiology using mendelian randomization: the STROBE-MR statement. JAMA 326:1614–1621
Schmidt AF, Finan C, Gordillo-Maranon M et al (2020) Genetic drug target validation using Mendelian randomisation. Nat Commun 11:3255
Hemani G, Zheng J, Elsworth B et al (2018) The MR-Base platform supports systematic causal inference across the human phenome. Elife 7:e34408
Golomb BA, Verden A, Messner AK, Koslik HJ, Hoffman KB (2018) Amyotrophic lateral sclerosis associated with statin use: a disproportionality analysis of the FDA’s Adverse Event Reporting System. Drug Saf 41:403–413
Mariosa D, Kamel F, Bellocco R et al (2020) Antidiabetics, statins and the risk of amyotrophic lateral sclerosis. Eur J Neurol 27:1010–1016
Sorensen HT, Riis AH, Lash TL, Pedersen L (2010) Statin use and risk of amyotrophic lateral sclerosis and other motor neuron disorders. Circ Cardiovasc Qual Outcomes 3:413–417
Chio A, Calvo A, Ilardi A et al (2009) Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology 73:1681–1685
Mariosa D, Hammar N, Malmström H et al (2017) Blood biomarkers of carbohydrate, lipid, and apolipoprotein metabolisms and risk of amyotrophic lateral sclerosis: a more than 20-year follow-up of the Swedish AMORIS cohort. Ann Neurol 81:718–728
Dupuis L, Corcia P, Fergani A et al (2008) Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 70:1004–1009
Goldstein MR, Mascitelli L, Pezzetta F (2008) Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. 71:956 (author reply 956–957)
Schmitt F, Hussain G, Dupuis L, Loeffler JP, Henriques A (2014) A plural role for lipids in motor neuron diseases: energy, signaling and structure. Front Cell Neurosci 8:25
Timmins HC, Saw W, Cheah BC et al (2017) Cardiometabolic health and risk of amyotrophic lateral sclerosis. Muscle Nerve 56:721–725
Kioumourtzoglou MA, Seals RM, Gredal O, Mittleman MA, Hansen J, Weisskopf MG (2016) Cardiovascular disease and diagnosis of amyotrophic lateral sclerosis: a population based study. Amyotroph Lateral Scler Frontotemporal Degener 17:548–554
Hegele RA, Tsimikas S (2019) Lipid-lowering agents targets beyond PCSK9. Circ Res 124:386–404
Zhang XQ, Yang YX, Zhang C et al (2022) Validation of external and internal exposome of the findings associated to cerebral small vessel disease: a Mendelian randomization study. J Cereb Blood Flow Metab 42:1078–1090
Nicolas A, Kenna KP, Renton AE et al (2018) Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron 97:1268-1283.e1266
Pierce BL, Burgess S (2013) Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol 178:1177–1184
Hemani G, Tilling K, Davey SG (2017) Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet 13:e1007081
Verbanck M, Chen CY, Neale B, Do R (2018) Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 50:693–698
Nikpay M, Goel A, Won HH et al (2015) A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet 47:1121–1130
Graham SE, Clarke SL, Wu KH et al (2021) The power of genetic diversity in genome-wide association studies of lipids. Nature 600:675–679
Richardson TG, Sanderson E, Palmer TM et al (2020) Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis. PLoS Med 17:e1003062
Benyamin B, He J, Zhao Q et al (2017) Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nat Commun 8:611
Ference BA, Ray KK, Catapano AL et al (2019) Mendelian Randomization Study of ACLY and Cardiovascular Disease. N Engl J Med 380:1033–1042
Williams DM, Bandres-Ciga S, Heilbron K et al (2020) Evaluating lipid-lowering drug targets for Parkinson’s disease prevention with mendelian randomization. Ann Neurol 88:1043–1047
Levin MG, Zuber V, Walker VM et al (2021) Prioritizing the role of major lipoproteins and subfractions as risk factors for peripheral artery disease. Circulation 144:353–364
Wang JQ, Li LL, Hu A et al (2022) Inhibition of ASGR1 decreases lipid levels by promoting cholesterol excretion. Nature 608:413–420
Reiner Z (2013) Managing the residual cardiovascular disease risk associated with HDL-cholesterol and triglycerides in statin-treated patients: a clinical update. Nutr Metab Cardiovasc Dis 23:799–807
Barter PJ, Brandrup-Wognsen G, Palmer MK, Nicholls SJ (2010) Effect of statins on HDL-C: a complex process unrelated to changes in LDL-C: analysis of the VOYAGER Database. J Lipid Res 51:1546–1553
Thanassoulis G, Williams K, Ye K et al (2014) Relations of change in plasma levels of LDL-C, non-HDL-C and apoB with risk reduction from statin therapy: a meta-analysis of randomized trials. J am heart assoc 3:e000759
Zeng P, Zhou X (2019) Causal effects of blood lipids on amyotrophic lateral sclerosis: a Mendelian randomization study. Hum Mol Genet 28:688–697
Ference BA, Kastelein JJP, Ginsberg HN et al (2017) Association of genetic variants related to CETP inhibitors and statins with lipoprotein levels and cardiovascular risk. JAMA 318:947–956
Nowak C, Arnlov J (2018) A Mendelian randomization study of the effects of blood lipids on breast cancer risk. Nat Commun 9:3957
Ference BA, Kastelein JJP, Ray KK et al (2019) Association of triglyceride-lowering LPL variants and LDL-C-lowering LDLR variants with risk of coronary heart disease. JAMA 321:364–373
Wang Q, Wang Y, Lehto K, Pedersen NL, Williams DM, Hagg S (2019) Genetically-predicted life-long lowering of low-density lipoprotein cholesterol is associated with decreased frailty: a Mendelian randomization study in UK biobank. EBioMedicine 45:487–494
Noyce AJ, Bandres-Ciga S, Kim J et al (2019) The Parkinson’s disease Mendelian randomization research portal. Mov Disord 34:1864–1872
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR (2016) Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol 45:1961–1974
Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD (2015) Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or Both: a 2 x 2 factorial Mendelian randomization study. J Am Coll Cardiol 65:1552–1561
Ference BA, Robinson JG, Brook RD et al (2016) Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med 375:2144–2153
Yan Z, Xu Y, Li K, Liu L (2023) Association between genetically proxied lipid-lowering drug targets, lipid traits, and amyotrophic lateral sclerosis: a mendelian randomization study. Acta Neurol Belg [online ahead of print]
Wang W, Zhang L, Xia K, Huang T, Fan D (2023) Mendelian randomization analysis reveals statins potentially increase amyotrophic lateral sclerosis risk independent of peripheral cholesterol-lowering effects. Biomedicines 11(5):1359
Li Z, Tian M, Jia H et al (2023) Genetic variation in targets of lipid-lowering drugs and amyotrophic lateral sclerosis risk: a Mendelian randomization study. Amyotroph Lateral Scler Frontotemporal Degener 25(1–2):197–206
Pan S, Kang H, Liu X et al (2022) Brain Catalog: a comprehensive resource for the genetic landscape of brain-related traits. Nucleic Acids Res 51(D1): D835–D844
Bandres-Ciga S, Noyce AJ, Hemani G et al (2019) Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Ann Neurol 85(4):470–481
Thompson A, Talbot K, Turner M (2022) Higher blood high density lipoprotein and apolipoprotein A1 levels are associated with reduced risk of developing amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 93:75–81
Xia K, Klose V, Högel J et al (2023) Lipids and amyotrophic lateral sclerosis: a two-sample Mendelian randomization study. Eur J Neurol 30:1899–1906
Julian TH, Boddy S, Islam M et al (2022) A review of Mendelian randomization in amyotrophic lateral sclerosis. Brain 145:832–842
Pfrieger FW (2003) Cholesterol homeostasis and function in neurons of the central nervous system. Cell Mol Life Sci 60:1158–1171
Wong JK, Roselle AK, Shue TM et al (2022) Apolipoprotein B-100-mediated motor neuron degeneration in sporadic amyotrophic lateral sclerosis. Brain Commun 4: fcac207
Andrikoula M, McDowell IF (2008) The contribution of ApoB and ApoA1 measurements to cardiovascular risk assessment. Diabetes Obes Metab 10:271–278
Tiniakou I, Drakos E, Sinatkas V et al (2015) High-density lipoprotein attenuates Th1 and th17 autoimmune responses by modulating dendritic cell maturation and function. J Immunol 194:4676–4687
Sengupta MB, Saha S, Mohanty PK, Mukhopadhyay KK, Mukhopadhyay D (2017) Increased expression of ApoA1 after neuronal injury may be beneficial for healing. Mol Cell Biochem 424:45–55
Ho WY, Chang JC, Lim K et al (2021) TDP-43 mediates SREBF2-regulated gene expression required for oligodendrocyte myelination. J Cell Biol 220(9):e201910213
Funding
This study was supported by the National Key Research and Development Program of China (Grant No. 2022YFC2703101 to Y.P.C), the National Natural Science Fund of China (Grant No. 82371422 and 81971188 to Y.P.C.), the National Natural Science Fund of Sichuan (Grant No. 2022NSFSC0749 to B.C.), and the Science and Technology Bureau Fund of Sichuan Province (Grant No. 2023YFS0269 to Y.P.C). We are grateful to all the studies that have made the public GWAS summary data available. We thank all the patients and their families for their generous contribution to this research.
Author information
Authors and Affiliations
Contributions
Z.J. and Y.P.C. contributed to the conception and design of the study; Z.J., X.J.G., W.M.S., Q.Q.D., K.F.Y., Y.L.R., Y.W., and B.C. contributed to the acquisition and analysis of data; Z.J. and Y.P.C. contributed to drafting the text and preparing the figures.
Corresponding author
Ethics declarations
Ethics Approval
This research involves analyzing publicly available data, for which ethical approval and individual consent were obtained from all original studies.
Patient Consent for Publication
Not required.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jiang, Z., Gu, XJ., Su, WM. et al. Discovery and Exploration of Lipid-Modifying Drug Targets for ALS by Mendelian Randomization. Mol Neurobiol 61, 6572–6583 (2024). https://doi.org/10.1007/s12035-024-04007-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-024-04007-9