
Abstract
Whether epigenetic modifications participate in the cell apoptosis after ischemic stroke remains unclear. Histone 3 tri-methylation at lysine 27 (H3K27me3) is a histone modification that leads to gene silencing and is involved in the pathogenesis of ischemic stroke. Since the expression of many antiapoptotic genes is inhibited in the ischemic brains, here we aimed to offer an epigenetic solution to cell apoptosis after stroke by reversing H3K27me3 levels after ischemia. GSK-126, a specific inhibitor of enhancer of zeste homolog 2 (EZH2), significantly decreased H3K27me3 levels and inhibited middle cerebral artery occlusion (MCAO) induced and oxygen glucose deprivation (OGD) induced cell apoptosis. Moreover, GSK-126 attenuated the apoptosis caused by oxidative stress, excitotoxicity, and excessive inflammatory responses in vitro. The role of H3K27me3 in regulating of the expression of the antiapoptotic molecule B cell lymphoma-2 like 1 (Bcl2l1) explained the antiapoptotic effect of GSK-126. In conclusion, we found that GSK-126 could effectively protect brain cells from apoptosis after cerebral ischemia, and this role of GSK-126 is closely related to an axis that regulates Bcl2l1 expression, beginning with the regulation of EZH2-dependent H3K27me3 modification.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Stroke is the second leading cause of death worldwide [1], and most cases of stroke were ischemic stroke [2]. Currently, the main approach for treating ischemic stroke was the restoration of blood flow by intravenous thrombolysis or intraarterial thrombectomy [3], but subsequent brain cell apoptosis was inevitable. As a result, approximately 40% of poststroke survivors were clinically disabled [4]. Apoptosis was a kind of programmed cell death that occurs in response to external damage, and it involves the regulation of gene expression levels [5, 6]. Histone methylation, particularly histone 3 tri-methylation at lysine 27 (H3K27me3), which regulates gene expression, was thought to be involved in cellular stress responses [7]. Elucidating the mechanism by which H3K27me3 regulates apoptosis has attracted the interest of researchers. Investigation of this mechanism may be beneficial for the clinical treatment of ischemic stroke.
H3K27me3 could silence gene expression by increasing DNA methylation or altering the binding of histones to heterochromatin [8, 9]. H3K27me3 levels were regulated by the polycomb repressive complex 2 (PRC2), or more precisely, by one of its components, namely, enhancer of zeste homolog 2 (EZH2) [10]. In contrast, H3K27me3 demethylation was mediated by lysine demethylase 6 (KDM6) family members and Jumonji domain-containing protein D3 (JMJD3) [11, 12]. Previously, reports have shown that histone methylation participates in the pathogenesis of cerebral ischemia, including oxidative stress, calcium overload, and inflammatory responses [13]. However, until now, evidence that histone methylation, including H3K27me3, regulates apoptosis during ischemic brain injury has been insufficient.
GSK-126 is a potent inhibitor of EZH2 methyltransferase activity, and it can reduce global H3K27me3 levels and reactivate silenced PRC2 target genes [14, 15]. In particular, we previously found that GSK-126-mediated decreases in H3K27me3 levels in hippocampal neurons benefited neuronal survival after cerebral ischemia [13]. Previous data also indicated that the neuroprotective role of GSK-126 was based on its ability to inhibit cell apoptosis. However, the underlying mechanism is still unclear. Interestingly, in the present research, GSK-126 not only effectively attenuated apoptosis after middle cerebral artery occlusion (MCAO) but also practically inhibited apoptosis induced by three ischemia-related models, including oxidative stress, excitotoxicity, and inflammation. This suggests that the protective effect of GSK-126 against cerebral ischemia may act directly on genes related to apoptosis. Combined with previous research, we assumed that the expression level of B cell lymphoma-2 like 1 (Bcl2l1), a gene that negatively regulates apoptosis, might be regulated by H3K27me3, and we assumed that GSK-126 might attenuate postischemic apoptosis via the EZH2-H3K27me3-Bcl2l1 axis.
Bcl2l1 is a member of the Bcl-2 family. Members of the Bcl-2 family (Bcl2, Bclw, Bcl2l1, and Mcl1) have similar names and functions, but not similar protein structures. In contrast to proapoptotic molecules such as Bax and Bak, which contain three BH domains (BH1, BH2, and BH3), Bcl2l1 has a BH4 domain [16]. The BH4 domain is necessary for pro-survival activity. Bcl2l1 could inhibit the binding of apoptotic protease activating factor-1 (Apaf-1) to pro-Caspase-9, thereby preventing Caspase-9 from activation [17]. As a result, Caspase-9 may fail to interact with Caspase-3 and activate the apoptotic program. Bcl2l1 plays an essential role in preventing cell apoptosis. Therefore, exploring the epigenetic regulatory mechanism of Bcl2l1 in the development of ischemic brain injury could contribute to providing novel insights into neuronal apoptosis after ischemic stroke.
Materials and Methods
Animals and Ethics
Male Sprague-Dawley rats weighing 220–250 g were purchased from the experimental animal center of Xuzhou Medical University (202209S066). The rats were housed in an environment of 12 light/12 dark cycles with free access to food and water at 23 °C. All experimental protocols were approved by the Ethics Committee of Xuzhou Medical University (date 06, 2022/No. 202209S066).
Establishment of the MCAO Model
The MCAO model was established as described previously [18]. Briefly, after anesthetizing the rats with 4% isoflurane and then maintaining anesthesia with 2% isoflurane, the neck skin was cut to expose the left carotid sheath, common carotid artery, and external and internal carotid arteries. The thread embolism (Cinontech, catalog no. 2634A4, Beijing, China) was inserted from the common carotid artery to the internal carotid artery (insertion depth: approximately 20 mm), and the thread embolism will be withdrawn to the common carotid artery after 2 h. The animals in the sham operation control group were subjected to the same procedure, but the carotid artery was not occluded. During the operation, a laser speckle flow imaging system (RFSLI ZW/RFLSI III, RWD Life Science Co., Shenzhen, China) was used to measure cerebral blood flow in real time. Subsequent experiments were performed after 24 h of reinfusion.
Drugs and Administration
For surgery, rats were anesthetized with isoflurane and placed on a stereotaxic table, and anesthesia was maintained with isoflurane using the gas delivery system of the localizer. The scalp was incised longitudinally to expose the fontanelle, and a small hole was drilled in the skull using the fontanelle as a reference (stereotaxic zero) with the following stereotaxic coordinates: medio-lateral (ML): 1.0 mm left of the midline of the skull and anterior-posterior (AP): 1.5 mm posterior to the bregma, which corresponds to the lateral ventricular region. A catheter was inserted into the small hole at a depth of 4 mm from the skull and then secured with dental cement. The rats were returned to their cages after surgery and allowed to rest for 3 days with adequate food and water to eliminate surgery-induced inflammatory reactions and repair the blood-brain barrier. On the fourth day, the rats were anesthetized with isoflurane and injected with the drug via intracerebroventricular injection (ICV).
GSK-126 was purchased from Vicmed Biotech (Xuzhou, China) and stored at − 20 °C. GSK-126 was dissolved in 20% β-cyclodextrin to a final concentration of 5 mg/ml and injected into the lateral ventricle daily for seven consecutive days (20 μl/day) before the MCAO model was established.
2,3,5-Triphenyl Tetrazolium Chloride Staining and Neurobehavioral Scores
Triphenyl tetrazolium chloride (TTC) reacts with succinate dehydrogenase in the mitochondria to produce red formazan, but in ischemic tissues, the activity of dehydrogenase is reduced, resulting in a pale white block. Rats were decapitated after anesthesia, and the brains were washed once with PBS. The brains were then cut into 2-mm consecutive sections and stained with 2% TTC in the dark for 30 min at 37 °C. Subsequently, brain sections were fixed with 4% paraformaldehyde at room temperature for 24 h and photographed. After TTC staining, we recorded with photos and calculated the volume of cerebral infarction by ImageJ. The modified neurological severity score (mNSS) was used to evaluate the neurological function of rats, with a maximum score of 18 [19, 20]. The test included motor, sensory, reflex, and balance tests, and a higher score indicated more severe injury.
TUNEL Assay
Cortical neuronal apoptosis was analyzed by TUNEL staining (Keygen Biotech, Nanjing, China). The experiments were performed according to the manufacturer’s instructions. Briefly, the rat brains were washed with PBS and then permeabilized with 1% Triton X-100 for 15 min at room temperature. Subsequently, they were incubated with TUNEL staining solution for 1 h in the dark and then washed with PBS. Finally, each sample was incubated with DAPI staining solution for 5 min at room temperature in the dark and observed by fluorescence microscopy (Olympus, Japan). Images were captured at 450 nm and 520 nm. Fluorescence intensity was analyzed using ImageJ, and the data were analyzed using GraphPad Prism 5.
Cell Culture and Modeling
HT22 cells were cultured under 37 °C and 5% CO2 in a medium containing 10% FBS. The cells were pretreated with GSK-126 (50 μM) for 24 h after being passaged for 24 h. Then, the cells were administrated with H2O2 (500 μM) for 12 h to construct the oxidative stress model, glutamate (20 mM) for 12 h to construct the excitotoxicity model, or LPS (15 μg/ml) for 12 h to construct the excessive inflammatory model, respectively. After pretreated with GSK-126, the cells were modeled by oxygen and glucose deprivation (OGD). For the OGD model, cells were cultured for 12 h with glucose-free medium (Gibco, America) in 1.5% O2.
Cell transfections were performed according to the protocols of Engreen Entranster™ H4000 (Engreen, Beijing, China). In brief, 2 μg of plasmids was dissolved in 100 μl of serum-free medium and 5 μl of Engreen Entranster™ H4000 was diluted in 100 μl of serum-free medium (Keygen Biotech, Nanjing, China). The mixture was added to the cell culture medium after 15 min of incubation. The coding DNA sequence (CDS) was obtained from NCBI. Plasmids for Bcl2l1 overexpression were cloned using the ClonExpress II One Step Cloning Kit (Vazyme, Nanjing, China); cDNA from the mouse hippocampal neuronal cell line HT22 was used as a template. The CDS of Bcl2l1 was inserted into the pcDNA3.1 vector by the restriction enzyme EcoRI. The primers for pcDNA3.1-Bcl2l1 are listed in Table 1.
Western Blots
Rat cortical samples and cells were lysed with RIPA buffer supplemented with 1% protease inhibitor. The mixture was allowed to incubate on ice for 30 min and then centrifuged at 12,000 rpm for 15 min. The protein concentrations were quantified by the BCA method, and the proteins were separated via 12% SDS-PAGE. The separated proteins were subsequently transferred to 0.22-μm PVDF membranes and then blocked with 5% fat-free milk for 2 h. The membranes were incubated with primary antibodies at 4 °C overnight. Following the primary antibody incubation, the membranes were washed with TBST three times and then incubated with HRP-conjugated goat anti-rabbit IgG or anti-mouse IgG secondary antibodies for 1 h at room temperature. All the antibodies used for western blots are listed in Table 2. Protein bands were visualized by a chemical fluorescence gel imaging system and analyzed by ImageJ.
Chromatin Immunoprecipitation
The rat cortex samples were first shredded into 1-mm3 size pieces, the samples were cross-linked with 1% formaldehyde for 5 min at room temperature, and then, glycine was added to terminate the cross-linking reaction. The cortices were washed three times with PBS and collected into tubes after low-speed centrifugation. After ultrasonic fragmentation, the chromosome will be cut into a size containing DNA fragment of 150–300 bp. Approximately 10% of the supernatant was used as input, and 1 μl of anti-H3K27me3 and 60 μl of Protein G were added to the remaining lysate and incubated overnight at 4 °C. The next day, the beads were washed five times with the designated buffer, and the complexes were eluted from beads after heating to 65 °C. The DNA cross-linking reaction was reversed at 67 °C for 5 h, followed by purification. The purified DNA sample was used as a template to detect the Bcl2l1 promoter by real-time PCR. The primers for Bcl2l1 for ChIP are listed in Table 1.
Real-Time PCR
Extraction of total mRNA was performed according to the instruction for the RNA Isolater Total RNA Extraction Reagent (Vazyme, Nanjing, China). Subsequently, mRNA was reverse transcribed to cDNA with a of HiFiScript cDNA Synthesis Kit (ComWin Biotech, Taizhou, China). Finally, RT-PCR was carried out with SYBR Green Master Mix (Vazyme, Nanjing, China) according to the user manual with a real-time PCR system (Thermo Fisher Scientific, America). β-Actin was used for normalization. All the RT-PCR primers used in this study are listed in Table 1.
Flow Cytometry
The percentage of apoptotic HT22 cells was evaluated using an annexin V-FITC/PI kit (Keygen Biotech, Nanjing, China). In brief, the cells were collected with EDTA-free trypsin and then washed twice with PBS. The supernatant was discarded, and the cell pellet was resuspended in binding buffer. Annexin V-FITC and propidium iodide were added to the suspensions, respectively. The suspensions are incubated at room temperature for 15 min in the dark and then analyzed.
Detection of Intracellular Reactive Oxygen Species
The intracellular reactive oxygen species (ROS) level of each group was measured with a ROS Assay Kit (Beyotime, Beijing, China). According to the manufacturer’s instructions, the cell culture medium was discarded, and the DCFH-DA diluted with serum-free medium was added. Cells were incubated at 37 °C and 5% CO2 in the dark for 20 min. Subsequently, cells were washed three times with serum-free medium and then observed by a fluorescence microscope (Olympus IX-71, Japan).
Intracellular Ca2+ Staining
The intracellular calcium ion concentration was measured by Fluo-3 AM (Beyotime, Beijing, China). Cells were cultured with medium containing 1 μM Fluo-3AM at 37 °C and 5% CO2 in the dark for 40 min. Then, PBS was used to wash once, and the cells were incubated under the same conditions for 20 min again. Finally, the cells were observed by a fluorescence microscope (Olympus IX-71, Japan).
Bisulfite Sequencing PCR
According to the manufacturer’s instructions, genomic DNA was extracted from cells (Yuanpinghao Biological Technology, Beijing, China). Then, DNA was modified using EpiTect® Bisulfite Kit (QIAGEN, Germany). After the DNA was treated with bisulfite, the fragments were amplified by PCR. The primers for modified Bcl2l1 are listed in Table 1. The PCR products were purified and ligated to the pMD18-T Vector (TaKaRa, Japan). The products were transformed into DH-5α; positive clones were selected, shaken, and sent to the Nanjing Springen Biotechnology Co., Ltd for sequencing.
Statistical Analysis
The statistical analysis of the data was performed using GraphPad Prism 5. All the experiments were performed in three replicates, and data are expressed as the mean ± standard error of the mean (SEM). One-way ANOVA and the unpaired t test were used to analyze statistical significance between the different groups.
Results
GSK-126 Attenuates MCAO-Induced Injury by Reducing H3K27me3 Levels
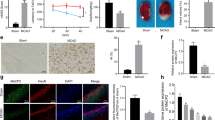
In our previous study, we found that GSK-126 exerted a protective effect by inhibiting H3K27me3 levels in a transient global cerebral ischemia model. In this study, the effects of GSK-126 in a MCAO model were first examined in rats. The flowchart of the experimental operation and the results are shown in Fig. 1. The results showed that GSK-126 significantly reduced the cerebral infarct area (P < 0.05; Fig. 1B, C and Fig. S1) and neurobehavioral scores (P < 0.01; Fig. 1D) after MCAO. This protective effect was mediated out by inhibiting H3K27me3 levels (P < 0.05; Fig. 1E, F). In addition, GSK-126 may exert its neuroprotective effect by reducing the level of brain cell apoptosis (P < 0.001; Fig. 1G, H). In conclusion, the results suggest that pretreatment with GSK-126 can effectively reduce H3K27me3 levels in the postischemic brain as well as ischemic brain injury caused by MCAO.

GSK-126 attenuates ischemic brain injury in rats. A Schematic diagram of GSK-126 administration and MCAO modeling. B TTC staining of rat brains in the MCAO model. C The ratio of infarct volume. D The behavioral function was detected by modified neurological severity score (mNSS). E The expression of H3K27me3 in the MCAO model was detected by western blots. F Gray intensity analysis of western blots results. The results were normalized by histone H3. G Apoptosis was detected using TUNEL staining. Scale bar = 20 μm. H Quantitative analysis of TUNEL-positive cells. *P < 0.05, **P < 0.01, ***P < 0.001. N = 3
GSK-126 Attenuates Damage Caused by Oxidative Stress
The main factors that influence apoptosis under postischemic conditions are (1) increased oxygen radical production, (2) intracellular calcium overload, and (3) excessive activation of inflammatory responses. To explore the mechanism underlying the anti-apoptotic effects of GSK-126, we investigated the effect of GSK-126 (50 μM) pretreatment for 24 h in three apoptosis models in vitro (Fig. S2). The first is the oxidative stress model, characterized by excessive production or reduced degradation of ROS. We used hydrogen peroxide (H2O2) to establish the oxidative stress model. Flow cytometry analysis of apoptosis showed that GSK-126 reduced apoptosis induced by H2O2 (P < 0.05; Fig. 2A, B). In addition, we found that GSK-126 decreased the levels of intracellular ROS (P < 0.001; Fig. 2C, D). The results of western blots showed that GSK-126 inhibited Caspase-3 activation by rescuing the expression of Bcl2/Bax (P < 0.05; Fig. 2E, F and Fig. S3A). In summary, GSK-126 could maintain normal cellular status via inhibiting intracellular ROS production and resist apoptosis caused by oxidative stress through increasing the Bcl2/Bax levels.

GSK-126 alleviates H2O2-induced apoptosis. HT22 cells were pretreated with GSK-126 (50 μM) for 24 h and then stimulated with H2O2 (500 μM) for 12 h. A H2O2-induced apoptosis was detected by flow cytometry. B Analysis of H2O2-induced apoptosis ratio. C The expression of ROS was detected by DCFH-DA probe. Scale bar = 100 μm. D Quantitative analysis of ROS level. E The expression of cleaved Caspase-3, Bcl2, and Bax in the oxidative stress model was detected by western blots. F Gray intensity analysis of western blots results. The results were normalized by β-actin. *P < 0.05, **P < 0.01, ***P < 0.001. N = 3
GSK-126 Attenuates Excitotoxic-Induced Apoptosis via Bcl2/Bax Pathways
Excitotoxicity induced by glutamate is also one of the most important pathogenic mechanisms in ischemic stroke, and it mainly functions through the activation of calcium-dependent death pathways. We established a cellular excitotoxicity model using glutamate treatment. Flow cytometry analysis revealed that GSK-126 reduced glutamate-induced apoptosis (P < 0.001; Fig. 3A, B). Using the Fluo-3AM probe to measure intracellular Ca2+ concentration, we found that GSK-126 significantly reversed the glutamate-induced increase in intracellular Ca2+ concentrations (P < 0.05; Fig. 3C). Consistent with previous results, GSK-126 also restored Bcl2/Bax levels and prevented Caspase-3 activation (P < 0.01; Fig. 3D, E and Fig. S3B). That is, GSK-126 may exert anti-apoptotic effects by altering the intracellular Ca2+ concentration.

GSK-126 mitigates glutamate-induced apoptosis. HT22 cells were pretreated with GSK-126 (50 μM) for 24 h and then stimulated with glutamate (20 mM) for 12 h. A Glutamate-induced apoptosis was detected by flow cytometry. B Analysis of glutamate-induced apoptosis ratio. C The intracellular Ca2+ concentration was detected by Fluo-3 AM probe. Scale bar = 100 μm. D The expression of cleaved Caspase-3, Bcl2, and Bax in the excitotoxicity model was detected by western blots. E Gray intensity analysis of western blots results. The results were normalized by β-actin. *P < 0.05, **P < 0.01, ***P < 0.001. N = 3
GSK-126 Protects Cells from Inflammatory Damage
Cerebral ischemia is accompanied by severe inflammatory response, which in turn further aggravates cerebral ischemia. Therefore, we first examined the degree of LPS-induced apoptosis and apoptosis in the inflammation model after pretreatment with GSK-126 by flow cytometry. GSK-126 effectively reduced LPS-induced apoptosis (P < 0.05; Fig. 4A, B). Furthermore, GSK-126 could still increase Bcl2/Bax ratio to minimize Caspase-3 activation (P < 0.05; Fig. 4D, E and Fig. S3C). The RT-qPCR assay revealed that the mRNA levels of IL-1β, IL-6, and TNF-α increased after stimulation with LPS (P < 0.05; Fig. 4C). When cells were pretreated with GSK-126, the mRNA levels of IL-1β, IL-6, and TNF-α were not elevated by LPS stimulation (P < 0.001; Fig. 4C).

GSK-126 protects HT22 cells from LPS-induced damage. HT22 cells were pretreated with GSK-126 (50 μM) for 24 h and then stimulated with LPS (15 μg/ml) for 12 h. A LPS-induced apoptosis was detected by flow cytometry. B Analysis of LPS-induced apoptosis ratio. C The mRNA levels of IL-1β, IL-6, and TNF-α were determined by RT-PCR. D The expression of cleaved Caspase-3, Bcl2, and Bax in the inflammation model was detected by western blots. E Gray intensity analysis of western blots results. The results were normalized by β-actin. *P < 0.05, **P < 0.01, ***P < 0.001. N = 3
GSK-126 Upregulates Bcl2l1 by Inhibiting H3K27me3 Enrichment Levels in the Bcl2l1 Promoter
These above results confirm that GSK-126 has a good protective effect on all three major factors that lead to apoptosis during ischemic brain injury. The mechanism underlying its effect on apoptosis may be direct targeting of the apoptotic signaling pathway. We previously found that the anti-apoptotic role of GSK-126 may be related to the regulation of H3K27me3 in genes that negatively regulate of the execution phase of apoptosis, including Dffa, Fzd3, Bcl2l1, Pam16, and Nmnat1 [13]. Further analysis showed that the expression level of Bcl2l1 was regulated by GSK-126 (P < 0.01; Fig. S4A). Therefore, we next examined the expression of Bcl2l1 in the MCAO model. The results showed that GSK-126 increased the protein levels of Bcl2l1 (P < 0.05; Fig. 5A, B). GSK-126 also reversed the reduction in Bcl2/Bax caused by MCAO, which reduced Caspase-3 activation (P < 0.001; Fig. 5A, B). Furthermore, regional IHC analysis revealed that GSK-126 could also reverse the MCAO-induced decrease in Bcl2l1 expression in the frontal parietal cortex, striatum, and hypothalamus (Fig. S5A). Conversely, in areas where blood supply was not provided by the middle cerebral artery, such as the hippocampus and SVZ, the expression levels of Bcl2l1 remained unaffected by MCAO modeling. GSK-126 also did not elevate Bcl2l1 expression levels in these regions following MCAO (Fig. S5B).

The expression of Bcl2l1 could be upregulated by GSK-126. A The expression of Bcl2l1, cleaved Caspase-3, Bcl2, and Bax in the MCAO model was detected by western blots. B Gray intensity analysis of western blots results. The results were normalized by β-actin. C The enrichment of H3K27me3 in the Bcl2l1 promoter region after MCAO was determined by ChIP assay. D The methylation level of Bcl2l1 promoter region in the OGD model was determined by bisulfite sequencing PCR (BSP). *P < 0.05, **P < 0.01, ***P < 0.001. N = 3
As expected, Bcl2l1 showed the same trend in models of oxygen glucose deprivation (OGD), oxidative stress, excitotoxicity, and excessive inflammatory response (Fig. S4). ChIP analysis revealed that H3K27me3 enrichment in the Bcl2l1 promoter region was significantly elevated after MCAO but decreased after pretreatment with GSK-126 (P < 0.01; Fig. 5C). However, the alteration of H3K27me3 enrichment levels in each group did not affect the DNA methylation levels in their promoter (Fig. 5D). These results suggested that H3K27me3 regulates the expression level of Bcl2l1 but not through DNA methylation.
Bcl2l1 Protects Cells from OGD-Induced Apoptosis
GSK-126/H3K27me3 can regulate the expression levels of many genes, and to identify the crucial role of Bcl2l1 in postischemic apoptosis, we constructed plasmids to overexpress Bcl2l1. Bcl2l1 expression was increased significantly after transfection (P < 0.05; Fig. 6A, B). In addition, according to the results of the flow cytometric assay, it was found that the apoptosis ratio was significantly reduced after increased Bcl2l1 expression compared with OGD (P < 0.05; Fig. 6C, D). It could also be observed that increased Bcl2l1 expression led to a decrease in the OGD-induced levels of cleaved Caspase-3 induced by OGD (P < 0.05; Fig. 6E, F). In conclusion, the experimental results suggest that Bcl2l1 is directly linked to the apoptosis levels, and the anti-apoptotic effect of GSK-126 may greatly depend on the regulation of Bcl2l1 expression.

Bcl2l1 overexpression alleviates OGD-induced damage. A The expression of Bcl2l1 after Bcl2l1 overexpression was detected by western blots. B Gray intensity analysis of western blots results. The results were normalized by β-actin. C OGD-induced apoptosis after Bcl2l1 overexpression determined by flow cytometry. D Analysis of apoptosis ratio. E The protein levels of Bcl2l1 and cleaved Caspase-3 were detected by western blots. F Gray intensity analysis of western blots results. The results were normalized by β-actin. *P < 0.05, **P < 0.01. N = 3
Discussion
Stroke is one of the most important fatal diseases in the world, and it can seriously affect human health; stroke is associated with a rapid onset and serious sequelae. Neuronal apoptosis is the leading cause of sequelae (death and disability) in ischemic stroke patients, and its mechanism is very complicated. Currently, the known causes of neuronal apoptosis after ischemia include oxidative stress [21], excitotoxicity [22], and overreactive inflammation [23]. After local ischemia in the brain, mitochondrial oxidative phosphorylation was inhibited or reduced, leading to an accumulation of superoxide radicals and ROS with decreased ATP [24]. During ischemic/hypoxic injury, the production of ROS overwhelms the antioxidant capacity. ROS can damage the plasma membrane and break DNA strands [25, 26]. Oxidative stress is often accompanied by reverse activation of the Na+/Ca2+ exchanger [27]. Once the ionic gradient was disrupted, neuronal depolarization was exacerbated and large amounts of excitatory neurotransmitters, especially glutamate, were released [28]. The binding of glutamate to NMDARs activates calcium channels, which leads to intracellular Ca2+ overload [29, 30]. The conjunction of oxidative stress and calcium overload leads to increased permeability of the inner mitochondrial membrane and loss of mitochondrial membrane potential, resulting in a caspase cascade reaction [31, 32]. In addition, ischemic brain injury leads to a massive release of inflammatory factors and disruption of the blood-brain barrier. These inflammatory factors promote thrombosis and remain elevated in the late stages of stroke [33]. After cerebral ischemia, NADPH oxidase and xanthine oxidase production were increased, leading to accumulation of ROS [34]. Additionally, glutamate binding to extra-synaptic NMDARs exacerbates neuronal damage by activating the phosphatase and tensin homolog (PTEN) and death-associated protein kinase 1 (DAPK1) signaling pathways [1]. During ischemia, oxidative stress and excessive Ca2+ levels promote the release of inflammatory factors. Once those factors are recognized by the pattern recognition receptors (PRRs), downstream signaling pathways, such as the nuclear factor (NF)-κB, mitogen activated protein kinase (MAPK), interferon regulatory factors (IRF), or the inflammasome signaling pathways, were activated [33]. The expression levels of many genes are altered during these processes and ultimately determine the survival of brain cells after ischemia. Therefore, studying how to regulate the expression of these genes will be helpful for exploring the antiapoptotic mechanism after stroke.
Histone methylation, especially H3K27me3, is involved in a variety of biological processes due to its role in regulating gene expression. H3K27me3 silences gene expression by promoting DNA methylation or heterochromatin compaction [35, 36]. And, H3K27me3 and EZH2 have been shown to be involved in the pathogenesis of ischemic stroke [37]. Yang et al. reported that sevoflurane could mitigate the apoptosis of hippocampal neurons after cerebral ischemia by affecting EZH2 [38]. The enrichment of H3K27me3 in the Bax promoter was reduced in cortical neurons with insufficient oxygen glucose supply, and when its expression increased, Bax bound to p53 and promoted neuronal apoptosis [39, 40]. In addition, H3K27me3 represses Reelin expression after cerebral ischemia, which ultimately leads to neuronal apoptosis [41]. However, the role of H3K27me3 in regulating the expression of antiapoptotic genes was still unclear. Due to the specific inhibitory effect of GSK-126 on EZH2 activity [14, 42], it is possible to study the role of H3K27me3 on apoptosis after ischemia using GSK-126 (Fig. 1E, F). We previously demonstrated that GSK-126 could effectively reverse the apoptosis of CA1 neurons after global cerebral ischemia by reversing the levels of H3K27me3 [13]. Previous research also revealed that decreased levels of H3K27me3 might increase the expression levels of genes that were related to negative regulation of the execution phase of apoptosis. Therefore, in the present study, we first determined that GSK-126 alleviated neuronal apoptosis after local cerebral ischemia by decreasing H3K27me3 in the brains of MCAO rats (Fig. 1), as well as alleviated apoptosis in three cell models including oxidative stress (Fig. 2), excitotoxicity (Fig. 3), and excessive inflammatory response (Fig. 4) models.
There is no doubt that H3K27me3 is associated with oxidative stress, excitotoxicity, and excessive inflammatory responses [43,44,45,46]. The question is whether H3K27me3 regulates the expression of antiapoptotic genes during these processes. In fact, it has been reported that the downregulation of apoptotic genes might be attributed to decreased H3K4me3 rather than increased H3K27me3 [47]. We also found that GSK-126 failed to regulate the expression of Bcl2 (Fig. S4D). However, we have previously reported that decreasing H3K27me3 may upregulate the expression levels of several genes that participate in the “negative regulation of the execution phase of apoptosis” [13], Bcl2l1 was one of these genes, which was confirmed by subsequent experiments (Fig. S4A). Bcl2l1 and Bcl2 are members of the Bcl-2 family that play antiapoptotic roles, but they are different from each other. Bcl2l1 closely controls the permeability of the outer mitochondrial membrane, thereby inhibiting the release of proapoptotic proteins, such as Cytc and AIF [48]. Moreover, Bcl2l1 is abundantly expressed in developing or adult neurons, and Bcl2l1 plays an important role in the survival of mouse cerebellar granule neuron progenitors [49, 50]. Although it had been suggested that Bcl2l1 can exert antiapoptotic effects in response to a variety of stresses [51, 52], it was unclear whether Bcl2l1 can protect neurons from apoptosis after cerebral ischemia. In this study, we found that the expression levels of Bcl2l1 were correlated with the enrichment levels of H3K27me3 in its promotor, but not with the levels of DNA methylation (Fig. 5). This seems contradictory since H3K27me3 was known to cause gene silence through DNA methylation, but DNA methylation was not the only way that H3K27me3 silenced gene expression. Margueron et al. reported that H3K27me3 may regulate gene expression either by altering chromatin structure (by modulating the DNA–histone interaction) or by contributing to the recruitment of further regulatory (DNA methylase, etc.) [53]. The presence of H3K27me3 enrichment indicates gene silencing [54]. Therefore, our data indicated that Bcl2l1 is regulated by H3K27me3, but its regulatory mechanism remained to be studied. The lower expression levels of Bcl2l1 play an insufficient role in anti-apoptotic mechanisms (Fig. 6). Once GSK-126 inhibited EZH2 activity and reversed H3K27me3 levels, Bcl2l1 expression levels rebounded. This alleviated the apoptosis of brain cells after MCAO. To this end, we believe that GSK-126 attenuates cell apoptosis during ischemic brain injury by modulating the EZH2-H3K27me3-Bcl2l1 axis (Fig. 7).

Schematic illustration of the molecular mechanism by which GSK-126 protects neuron from apoptosis. Oxidative stress, calcium overload and inflammatory responses were the main causes of ischemic stroke. The levels of H3K27me3 were increased after ischemic stroke via EZH2, and H3K27me3 enriched in the Bcl2l1 promoter region to repress its expression, which in turn led to neuronal apoptosis. In contrast, GSK-126 could suppress H3K27me3 levels by specifically inhibiting EZH2 activity. When the enrichment of H3K27me3 in the Bcl2l1 promoter region was reduced, Bcl2l1 expression rebounded to promote neuronal survival
Conclusion
In conclusion, we found that GSK-126 alleviated the apoptosis induced by local cerebral ischemia or related conditions in vivo and in vitro. Importantly, the anti-apoptotic effect of GSK-126 was mediated by modulating the EZH2-H3K27me3-Bcl2l1 axis. These findings provide novel insights into the role of histone modification in ischemic stroke and also provide a new theoretical basis for the research and development of drugs in ischemic stroke.
Data Availability
All data used to support the results of this study are available from the corresponding authors upon request.
References
Qin C, Yang S, Chu YH, Zhang H, Pang XW, Chen L, Zhou LQ, Chen M et al (2022) Signaling pathways involved in ischemic stroke: molecular mechanisms and therapeutic interventions. Signal transduct tar 7(1):215. https://doi.org/10.1038/s41392-022-01064-1
Mishra A, Malik R, Hachiya T, Jürgenson T, Namba S, Posner DC, Kamanu FK, Koido M et al (2022) Stroke genetics informs drug discovery and risk prediction across ancestries. Nature 611(7934):115–123. https://doi.org/10.1038/s41586-022-05165-3
Renú A, Millán M, San Román L, Blasco J, Martí-Fàbregas J, Terceño M, Amaro S, Serena J et al (2022) Effect of Intra-arterial alteplase vs placebo following successful thrombectomy on functional outcomes in patients with large vessel occlusion acute ischemic stroke: the CHOICE randomized clinical trial. Jama-J Am Med Assoc 327(9):826–835. https://doi.org/10.1001/jama.2022.1645
He W, Mei Q, Li J, Zhai Y, Chen Y, Wang R, Lu E, Zhang XY et al (2021) Preferential targeting cerebral ischemic lesions with cancer cell-inspired nanovehicle for ischemic stroke treatment. Nano Lett 21(7):3033–3043. https://doi.org/10.1021/acs.nanolett.1c00231
Cheng X, Ferrell JE Jr (2018) Apoptosis propagates through the cytoplasm as trigger waves. Science 361(6402):607–612. https://doi.org/10.1126/science.aah4065
Sorice M (2022) Crosstalk of autophagy and apoptosis. Cells 11(9). https://doi.org/10.3390/cells11091479
Basenko EY, Sasaki T, Ji L, Prybol CJ, Burckhardt RM, Schmitz RJ, Lewis ZA (2015) Genome-wide redistribution of H3K27me3 is linked to genotoxic stress and defective growth. P natl acad sci USA 112(46):E6339–E6348. https://doi.org/10.1073/pnas.1511377112
Sievers P, Sill M, Schrimpf D, Stichel D, Reuss DE, Sturm D, Hench J, Frank S et al (2021) A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro-Oncology 23(1):34–43. https://doi.org/10.1093/neuonc/noaa251
Kraft K, Yost KE, Murphy SE, Magg A, Long Y, Corces MR, Granja JM, Wittler L et al (2022) Polycomb-mediated genome architecture enables long-range spreading of H3K27 methylation. Proc Natl Acad Sci USA 119(22):e2201883119. https://doi.org/10.1073/pnas.2201883119
Cerase A, Smeets D, Tang YA, Gdula M, Kraus F, Spivakov M, Moindrot B, Leleu M et al (2014) Spatial separation of Xist RNA and polycomb proteins revealed by superresolution microscopy. P Natl Acad Sci USA 111(6):2235–2240. https://doi.org/10.1073/pnas.1312951111
Jadhav U, Manieri E, Nalapareddy K, Madha S, Chakrabarti S, Wucherpfennig K, Barefoot M, Shivdasani RA (2020) Replicational dilution of H3K27me3 in mammalian cells and the role of poised promoters. Mol Cell 78(1):141–151.e145. https://doi.org/10.1016/j.molcel.2020.01.017
Romero OA, Vilarrubi A, Alburquerque-Bejar JJ, Gomez A, Andrades A, Trastulli D, Pros E, Setien F et al (2021) SMARCA4 deficient tumours are vulnerable to KDM6A/UTX and KDM6B/JMJD3 blockade. Nat Commun 12(1):4319. https://doi.org/10.1038/s41467-021-24618-3
Wang Z, Su Y, Zhang L, Lan T, Li L, Qi S (2022) GSK-126 protects CA1 neurons from H3K27me3-mediated apoptosis in cerebral ischemia. Mol Neurobiol 59(4):2552–2562. https://doi.org/10.1007/s12035-021-02677-3
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP et al (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492(7427):108–112. https://doi.org/10.1038/nature11606
Li Y, Gan Y, Liu J, Li J, Zhou Z, Tian R, Sun R, Liu J et al (2022) Downregulation of MEIS1 mediated by ELFN1-AS1/EZH2/DNMT3a axis promotes tumorigenesis and oxaliplatin resistance in colorectal cancer. Signal Transduct Target Ther 7(1):87. https://doi.org/10.1038/s41392-022-00902-6
Wu H, Xue D, Chen G, Han Z, Huang L, Zhu C, Wang X, Jin H et al (2014) The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 10(10):1712–1725. https://doi.org/10.4161/auto.29568
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281(5381):1322–1326. https://doi.org/10.1126/science.281.5381.1322
Qi SH, Liu Y, Hao LY, Guan QH, Gu YH, Zhang J, Yan H, Wang M et al (2010) Neuroprotection of ethanol against ischemia/reperfusion-induced brain injury through decreasing c-Jun N-terminal kinase 3 (JNK3) activation by enhancing GABA release. Neuroscience 167(4):1125–1137. https://doi.org/10.1016/j.neuroscience.2010.02.018
Nalamolu KR, Chelluboina B, Fornal CA, Challa SR, Pinson DM, Wang DZ, Klopfenstein JD, Veeravalli KK (2021) Stem cell treatment improves post stroke neurological outcomes: a comparative study in male and female rats. Stroke Vasc Neurol 6(4):519–527. https://doi.org/10.1136/svn-2020-000834
Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M (2001) Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke 32(11):2682–2688. https://doi.org/10.1161/hs1101.098367
Ren ZL, Wang CD, Wang T, Ding H, Zhou M, Yang N, Liu YY, Chan P (2019) Ganoderma lucidum extract ameliorates MPTP-induced parkinsonism and protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis. Acta Pharmacol Sin 40(4):441–450. https://doi.org/10.1038/s41401-018-0077-8
Tian Q, Liu S, Han SM, Zhang W, Qin XY, Chen JH, Liu CL, Guo YJ et al (2023) The mechanism and relevant mediators associated with neuronal apoptosis and potential therapeutic targets in subarachnoid hemorrhage. Neural Regen Res 18(2):244–252. https://doi.org/10.4103/1673-5374.346542
Chen S, Peng J, Sherchan P, Ma Y, Xiang S, Yan F, Zhao H, Jiang Y et al (2020) TREM2 activation attenuates neuroinflammation and neuronal apoptosis via PI3K/Akt pathway after intracerebral hemorrhage in mice. J Neuroinflammation 17(1):168. https://doi.org/10.1186/s12974-020-01853-x
Huang G, Zang J, He L, Zhu H, Huang J, Yuan Z, Chen T, Xu A (2022) Bioactive nanoenzyme reverses oxidative damage and endoplasmic reticulum stress in neurons under ischemic stroke. ACS Nano 16(1):431–452. https://doi.org/10.1021/acsnano.1c07205
Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, Cui J, Bai L et al (2017) CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun 8(1):202. https://doi.org/10.1038/s41467-017-00227-x
Wu R, Högberg J, Adner M, Ramos-Ramírez P, Stenius U, Zheng H (2020) Crystalline silica particles cause rapid NLRP3-dependent mitochondrial depolarization and DNA damage in airway epithelial cells. Part Fibre Toxicol 17(1):39. https://doi.org/10.1186/s12989-020-00370-2
Bernardi P, Rasola A, Forte M, Lippe G (2015) The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev 95(4):1111–1155. https://doi.org/10.1152/physrev.00001.2015
Chamorro Á, Dirnagl U, Urra X, Planas AM (2016) Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol 15(8):869–881. https://doi.org/10.1016/S1474-4422(16)00114-9
Tuo QZ, Zhang ST, Lei P (2022) Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev 42(1):259–305. https://doi.org/10.1002/med.21817
Gielen M, Siegler Retchless B, Mony L, Johnson JW, Paoletti P (2009) Mechanism of differential control of NMDA receptor activity by NR2 subunits. Nature 459(7247):703–707. https://doi.org/10.1038/nature07993
Ham PB, Raju R (2017) Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog Neurobiol 157:92–116. https://doi.org/10.1016/j.pneurobio.2016.06.006
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11(3):577–590. https://doi.org/10.1016/s1097-2765(03)00050-9
Simats A, Liesz A (2022) Systemic inflammation after stroke: implications for post-stroke comorbidities. Embo mol med 14(9):e16269. https://doi.org/10.15252/emmm.202216269
Jelinek M, Jurajda M, Duris K (2021) Oxidative stress in the brain: basic concepts and treatment strategies in stroke. Antioxidants 10(12). https://doi.org/10.3390/antiox10121886
Zhang H, Liu Y, Xie Y, Zhu Y, Liu J, Lu F (2022) H3K27me3 shapes DNA methylome by inhibiting UHRF1-mediated H3 ubiquitination. Sci China Life Sci 65(9):1685–1700. https://doi.org/10.1007/s11427-022-2155-0
Eeftens JM, Kapoor M, Michieletto D, Brangwynne CP (2021) Polycomb condensates can promote epigenetic marks but are not required for sustained chromatin compaction. Nat Commun 12(1):5888. https://doi.org/10.1038/s41467-021-26147-5
Zhao H, Li G, Wang R, Tao Z, Zhang S, Li F, Han Z, Li L et al (2019) MiR-424 prevents astrogliosis after cerebral ischemia/reperfusion in elderly mice by enhancing repressive H3K27me3 via NFIA/DNMT1 signaling. FEBS J 286(24):4926–4936. https://doi.org/10.1111/febs.15029
Yang L, Chen H, Guan L, Xu Y (2022) Sevoflurane offers neuroprotection in a cerebral ischemia/reperfusion injury rat model through the E2F1/EZH2/TIMP2 regulatory axis. Mol Neurobiol 59(4):2219–2231. https://doi.org/10.1007/s12035-021-02602-8
Estarás C, Akizu N, García A, Beltrán S, de la Cruz X, Martínez-Balbás MA (2012) Genome-wide analysis reveals that Smad3 and JMJD3 HDM co-activate the neural developmental program. Development 139(15):2681–2691. https://doi.org/10.1242/dev.078345
Nowoslawski L, Klocke BJ, Roth KA (2005) Molecular regulation of acute ethanol-induced neuron apoptosis. J Neuropathol Exp Neurol 64(6):490–497. https://doi.org/10.1093/jnen/64.6.490
Zhu X, Li J, You D, Xiao Y, Huang Z, Yu W (2022) Neuroprotective effect of E3 ubiquitin ligase RNF8 against ischemic stroke via HDAC2 stability reduction and reelin-dependent GSK3β inhibition. Mol Neurobiol 59(8):4776–4790. https://doi.org/10.1007/s12035-022-02880-w
Duan R, Du W, Guo W (2020) EZH2: a novel target for cancer treatment. J Hematol Oncol 13(1):104. https://doi.org/10.1186/s13045-020-00937-8
Luo Y, Fang Y, Kang R, Lenahan C, Gamdzyk M, Zhang Z, Okada T, Tang J et al (2020) Inhibition of EZH2 (enhancer of zeste homolog 2) attenuates neuroinflammation via H3k27me3/SOCS3/TRAF6/NF-κB (trimethylation of histone 3 lysine 27/suppressor of cytokine signaling 3/tumor necrosis factor receptor family 6/nuclear Factor-κB) in a rat model of subarachnoid hemorrhage. Stroke 51(11):3320–3331. https://doi.org/10.1161/STROKEAHA.120.029951
Liu H, Chen Z, Weng X, Chen H, Du Y, Diao C, Liu X, Wang L (2020) Enhancer of zeste homolog 2 modulates oxidative stress-mediated pyroptosis in vitro and in a mouse kidney ischemia-reperfusion injury model. FASEB J 34(1):835–852. https://doi.org/10.1096/fj.201901816R
Wan SS, Pan YM, Yang WJ, Rao ZQ, Yang YN (2020) Inhibition of EZH2 alleviates angiogenesis in a model of corneal neovascularization by blocking FoxO3a-mediated oxidative stress. FASEB J 34(8):10168–10181. https://doi.org/10.1096/fj.201902814RRR
Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M (2003) A key role for TRPM7 channels in anoxic neuronal death. Cell 115(7):863–877. https://doi.org/10.1016/s0092-8674(03)01017-1
Jiang W, Chen L, Zheng S (2021) Global reprogramming of apoptosis-related genes during brain development. Cells 10(11). https://doi.org/10.3390/cells10112901
Klanova M, Klener P (2020) BCL-2 proteins in pathogenesis and therapy of B-cell non-Hodgkin lymphomas. Cancers (Basel) 12(4). https://doi.org/10.3390/cancers12040938
Park HA, Licznerski P, Alavian KN, Shanabrough M, Jonas EA (2015) Bcl-xL is necessary for neurite outgrowth in hippocampal neurons. Antioxid Redox Signal 22(2):93–108. https://doi.org/10.1089/ars.2013.5570
Veleta KA, Cleveland AH, Babcock BR, He YW, Hwang D, Sokolsky-Papkov M, Gershon TR (2021) Antiapoptotic Bcl-2 family proteins BCL-xL and MCL-1 integrate neural progenitor survival and proliferation during postnatal cerebellar neurogenesis. Cell Death Differ 28(5):1579–1592. https://doi.org/10.1038/s41418-020-00687-7
You GR, Chang JT, Li YL, Huang CW, Tsai YL, Fan KH, Kang CJ, Huang SF et al (2022) MYH9 facilitates cell invasion and radioresistance in head and neck cancer via modulation of cellular ROS levels by activating the MAPK-Nrf2-GCLC pathway. Cells 11(18). https://doi.org/10.3390/cells11182855
Kakinohana M, Marutani E, Tokuda K, Kida K, Kosugi S, Kasamatsu S, Magliocca A, Ikeda K et al (2019) Breathing hydrogen sulfide prevents delayed paraplegia in mice. Free radical bio med 131:243–250. https://doi.org/10.1016/j.freeradbiomed.2018.12.003
Margueron R, Reinberg D (2011) The Polycomb complex PRC2 and its mark in life. Nature 469(7330):343–349. https://doi.org/10.1038/nature09784
Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I et al (2007) High-resolution profiling of histone methylations in the human genome. Cell 129(4):823–837. https://doi.org/10.1016/j.cell.2007.05.009
Acknowledgements
We are grateful to Prof. Suhua Qi for her help in completing the animal model. We are grateful to American Journal Experts (AJE) for language editing of this manuscript. We thank the National Natural Sciences Foundation of China and the Science and Technology Innovation Project of Xuzhou for their financial support.
Funding
This work was supported by the National Natural Sciences Foundation of China (grant number 81801168) and the Science and Technology Innovation Project of Xuzhou (grant number KC22123).
Author information
Authors and Affiliations
Contributions
ZW and LL conceived and designed the experiments in this study. TZ, LZ, and LH performed the experiments. YL, LD, and ZW analyzed the data. TZ, LZ, and ZW drafted the manuscript. LD and LL contributed to editing the manuscript. All authors approved the final version of the manuscript. All authors did the critical revised of the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
All animal procedures were approved by the Ethics Committee of Xuzhou Medical University (date: 06, 2022/No. 202209S066). Animals were cared for in accordance with the Guidelines for the Care and Use of Laboratory Animals.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information

Supplemental Figure 1
TTC staining. The cerebral infract aera after MCAO in rats was detected by TTC staining. N=3. (PNG 3900 kb)

Supplemental Figure 2
The expression of H3K27me3 after GSK-126 treated for 24 h. (A) The expression of H3K27me3 and Histone H3 was detected by western blots. (B) Gray intensity analysis of western blots results. The results were normalized by Histone H3. *p < 0.05, ***p < 0.001. N=3. (PNG 393 kb)

Supplemental Figure 3
Western blots assay for Caspase-3 expression. (A) The expression of Caspase-3 after H2O2 stimulation. (B) The expression of Caspase-3 after glutamate stimulation. (C) The expression of Caspase-3 after LPS stimulation. (PNG 1029 kb)

Supplemental Figure 4
GSK-126 restored the levels of Bcl2l1 after ischemia. (A) The mRNA levels of Bcl2l1, Dffa, Fzd3, Nmnat1, and Pam16 in the MCAO model were determined by RT-PCR. (B) The expression of Cleaved Caspase-3, Bcl2l1, Bcl2, Bax, and H3K27me3 in the OGD model was detected by western blots. (C) Gray intensity analysis of western blots results. The results were normalized by β-actin. (D) The mRNA levels of Bcl2 in the OGD model were determined by RT-PCR. (E) The expression of Bcl2l1 in the oxidative stress model was detected by western blots. (F) The expression of Bcl2l1 in the excitotoxicity model was detected by western blots. (G) The expression of Bcl2l1 in the inflammation model was detected by western blots. *p < 0.05, **p < 0.01, ***p < 0.001. N=3. (PNG 1411 kb)

Supplemental Figure 5
Regional immunohistochemistry of Bcl2l1 expression levels in rat brains. (A) Representative Bcl2l1 expression levels in the frontal parietal cortex, hypothalamus, and striatum. (B) Immunohistochemical detection of Bcl2l1 expression in the hippocampus and SVZ. N=5. (PNG 45310 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Zhou, T., Zhang, L., He, L. et al. GSK-126 Attenuates Cell Apoptosis in Ischemic Brain Injury by Modulating the EZH2-H3K27me3-Bcl2l1 Axis. Mol Neurobiol 61, 3369–3383 (2024). https://doi.org/10.1007/s12035-023-03808-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03808-8