
Abstract
The overproduction of neurotoxic amyloid-β (Aβ) peptides in the brain is a hallmark of Alzheimer’s disease (AD). To determine the role of intracellular zinc ion (iZn2+) dysregulation in mediating Aβ-related neurotoxicity, this study aimed to investigate whether N, N, N′, N′‑tetrakis (2‑pyridylmethyl) ethylenediamine (TPEN), a Zn2+‑specific chelator, could attenuate Aβ25–35‑induced neurotoxicity and the underlying mechanism. We used the 3-(4, 5-dimethyl-thiazol-2-yl)-2, 5-diphenyltetrazolium bromide assay to measure the viability of primary hippocampal neurons. We also determined intracellular Zn2+ and Ca2+ concentrations, mitochondrial and lysosomal functions, and intracellular reactive oxygen species (ROS) content in hippocampal neurons using live-cell confocal imaging. We detected L-type voltage-gated calcium channel currents (L-ICa) in hippocampal neurons using the whole‑cell patch‑clamp technique. Furthermore, we measured the mRNA expression levels of proteins related to the iZn2+ buffer system (ZnT-3, MT-3) and voltage-gated calcium channels (Cav1.2, Cav1.3) in hippocampal neurons using RT-PCR. The results showed that TPEN attenuated Aβ25–35‑induced neuronal death, relieved the Aβ25–35‑induced increase in intracellular Zn2+ and Ca2+ concentrations; reversed the Aβ25–35‑induced increase in ROS content, the Aβ25–35‑induced increase in the L-ICa peak amplitude at different membrane potentials, the Aβ25–35‑induced the dysfunction of the mitochondria and lysosomes, and the Aβ25–35‑induced decrease in ZnT-3 and MT-3 mRNA expressions; and increased the Cav1.2 mRNA expression in the hippocampal neurons. These results suggest that TPEN, the Zn2+-specific chelator, attenuated Aβ25–35‑induced neuronal damage, correlating with the recovery of intracellular Zn2+ and modulation of abnormal Ca2+-related signaling pathways.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that leads to cognitive impairment accompanied by neuronal dysfunction or death in the elderly [1, 2]. AD patients show atrophy of the cerebral cortex and hippocampus in the brain, which leads to impairment of learning and the ability to form new memories [3]. Amyloid-β (Aβ) aggregates and accumulates in the brain to form amyloid plaques, which are believed to play an important role in the pathogenesis of AD. The neurotoxicity of Aβ can trigger a deleterious cascade of events that accelerate AD progression, including neuronal excitotoxicity damage, intracellular ion dyshomeostasis, oxidative stress, loss of synapses, and cell apoptosis [4,5,6].
Zinc ions (Zn2+) are essential trace elements in humans and other animals that participate in a variety of physiological processes, such as catalytic reactions of enzymes, intracellular signaling, metabolism, differentiation, cellular growth, and neurodevelopment [7]. However, excess free Zn2+ has been shown to be neurotoxic and can cause neuronal damage under some pathological conditions [8]. Furthermore, an increase in free Zn2+ concentration in the brain or Zn2+ dysregulation in neurons is related to the occurrence and development of AD [9, 10]. Several autopsy studies have shown Zn2+ increase in the hippocampus and amygdala of AD patients [11]. Excess Zn2+ can promote the formation of Aβ oligomers by disrupting the helical structure at the N-terminus and inducing β-sheet formation at the C-terminus, along with the formation of intra- and inter-chain salt bridges [12]. Other than exogenous Zn2+, the free Zn2+ stored in presynaptic vesicles of some glutamatergic neurons is released into the synaptic cleft along with neurotransmitters in response to synaptic activity, and this could also promote the aggregation of Aβ [13]. Therefore, maintaining Zn2+ homeostasis is a potential strategy for AD treatment.
Calcium ions (Ca2+), an important second messenger, are involved in many different neurophysiological functions, including the modulation of membrane excitability and mitochondrial function, synaptic transmission, neuronal intra- and paracellular signaling, reactive oxygen/nitrogen species (ROS/RNS) formation, and apoptosis/necrosis [14, 15]. Ca2+ can enter neurons from the extracellular space via membrane-embedded Ca2+-permeable channels, such as voltage-gated calcium channels (VGCCs), non-specific cation channels, N-methyl-d-aspartate receptors (NMDAR), and transient receptor potential channels (TRPCs) [16]. In neurons, intracellular Ca2+ (iCa2+) is mainly stored in the endoplasmic reticulum (ER) and mitochondria, and cytosolic Ca2+ is maintained at low nanomolar concentrations (50–300 nM) when neurons are at rest or having minimal activity [15, 17]. The loss of the ability to handle iCa2+ is one of the central features of aging neurons. The calcium hypothesis suggests that neuronal iCa2+ dysregulation plays an important role in AD [18]. In different experimental models of AD, Ca2+ accumulates in neurons, and iCa2+ concentration increases, which is toxic to neurons and triggers subsequent pathological processes that drive AD development [17, 19]. Further evidence suggests that Aβ can increase iCa2+ concentrations via interactions with NMDAR and VGCCs [20, 21]. Collectively, drugs or strategies to correct iCa2+ dysregulation are promising therapeutic approaches for AD treatment.
Mitochondria are organelles in buffering iCa2+ levels by taking up and releasing Ca2+[22]. At synaptic terminals, mitochondria can sequester excess iCa2+ and release Ca2+ to prolong their residual levels [23] and is involved in regulating neurotransmission and certain types of short-term synaptic plasticity [24]. Aged and dysfunctional mitochondria are not only defective in their abilities for ATP production and iCa2+ buffering, but can also release harmful ROS that induces neuronal apoptosis [25]. Mitochondrial dysfunction has been suggested as a hallmark of neurodegenerative diseases such as AD, Parkinson’s disease (PD), amyotrophic lateral sclerosis, and Huntington’s disease (HD) [26]. According to the mitochondrial cascade hypothesis of AD, mitochondrial dysfunction associated with elevated ROS levels accumulates for many years in susceptible patients and leads to synaptic defects, cognitive dysfunction, tau protein and Aβ aggregation, and neuroinflammation at certain time points [27, 28].
Collectively, Aβ-induced deleterious neuronal cascades might be involved in Zn2+ and iCa2+ dysregulation, mitochondrial dysfunction, and oxidative stress damage. Our previous studies have shown that N, N, N′, N′-tetrakis-(2‑pyridylmethyl)-ethylenediamine (TPEN), a Zn2+‑specific chelator with cell membrane permeability, could attenuate soluble Aβ‑induced neuronal damage by restoring intracellular Zn2+ (iZn2+) concentration and the electrophysiological properties of voltage-gated sodium channels (Nav) and voltage-gated potassium channels (Kv) [29]. However, the detailed mechanisms of increased iZn2+ or abnormal Zn2+ homeostasis involved in Aβ neurotoxicity need to be further studied. Therefore, in this study, we first established an in vitro model of AD by exposing primary hippocampal neurons to soluble Aβ25–35, and then detected the potential protective effect and possible mechanism of TPEN on Aβ25-35-induced neuronal damage through in vitro experiments. Herein, we aimed to elucidate the mechanisms of iZn2+ dysregulation in Aβ-induced neurotoxicity, enrich the pathophysiological theory of AD, and provide a basis for preventing and combating AD using Zn2+-specific chelators.
Materials and Methods
Chemicals and Animals
Dulbecco’s modified Eagle medium/F12 + Glutamax™-1, Neurobasal™-A medium, Glutamax™, fetal bovine serum, B27 supplements, antibiotics (penicillin and streptomycin), and 0.25% trypsin–EDTA were purchased from Gibco (Grand Island, NY, USA). Hank’s balanced salt solution (HBSS) was purchased from SolarBio (Beijing, China). DNase, cytosine β-d-arabinofuranoside (Ara-C), TPEN, poly-l-lysine, TEA-Cl, 4-AP, and tetrodotoxin (TTX) were purchased from Sigma-Aldrich (MO, USA). We obtained 3-(4, 5-dimethyl-thiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) from Amresco Inc. (Solon, OH, USA). Chemical constructs of Aβ peptides were synthesized by China Peptides Co., Ltd. (Shanghai, China) using the Aβ25–35 sequences of human APP. Aβ25–35 was dissolved in ddH2O to prepare a stock solution at a concentration of 100 mM. The concentration of Aβ25–35 used in this study was 20 μM. Neonatal Sprague–Dawley rats were purchased from SPF Biotechnology Co., Ltd. (Beijing, China). All experimental protocols were approved by the Ethics Committee of Nankai University.
Primary Hippocampal Neuronal Culture
Primary hippocampal neurons were prepared from early postnatal (postnatal day 0–1) Sprague–Dawley rats (either sex) using protocols described previously [29]. Briefly, the hippocampal tissues were dissected in an ice-cold dissociation buffer (HBSS), minced with forceps, and digested with 0.25% trypsin–EDTA (Invitrogen, UK) at 37 °C for 12 min. The digested tissues were triturated in Dulbecco’s modified Eagle medium/F12 (Gibco, UK) medium supplemented with 10% fetal bovine serum (Gibco, UK) and 50 μg/mL DNase (Sigma, USA) using a fire-polished plastic pipette. The cell suspension was centrifuged at 100 × g for 5 min, and the cells were re-suspended in the following plating medium: Dulbecco’s modified Eagle medium/F12 medium supplemented with 10% fetal bovine serum, 5 unit/mL penicillin, and 50 µg/mL streptomycin (all from Gibco, UK). The cell suspension was then filtered through a 74-µm screen mesh and was plated into poly l-lysine-coated 96-well plates or 35-mm culture dishes at a density of 120 cells/mm2 in the plating medium. After 4–6 h, the plating medium was replaced with a maintenance medium, which consisted of Neurobasal-A medium supplemented with 2% B27, 1% Glutamax, 50 μg/mL streptomycin, and 5 unit/mL penicillin (all from Gibco, UK). On day 3, Ara-C (Sigma, USA) was added at a final concentration of 1–5 μM to inhibit glial overgrowth. Cells were cultured in a humidified 5% CO2 incubator at 37 °C, with half of the maintenance medium replaced every 3 days. Neurons were cultured for 8–12 days in vitro before the experiments.
Experimental Design
The cultured hippocampal neurons were divided into three groups: control, Aβ25–35, and Aβ25–35 + TPEN. In the Aβ25–35 group, hippocampal neurons were treated with Aβ25–35 in the maintenance medium at a final concentration of 20 μM for 24 h to establish an in vitro model of AD. According to our previous experimental results [29], the most appropriate concentration of TPEN was 100 nM. In the Aβ25–35 + TPEN group, hippocampal neurons were treated with TPEN in the maintenance medium at a final concentration of 100 nmol L−1 for 30 min before and during the exposure to Aβ25–35.
Measurement of Cell Viability Using the MTT Assay
Cultured neuron viability was determined using the MTT assay. The cells were plated in 96-well plates. Following cell treatment, the maintenance medium was removed, and the cells were incubated with MTT solution (5 mg/mL MTT, prepared in HBSS) at 37 °C for 4 h. Next, the supernatant was discarded, and the formazan particles were dissolved in 100 µL of DMSO. Absorbance was measured at 570 nm using a BIORAD680 plate reader (Thermo, Waltham, MA, USA). The experiments were repeated at least three times, and the results were expressed as the percentage of viable hippocampal neurons compared to the control group.
Single Live–Cell Confocal Imaging
Neurons were seeded in 35-mm glass-bottomed confocal dishes (Nest, China). After the corresponding treatments, neurons were rinsed with HBSS. For Ca2+ or Zn2+ imaging, neurons were incubated in HBSS containing 2 µM Fluo4-AM (Beyotime Biotechnology, China), 2 µM FluoZin3-AM (Life Technologies, USA), and 0.02% (w/v) pluronic acid (Solarbio) at 37 °C in the dark for 30–60 min. In order to detect the function of the mitochondria and lysosomes, neurons were incubated in HBSS containing 200 nM MitoTracker Red CMXRos, 75 nM LysoTracker Red, or 1 µM Hoechst 33,342 (all from Beyotime Biotechnology) at 37 °C in the dark for 10–25 min. Cellular ROS generation was measured using DCFH-DA (Beyotime Biotechnology, Shanghai, China). Neurons were incubated in HBSS containing 10 µM DCFH-DA at 37 °C in the dark for 20 min. Neurons were then rinsed and kept in HBSS before images were captured. The fluorescence of Fluo4-AM (laser, 488 nm; emission, 512–520 nm), FluoZin3-AM (laser, 488 nm; emission, 516 nm), MitoTracker Red CMXRos (laser, 579 nm; emission, 599 nm), LysoTracker Red (laser, 577 nm; emission, 590 nm), DCFH-DA (laser, 488 nm; emission, 525 nm), and Hoechst 33,342 (laser, 350 nm; emission, 461 nm) was measured with a confocal laser-scanning microscopic system (TCSSP5, Leica, Germany). Image-Pro Plus software was used to analyze the fluorescence intensity of the images.
Whole‑Cell Patch-Clamp Recording of Cultured Hippocampal Neurons
Whole-cell patch-clamp recordings were performed to record L-ICa in primary hippocampal neurons at room temperature (22–25 °C). The recording micropipettes were pulled using a micropipette horizontal puller (P-97, Sutter Instrument Company, USA) from a borosilicate capillary glass. After being filled with the intracellular solution, the pipettes were chosen for the recordings, with the tip resistance ranging from 3 to 5 MΩ. Cultures were mounted on the stage of an inverted microscope (BX51W1, Olympus, Japan) and incubated with the extracellular solution. Hippocampal neurons were patched in whole-cell voltage clamp mode and recorded using a Multiclamp 700B amplifier (Molecular Devices, Foster City, CA, USA). Digidata 1440A (Molecular Devices) was used to digitize the data. pClamp 10.1 software (Molecular Devices) was used for data acquisition. Recordings were accepted only if the series resistance was less than 20 MΩ and if it did not vary by over 20% throughout the experiment.
To record L-ICa, the intracellular solution contained 110 mM CsCl, 30 mM TEA-Cl, 10 mM EGTA, 10 mM HEPES, and 3 mM Na2ATP·3H2O (pH 7.3 with CsOH), and the extracellular solution contained 110 mM choline chloride, 10 mM BaCl2, 1 mM MgCl2·6H2O, 10 mM HEPES, 10 mM glucose, 4 mM 4-AP, 20 mM TEA-Cl, and 0.001 mM TTX (pH 7.4 with Tris).
To eliminate the influence of neuronal size, the currents were normalized to the cell membrane capacitance and presented as current densities (pA/pF).
Quantitative Real‑Time PCR
Total RNA was extracted from cultured neurons using the TransZol Up Plus RNA Kit (TransGen Biotech, Beijing, China), according to the manufacturer’s instructions. The mRNA was reverse-transcribed into cDNA using HiScript® II Q RT SuperMix for qPCR (Vazyme, Nanjing, China). The primer pair sequences used are listed in Table 1. RT-PCR was performed on a RealPlex2 Mastercycler (Eppendorf, Stevenage, UK) using 2 × Universal SYBR Green Fast qPCR Mix (ABclonal, Wuhan, China). Cycling conditions were as follows: 95 °C for 3 min and 40 cycles of 95 °C for 5 s, then 60 °C for 30 s. At the end of the cycling process, melting curve analysis was performed on each sample to confirm amplification specificity. β-actin mRNA was used as an endogenous control. All samples were run in triplicate. RT-PCR was repeated at least three times for each gene. Fold changes in the target genes were calculated using the comparative (2−△△CT) method [30].
Statistical Analysis
The experimental results were analyzed using Clampft 10.3 (Molecular Devices), Origin 8.5, and SPSS (IBM SPSS Statistics for Windows, Version 20.0. Armonk, NY: IBM Corp.). All data were expressed as mean ± standard error of the mean (SEMs). Statistical significance was assessed by one-way analysis of variance, with significance at the level of p < 0.05 and extreme significance at the level of p < 0.01.
Results
TPEN Increased the Cell Viability of Hippocampal Neurons Exposed to Aβ25–35
The effect of TPEN on the viability of Aβ25–35-treated hippocampal neurons was examined using the MTT assay. As shown in Fig. 1, exposure to 20 μM Aβ25–35 for 24 h led to significant neuronal death in control neurons (Aβ25–35 treatment vs. control: p < 0.01). However, Aβ25–35-induced neuronal death was markedly attenuated by treatment with 100 nM TPEN, although neuronal death could not be completely prevented (Aβ + TPEN vs. Aβ, p < 0.01; Aβ + TPEN vs. control, p < 0.01).

Effects of TPEN on the viability of the hippocampal neurons treated with Aβ25–35. The data are presented as means ± SEMs; **p < 0.01 versus the control group; ##p < 0.01 versus the Aβ group; n = 20. Aβ, amyloid-β
TPEN inhibited Aβ25–35-induced iZn2+ and iCa2+ concentration increase in hippocampal neurons
To investigate the concentrations of iZn2+ and iCa2+ in primary hippocampal neurons, we used cell-permeant Zn2+- and Ca2+-selective fluorescent indicators, that is, FluoZin-3 and Fluo-4 AM, respectively, to examine the iZn2+ and iCa2+ concentrations in individual hippocampal neurons. Live-cell confocal imaging showed that both FluoZin-3 and Fluo-4 AM fluorescence intensities were significantly increased after Aβ25-35 treatment, which indicated that both iZn2+ and iCa2+ concentrations were increased after being treated with Aβ25-35 (Figs. 2 and 3); however, TPEN treatment strongly suppressed the Aβ25–35-induced iZn2+ and iCa2+ concentration increases (Figs. 2 and 3), especially the iZn2+ concentration, to the point where there was no difference between the Aβ + TPEN and control groups (p > 0.05).

Effects of TPEN on the intracellular Zn2+ concentration of the hippocampal neurons treated with Aβ25–35. a Representative confocal images showing FluoZin-3 (green) staining under the different treatments. The scale bar is 50 μm. b Mean fluorescence intensity of FluoZin-3 in the different groups. The data shown in b were obtained from three independent experiments, each examining 15–20 neurons for each condition. The data are presented as means ± SEMs; **p < 0.01 versus the control group; ##p < 0.01 versus the Aβ group. Aβ, amyloid-β

Effects of TPEN on the intracellular Ca2+ concentration of the hippocampal neurons treated with Aβ25–35. a Representative confocal images showing Fluo-4 AM (green) staining under the different treatments. The scale bar is 50 μm. b Mean fluorescence intensity of Fluo-4 AM in the different groups. The data shown in b were obtained from three independent experiments, each examining 15–20 neurons for each condition. The data are presented as means ± SEMs; **p < 0.01 versus the control group; ##p < 0.01 versus the Aβ group. Aβ, amyloid-β
TPEN Inhibited Aβ25–35-Induced Increase of L-ICa Density in Hippocampal Neurons
L-type voltage-gated calcium channel currents (L-ICa) in primary hippocampal neurons were examined using whole-cell patch-clamp recordings. To record L-ICa, hippocampal neuron potentials were held at − 50 mV, and current traces were evoked using a 250-ms constant depolarizing pulse from − 60 to + 40 mV in increments of 10 mV (Fig. 4a). Consequently, Aβ25–35 significantly increased the maximum current density of L-ICa compared to the control (from − 17.40 ± 1.08 pA/pF to − 27.61 ± 1.75 pA/pF, p < 0.01; Fig. 4b). Furthermore, the L-ICa increased at different membrane potentials after exposure to Aβ25–35, which were visible from the current–voltage (I–V) curves (Fig. 4c), compared to that after exposure to the control (p < 0.05). However, TPEN treatment not only completely reversed the increase in the maximum L-ICa current density caused by Aβ25–35, but also prevented the Aβ25–35-induced downward shift of the I-V curves (Aβ + TPEN vs. Aβ, p < 0.05; Aβ + TPEN vs. control, p > 0.05; Fig. 4b, c).

Effects of TPEN on the amplitude properties of L-ICa in the hippocampal neurons treated with Aβ25–35. a Typical example of L-ICa traces obtained in the hippocampal neurons (left) and record protocol (right). b Maximum current density of L-ICa in the different treatments. c Current–voltage (I-V) curves of L-ICa in the different treatments. The data are presented as means ± SEMs; *p < 0.05 and **p < 0.01 versus the control group; #p < 0.05 and ##p < 0.01 versus the Aβ group; n = 8 for the control group; n = 12 for the Aβ group; n = 14 for the Aβ + TPEN group. Aβ, amyloid-β; L-ICa, L-type voltage-gated Ca2+ channel current
TPEN Reversed Aβ25–35-Induced Changes in the mRNA Expression of Proteins Related to the iZn2+ Buffer System and Cav in Hippocampal Neurons
We detected the mRNA expression levels of proteins related to the iZn2+ buffer system (ZnT-3, MT-3) and voltage-gated Ca2+ channels (Cav1.2, Cav1.3) in hippocampal neurons using RT-PCR. The results are shown in Fig. 5. After Aβ25–35 treatment, the mRNA levels of ZnT-3 and MT-3 were significantly decreased (Aβ vs. control, p < 0.05), but the mRNA level of Cav1.2 was significantly increased (Aβ vs. control, p < 0.05); however, TPEN treatment completely reversed these Aβ25-35‑induced changes (Aβ + TPEN vs. Aβ, p < 0.05; Aβ + TPEN vs. control, p > 0.05), whereas there was no difference in the mRNA level of Cav1.3 among all groups (p > 0.05).

Effects of TPEN on the mRNA expression of ZnT-3, MT-3, Cav1.2, and Cav1.3 in the hippocampal neurons treated with Aβ25–35. a Quantification of ZnT-3 mRNA levels. b Quantification of MT-3 mRNA levels. c Quantification of Cav1.2 mRNA levels. d Quantification of Cav1.3 mRNA levels. β-actin was used as an internal control. The data are presented as means ± SEMs; *p < 0.05 and **p < 0.01 versus the control group; #p < 0.05 and ##p < 0.01 versus the Aβ group; n = 5 or 6; Aβ, amyloid-β
TPEN Inhibited Aβ25–35-Induced Mitochondrial and Lysosomal Dysfunctions in Hippocampal Neurons
We performed single live-cell confocal imaging to examine mitochondrial and lysosomal functions using MitoTracker Red CMXRos and LysoTracker Red. As shown in Figs. 6 and 7, Aβ25–35 treatment noticeably reduced both MitoTracker Red CMXRos and LysoTracker Red fluorescence intensities, suggesting both mitochondrial and lysosomal dysfunctions. However, TPEN treatment completely reversed Aβ25–35-induced effects (Aβ + TPEN vs. Aβ, p < 0.01; Aβ + TPEN vs. control, p > 0.05).

Effects of TPEN on the mitochondrial membrane potential of the hippocampal neurons treated with Aβ25–35. a Representative confocal images showing MitoTracker Red CMXRos staining under the different treatments. The scale bar is 10 μm. b Mean fluorescence intensity of MitoTracker Red CMXRos in the different groups. The data shown in b were obtained from three independent experiments, each examining 15–20 neurons for each condition. The data are presented as means ± SEMs; **p < 0.01 versus the control group; ##p < 0.01 versus the Aβ group. Aβ, amyloid-β

Effects of TPEN on the lysosomal function of the hippocampal neurons treated with Aβ25–35. a Representative confocal images showing LysoTracker Red staining under the different treatments. The scale bar is 25 μm. b Mean fluorescence intensity of LysoTracker Red in the different groups. The data shown in b were obtained from three independent experiments, each examining 15–20 neurons for each condition. The data are presented as means ± SEMs; **p < 0.01 versus the control group; ##p < 0.01 versus the Aβ group. Aβ, amyloid-β
TPEN Inhibited Aβ25–35-Induced Cellular ROS Increase in Hippocampal Neurons
The levels of ROS in hippocampal neurons were detected using a DCFH-DA fluorescent probe. Figure 8 shows that Aβ25–35 treatment increased the fluorescence intensity of DCFH-DA, which was abolished by TPEN treatment. These results indicate that TPEN inhibited the Aβ25–35-induced increase in cellular ROS in hippocampal neurons.

Effects of TPEN on the level of ROS in the hippocampal neurons treated with Aβ25–35. a Representative confocal images showing DCFH (green) staining under the different treatments. The scale bar is 25 μm. b Mean fluorescence intensity of DCFH (green) in the different groups. The data shown in b were obtained from three independent experiments, each examining 15–20 neurons for each condition. The data are presented as means ± SEMs; **p < 0.01 versus the control group; ##p < 0.01 versus the Aβ group. Aβ, amyloid-β
Discussion
This study showed that TPEN attenuated Aβ25–35-induced neuronal death, alleviated Aβ25–35-induced increase in iZn2+ and iCa2+ concentrations, reversed the Aβ25–35-induced increase in the peak amplitudes of L-ICa at different membrane potentials, inhibited Aβ25–35-induced mitochondrial and lysosomal dysfunctions, and suppressed the Aβ25–35-induced increase in intracellular ROS level. These results suggest that Aβ25–35-induced neuronal damage correlates with iZn2+ and iCa2+ dysregulation.
Under physiological conditions, the basal level of extracellular Zn2+ is in the low nanomolar (~ 10 nM) range in the hippocampus, which increases with age [31, 32]. Extracellular Zn2+ is mainly released from zincergic terminals during synaptic activity, which can interact with postsynaptic-related receptors and ion channels (NMDAR, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPAR), γ-aminobutyric acid (GABA) receptors, glycine receptors, and VGCCs), thus mediating the neuronal excitability and synaptic plasticity [33, 34]. The basal level of iZn2+ is estimated to be less than 1 nM (~ 100 pM), and the body is more sensitive to changes in iZn2+ than to that of extracellular Zn2+ [35]. Excess iZn2+ becomes neurotoxic, stimulating neurons to generate ROS and triggering oxidative stress, which eventually leads to cell death [36]. When the Aβ concentration in the extracellular compartment exceeds 100 pM, Aβ can rapidly bind to extracellular Zn2+ with high affinity through its histidine residues or carboxyl groups (His6, His13, His14, and AsP1), inducing the formation of Zn-Aβ complexes [37], and rapidly incorporated into neurons to form Zn2+-permeable ion channels [38]. Moreover, free Zn2+ can be released from Zn-Aβ oligomers, resulting in increased intracellular Zn2+ and Aβ concentrations and inducing neuronal death and cognitive decline [39]. Since there is an age-related increase in extracellular Zn2+ concentration, Aβ-induced iZn2+ neurotoxicity accelerates with aging [38]. In addition, in vivo experiments showed that extracellular Zn2+ is essential for Aβ-induced cognitive decline [37]. Furthermore, extracellular Zn2+ can also enter cells via Ca2+- and Zn2+-permeable VGCCs, NMDAR, and GluR2-deficient AMPA receptors [40]. Excess influx of extracellular Zn2+ is more likely to induce glutamate excitotoxicity than the influx of extracellular Ca2+ because the iZn2+ concentration (~ 100 pM) is much lower than the iCa2+ concentration (~ 300 nM) but has higher neurotoxicity [41,42,43]. Similar to our previous results [29], the present experiment also showed a decrease in neuronal viability and an increase in iZn2+ concentration after exposure to 20 μM Aβ25–35 for 24 h, whereas TPEN pretreatment markedly attenuated Aβ25–35-induced neuronal death and reversed the Aβ25–35-induced iZn2+ concentration increase. These results suggest that iZn2+ dysregulation mediates the neurotoxicity of Aβ25–35. Additionally, systemic and cellular Zn2+ homeostasis in mammals is usually regulated by the expression and action of iZn2+-buffering systems, including zinc transporters (ZnTs), metallothioneins (MTs), and Zrt, Irt-related proteins (ZIPs) [44]. Zinc transporter 3 (ZnT-3) is mainly expressed in the cerebral cortex and hippocampus and is responsible for the transport of cytoplasmic Zn2+ into presynaptic vesicles [45]. Similarly, metallothionein 3 (MT-3) is principally expressed in the zincergic neurons. As an intracellular Zn2+ reservoir, MT-3 is to bind cytoplasmic Zn2+ with high affinity and releases Zn2+ when needed [46]. Both ZnT-3 and MT-3 are helpful for iZn2+ homeostasis in the brain. However, multiple studies have shown that AD development is accompanied by decreased expression of ZnT-3 and MT-3 [47, 48]. Furthermore, lysosomal dysfunction was observed in MT-3 knockdown astrocytes, consistent with the reduced autophagy observed during AD [49]. Our RT-PCR results showed that Aβ25-35 significantly decreased ZnT-3 and MT-3 mRNA levels in hippocampal neurons, whereas TPEN completely reversed these changes. These results suggest that Aβ can induce iZn2+ increase by reducing the mRNA expression levels of MT-3 and ZnT-3 in hippocampal neurons. Furthermore, TPEN partially counteracted Aβ-induced neuronal damage by restoring iZn2+ homeostasis, suggesting that specific Zn2+ chelators have potential therapeutic value against AD.
Presenilin, Aβ oligomers, and hyperphosphorylated tau can cause iCa2+ dyshomeostasis [50], which is involved in numerous molecular mechanisms, including increased Ca2+ influx by hyperactivated NMDAR and VGCCs, and promoting Ca2+ release from the ER by increasing inositol 1,4,5-trisphosphate receptors (IP3R) and ryanodine receptors (RYR) activities [51, 52]. Excessive Ca2+ influx can trigger multiple deleterious events including increased ROS, metabolic derangement, excessive inflammation, excitotoxicity, mitochondrial dysfunction, and impaired autophagy, all prominent features of AD pathogenesis [16, 19, 53]. Therefore, restoration and maintenance of iCa2+ homeostasis are helpful for the prevention and treatment of AD. In the VGCCs family, L-type voltage-gated calcium channels (LTCCs) can regulate neuronal synaptic plasticity and neuronal excitability by gating Ca2+ influx in response to membrane depolarization [54]. There are four known LTCCs (Cav1.1—Cav1.4), but only two, CaV1.2 and CaV1.3, are highly expressed in the brain [55]. There is increasing evidence of enhanced LTCCs activity in neurodegenerative diseases, such as PD and AD [56, 57]. The enhanced LTCCs activities on neurons are linked to altered mitochondrial function, with detrimental effects most commonly attributed to mitochondrial Ca2+ overload [58]. Moreover, LTCC antagonists as potential drugs may prevent the development of dementia [59]. In this study, Aβ25–35 significantly increased the iCa2+ concentration in hippocampal neurons and upregulated the expression levels of Cav1.2 mRNA, which could contribute to the over-activation of LTCCs. Patch-clamp data showed that Aβ25–35 markedly increased the maximum current density of L-ICa and elevated L-ICa at different membrane potentials, resulting in increased Ca2+ influx and increased neuronal excitability. However, TPEN attenuated the Aβ25–35-induced iCa2+ concentration increase and reversed the changes in the electrophysiological properties of L-ICa as well as the Cav1.2 mRNA expression levels caused by Aβ25–35, which suggest that Aβ25–35-induced iZn2+ dysregulation mediated the increase in iCa2+ concentration in hippocampal neurons. We have previously shown that Aβ25–35-induced increase in iZn2+ concentration had a significant inhibitory effect on voltage-gated K+ currents in hippocampal neurons [29]. Notably, several studies have shown that blockade of K+ channels can increase Ca2+ influx and lead to iCa2+ accumulation [60, 61]. Therefore, an imbalance in the homeostasis of iZn2+ may lead to iCa2+ increase by inhibiting K+ channels. Moreover, high concentrations of Zn2+ in the cytoplasm can activate RYR and promote Ca2+ release from the ER into the cytoplasm, leading to an increase in iCa2+ concentration, which eventually causes neuronal excitotoxic damage [62]. This may also explain the simultaneous dysregulation of iZn2+ and iCa2+ under pathological conditions; however, the specific mechanism involved needs to be further studied.
ROS are primarily produced in the mitochondria as physiological byproducts of various cellular processes and are normally detoxified by enzymes and antioxidants to help maintain neuronal homeostasis [10]. However, mitochondrial dysfunction can lead to excessive ROS production, triggering a state of oxidative stress that can lead to neuronal death. Furthermore, ROS can also contribute to mitochondrial dysfunction either indirectly through the initiation of toxic signaling cascades that target mitochondria or through direct damage to mitochondrial DNA [63]. Notably, ROS generation is closely related to iZn2+. Mitochondrial Zn2+ overload inhibits the mitochondrial electron transport chain and reduces mitochondrial membrane potential, resulting in increased ROS production and decreased cellular ATP levels [64]. Excess iZn2+ also activates other intracellular generators of injurious oxidants, such as lipoxygenases, NADPH oxidases, peroxisomes, and nitric oxide synthases, and promotes the production of neurotoxic peroxynitrite (ONOO −), leading to oxidative stress [65]. Interestingly, oxidative stress can promote MT-3 to release additional free Zn2+ into the cytoplasm, further exacerbating the increase in iZn2+ concentration [10]. In this study, single live-cell imaging showed that Aβ25–35 markedly impaired mitochondrial and lysosomal functions and increased intracellular ROS levels, whereas TPEN significantly inhibited the damages of mitochondrial and lysosomal functions caused by Aβ25-35, and reduced intracellular ROS levels. The above statements also showed that Aβ25–35 significantly increased both the iCa2+ concentration and intracellular ROS levels in hippocampal neurons, indicating an interlinkage between Ca2+ and ROS production. Indeed, accumulating evidence suggests that increased iCa2+ levels may lead to ROS accumulation because of Aβ induction [66, 67]. However, the interactions between Ca2+ and ROS signaling systems can be stimulatory or inhibitory, depending on the type of target proteins, ROS species, dose, and exposure time [68]. Therefore, it needs more evidences to clarify the interactions between Ca2+ and ROS signaling systems and the condition of causing interaction.
Excess iZn2+ is closely associated with lysosomal dysfunction. As acidic organelles containing hydrolase enzymes, lysosomes are indispensable for autophagy, which delivers unnecessary cellular materials to lysosomes for degradation [69]. Decreased autophagy is a distinguishing feature of AD. In APP/PS1-expressing Chinese hamster ovary cells, clioquinol, a Zn2+ ionophore, not only attenuated the accumulation of Aβ1-42, but also protected cells by overcoming autophagy arrest induced by chloroquine, suggesting that the accumulation of Zn2+ in lysosomes was related to Aβ aggregation [70]. Moreover, it has been suggested that cell death may be due to oxidative stress-induced accumulation of Zn2+ and Aβ within lysosomes, leading to lysosomal membrane permeabilization and the subsequent release of Zn2+, Aβ, and other toxic molecules into the cytosol [10, 71]. Taken together, these observations suggest that Aβ-induced iZn2+ increase can trigger oxidative stress by impairing mitochondrial and lysosomal functions in neurons, as well as various other pathways, ultimately leading to neuronal damage.
Conclusions
The present study demonstrated that Aβ25-35 could increase the concentration of iZn2+ by decreasing the expression levels of ZnT-3 and MT-3 mRNA in hippocampal neurons, eventually resulting in more serious neuronal death. Moreover, Aβ25-35-induced iZn2+ increase significantly upregulated the expression level of Cav1.2 mRNA in hippocampal neurons, leading to increased L-ICa activities, resulting in increased neuronal excitability, thereby mediating Aβ25-35-induced neuronal excitotoxic damage. Furthermore, the excessive iZn2+ induced by Aβ25-35 synergistically increased iCa2+ in hippocampal neurons, leading to the damage of mitochondrial and lysosomal functions, resulting in an increase in ROS levels in neurons, thus mediating the neuronal damage by Aβ25-35. These findings support the notion of a causal link between Aβ accumulation, iZn2+ and iCa2+ dyshomeostasis, and the dysregulation of crucial brain signaling molecules (Fig. 9). Whereas TPEN attenuated Aβ25-35-induced neuronal damage by recovering the concentrations of iZn2+ and iCa2+ in neurons, decreasing L-ICa activity and intracellular ROS levels, reducing oxidative stress, and restoring mitochondrial and lysosomal functions. Therefore, maintaining iZn2+ homeostasis may be an effective way to alleviate Aβ-induced neuronal damage in AD. TPEN, a specific-free Zn2+ chelator, has been shown to be neuroprotective by counteracting the neurotoxicity of Aβ and may represent a potential cell-targeted therapy in Zn2+-related diseases. However, our conclusions are somewhat limited due to the relatively simple and fixed assay conditions. To confirm these primary findings, more dose-dependent and in vivo experiments are required.

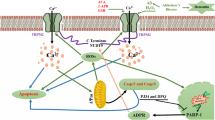
Synergistic contribution of Aβ, iZn2+ and iCa2+ dysregulation, oxidative stress, and excitotoxicity in AD. The pictogram summarizes the synergistic interaction between AD-related molecular changes, and the cation dysregulation triggered by altered glutamatergic neurotransmission. Accompanying the uptake of the Zn-Aβ complexes into neurons, excess free iZn2+ is released from the Zn-Aβ complexes (1). Extracellular Zn2+ and Ca2+ can enter cells via VGCCs, NMDAR, and GluR2-deficient AMPAR (2). Aβ adducts can also directly activate NMDAR and GluR2-deficient AMPAR, further promoting the glutamatergic overdrive, inducing iZn2+ and iCa2+ accumulation (3). LTCC overactivation promotes iCa2+ accumulation, increases neuronal excitability, and triggers mitochondrial Ca2+ overload (4). The diminished capacity of the iZn2+-buffering system (MT-3, ZnT-3) also contributes to Aβ-induced iZn2+ increase (5). The accumulation of excess iZn2+ is potent neurotoxic and acts synergistically with iCa2+, further promoting mitochondrial and lysosomal dysfunction, ROS generation, and the release of apoptotic molecules (6). Excessive iZn2+ rises may also trigger intraneuronal Aβ aggregation (7). Aβ adducts may further contribute to organelle impairment and ROS production (7). All these processes, set in motion at the level of synaptic spines, may be the primum movens of synaptic dysfunction and neuronal death
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code Availability
Not applicable.
References
Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44(1):181–193. https://doi.org/10.1016/j.neuron.2004.09.010
Swerdlow RH (2007) Pathogenesis of Alzheimer’s disease. Clin Interv Aging 2(3):347–359
Yamasaki T, Muranaka H, Kaseda Y, Mimori Y, Tobimatsu S (2012) Understanding the pathophysiology of Alzheimer’s disease and mild cognitive impairment: a mini review on fMRI and ERP studies. Neurol Res Int 2012:719056. https://doi.org/10.1155/2012/719056
Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y et al (2009) Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29(11):3453–3462. https://doi.org/10.1523/jneurosci.5215-08,2009
Praticò D (2008) Oxidative stress hypothesis in Alzheimer’s disease: a reappraisal. Trends Pharmacol Sci 29(12):609–615. https://doi.org/10.1016/j.tips.2008.09.001
Mucke L, Selkoe DJ (2012) Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med 2(7):a006338. https://doi.org/10.1101/cshperspect.a006338
Narayanan SE, Rehuman NA, Harilal S, Vincent A, Rajamma RG, Behl T, Uddin MS, Ashraf GM, Mathew B (2020) Molecular mechanism of zinc neurotoxicity in Alzheimer’s disease. Environ Sci Pollut Res Int 27(35):43542–43552. https://doi.org/10.1007/s11356-020-10477-w
Tyszka-Czochara M, Grzywacz A, Gdula-Argasińska J, Librowski T, Wiliński B, Opoka W (2014) The role of zinc in the pathogenesis and treatment of central nervous system (CNS) diseases. Implications of zinc homeostasis for proper CNS function. Acta Pol Pharm 71(3):369–377
Tamano H, Oneta N, Shioya A, Adlard PA, Bush AI, Takeda A (2019) In vivo synaptic activity-independent co-uptakes of amyloid β(1–42) and Zn(2+) into dentate granule cells in the normal brain. Sci Rep 9(1):6498. https://doi.org/10.1038/s41598-019-43012-0
McCord MC, Aizenman E (2014) The role of intracellular zinc release in aging, oxidative stress, and Alzheimer’s disease. Front Aging Neurosci 6:77. https://doi.org/10.3389/fnagi.2014.00077
Deibel MA, Ehmann WD, Markesbery WR (1996) Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: possible relation to oxidative stress. J Neurol Sci 143(1–2):137–142. https://doi.org/10.1016/s0022-510x(96)00203-1
Hane F, Leonenko Z (2014) Effect of metals on kinetic pathways of amyloid-β aggregation. Biomolecules 4(1):101–116. https://doi.org/10.3390/biom4010101
Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J (2009) A role for synaptic zinc in activity-dependent Abeta oligomer formation and accumulation at excitatory synapses. J Neurosci 29(13):4004–4015. https://doi.org/10.1523/jneurosci.5980-08.2009
Berridge MJ (1998) Neuronal calcium signaling. Neuron 21(1):13–26. https://doi.org/10.1016/s0896-6273(00)80510-3
Supnet C, Bezprozvanny I (2010) The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 47(2):183–189. https://doi.org/10.1016/j.ceca.2009.12.014
Popugaeva E, Chernyuk D, Bezprozvanny I (2020) Reversal of calcium dysregulation as potential approach for treating Alzheimer’s disease. Curr Alzheimer Res 17(4):344–354. https://doi.org/10.2174/1567205017666200528162046
Popugaeva E, Pchitskaya E, Bezprozvanny I (2018) Dysregulation of intracellular calcium signaling in Alzheimer’s disease. Antioxid Redox Signal 29(12):1176–1188. https://doi.org/10.1089/ars.2018.7506
Alzheimer’s Association Calcium Hypothesis Workgroup (2017)Calcium hypothesis of Alzheimer's disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer's Dement 13(2):178–182. https://doi.org/10.1016/j.jalz.2016.12.006
Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH et al (2019) Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat Commun 10(1):3885. https://doi.org/10.1038/s41467-019-11813-6
Foster TC, Kyritsopoulos C, Kumar A (2017) Central role for NMDA receptors in redox mediated impairment of synaptic function during aging and Alzheimer’s disease. Behav Brain Res 322(Pt B):223–232. https://doi.org/10.1016/j.bbr.2016.05.012
Anekonda TS, Quinn JF, Harris C, Frahler K, Wadsworth TL, Woltjer RL (2011) L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol Dis 41(1):62–70. https://doi.org/10.1016/j.nbd.2010.08.020
Cai Q, Jeong YY (2020) Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells 9(1):150(1-28). https://doi.org/10.3390/cells9010150
Tang Y, Zucker RS (1997) Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron 18(3):483–491. https://doi.org/10.1016/s0896-6273(00)81248-9
Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH (2008) Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132(1):137–148. https://doi.org/10.1016/j.cell.2007.11.024
Tönnies E, Trushina E (2017) Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J Alzheimer’s Dis JAD 57(4):1105–1121. https://doi.org/10.3233/jad-161088
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795. https://doi.org/10.1038/nature05292
Swerdlow RH, Burns JM, Khan SM (2010) The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimer’s Dis JAD 20(Suppl 2):S265-279. https://doi.org/10.3233/jad-2010-100339
Swerdlow RH, Burns JM (1842) Khan SM (2014) The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochem Biophys Acta 8:1219–1231. https://doi.org/10.1016/j.bbadis.2013.09.010
Chen WB, Wang YX, Wang HG, An D, Sun D, Li P, Zhang T, Lu WG et al (2021) TPEN attenuates amyloid-β(25–35)-induced neuronal damage with changes in the electrophysiological properties of voltage-gated sodium and potassium channels. Mol Brain 14(1):124. https://doi.org/10.1186/s13041-021-00837-z
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Tamano H, Nishio R, Shakushi Y, Sasaki M, Koike Y, Osawa M, Takeda A (2017) In vitro and in vivo physiology of low nanomolar concentrations of Zn2+ in artificial cerebrospinal fluid. Sci Rep 7:42897. https://doi.org/10.1038/srep42897
Frederickson CJ, Giblin LJ, Krezel A, McAdoo DJ, Mueller RN, Zeng Y, Balaji RV, Masalha R et al (2006) Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp Neurol 198(2):285–293. https://doi.org/10.1016/j.expneurol.2005.08.030
Takeda A, Nakamura M, Fujii H, Tamano H (2013) Synaptic Zn2+ homeostasis and its significance. Metallomics 5(5):417–423. https://doi.org/10.1039/c3mt20269k
Krall RF, Tzounopoulos T, Aizenman E (2021) The function and regulation of zinc in the brain. Neuroscience 457:235–258. https://doi.org/10.1016/j.neuroscience.2021.01.010
Colvin RA, Bush AI, Volitakis I, Fontaine CP, Thomas D, Kikuchi K, Holmes WR (2008) Insights into Zn2+ homeostasis in neurons from experimental and modeling studies. Am J Physiol Cell Physiol 294(3):C726-742. https://doi.org/10.1152/ajpcell.00541.2007
Shuttleworth CW, Weiss JH (2011) Zinc: new clues to diverse roles in brain ischemia. Trends Pharmacol Sci 32(8):480–486. https://doi.org/10.1016/j.tips.2011.04.001
Takeda A, Tamano H, Tempaku M, Sasaki M, Uematsu C, Sato S, Kanazawa H, Datki ZL et al (2017) Extracellular Zn2+ is essential for amyloid β1-42-induced cognitive decline in the normal brain and its rescue. J Neurosci 37(30):7253–7262. https://doi.org/10.1523/jneurosci.0954-17.2017
Takeda A, Koike Y, Osaw M, Tamano H (2018) Characteristic of extracellular Zn2+ influx in the middle-aged dentate gyrus and its involvement in attenuation of LTP. Mol Neurobiol 55(3):2185–2195. https://doi.org/10.1007/s12035-017-0472-z
Solomonov I, Korkotian E, Born B, Feldman Y, Bitler A, Rahimi F, Li H, Bitan G, Sagi I (2012) Zn2+-Aβ40 complexes form metastable quasi-spherical oligomers that are cytotoxic to cultured hippocampal neurons. J Biol Chem 287(24):20555–20564. https://doi.org/10.1074/jbc.M112.344036
Ji SG, Medvedeva YV, Wang HL, Yin HZ, Weiss JH (2019) Mitochondrial Zn2+ accumulation: a potential trigger of hippocampal ischemic injury. Neuroscientist 25(2):126–138. https://doi.org/10.1177/1073858418772548
Forostyak O, Forostyak S, Kortus S, Sykova E, Verkhratsky A, Dayanithi G (2016) Physiology of Ca2+ signalling in stem cells of different origins and differentiation stages. Cell Calcium 59(2–3):57–66. https://doi.org/10.1016/j.ceca.2016.02.001
Colbourne F, Grooms SY, Zukin RS, Buchan AM, Bennett MV (2003) Hypothermia rescues hippocampal CA1 neurons and attenuates down-regulation of the AMPA receptor GluR2 subunit after forebrain ischemia. Proc Natl Acad Sci USA 100(5):2906–2910. https://doi.org/10.1073/pnas.2628027100
Stork CJ, Li YV (2009) Rising zinc: a significant cause of ischemic neuronal death in the CA1 region of rat hippocampus. J Cereb Blood Flow Metab 29(8):1399–1408. https://doi.org/10.1038/jcbfm.2009.64
Chasapis CT, Loutsidou AC, Spiliopoulou CA, Stefanidou ME (2012) Zinc and human health: an update. Arch Toxicol 86(4):521–534. https://doi.org/10.1007/s00204-011-0775-1
Baltaci AK, Yuce K (2018) Zinc transporter proteins. Neurochem Res 43(3):517–530. https://doi.org/10.1007/s11064-017-2454-y
Baltaci AK, Yuce K, Mogulkoc R (2018) Zinc metabolism and metallothioneins. Biol Trace Elem Res 183(1):22–31. https://doi.org/10.1007/s12011-017-1119-7
Yu WH, Lukiw WJ, Bergeron C, Niznik HB, Fraser PE (2001) Metallothionein III is reduced in Alzheimer’s disease. Brain Res 894(1):37–45. https://doi.org/10.1016/s0006-8993(00)03196-6
Beyer N, Coulson DT, Heggarty S, Ravid R, Irvine GB, Hellemans J, Johnston JA (2009) ZnT3 mRNA levels are reduced in Alzheimer’s disease post-mortem brain. Mol Neurodegener 4:53. https://doi.org/10.1186/1750-1326-4-53
Lee SJ, Koh JY (2010) Roles of zinc and metallothionein-3 in oxidative stress-induced lysosomal dysfunction, cell death, and autophagy in neurons and astrocytes. Molecular brain 3(1):30. https://doi.org/10.1186/1756-6606-3-30
Calvo-Rodriguez M, Bacskai BJ (2021) Mitochondria and calcium in Alzheimer’s disease: from cell signaling to neuronal cell death. Trends Neurosci 44(2):136–151. https://doi.org/10.1016/j.tins.2020.10.004
Arispe N, Rojas E, Pollard HB (1993) Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci USA 90(2):567–571. https://doi.org/10.1073/pnas.90.2.567
Ferreiro E, Oliveira CR, Pereira C (2004) Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. J Neurosci Res 76(6):872–880. https://doi.org/10.1002/jnr.20135
Wang R, Reddy PH (2017) Role of glutamate and NMDA receptors in Alzheimer’s disease. J Alzheimer’s Dis JAD 57(4):1041–1048. https://doi.org/10.3233/jad-160763
Navakkode S, Liu C, Soong TW (2018) Altered function of neuronal L-type calcium channels in ageing and neuroinflammation: Implications in age-related synaptic dysfunction and cognitive decline. Ageing Res Rev 42:86–99. https://doi.org/10.1016/j.arr.2018.01.001
Berger SM, Bartsch D (2014) The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res 357(2):463–476. https://doi.org/10.1007/s00441-014-1936-3
Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S (2010) L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol 67(5):600–606. https://doi.org/10.1002/ana.21937
Webster NJ, Ramsden M, Boyle JP, Pearson HA, Peers C (2006) Amyloid peptides mediate hypoxic increase of L-type Ca2+ channels in central neurones. Neurobiol Aging 27(3):439–445. https://doi.org/10.1016/j.neurobiolaging.2005.02.002
Hotka M, Cagalinec M, Hilber K, Hool L, Boehm S, Kubista H (2020) L-type Ca(2+) channel-mediated Ca(2+) influx adjusts neuronal mitochondrial function to physiological and pathophysiological conditions. Sci Signal 13(618):eaaw6923(1-15). https://doi.org/10.1126/scisignal.aaw6923
Forette F, Seux ML, Staessen JA, Thijs L, Babarskiene MR, Babeanu S, Bossini A, Fagard R et al (2002) The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Arch Int Med 162(18):2046–2052. https://doi.org/10.1001/archinte.162.18.2046
Pannicke T, Iandiev I, Uckermann O, Biedermann B, Kutzera F, Wiedemann P, Wolburg H, Reichenbach A et al (2004) A potassium channel-linked mechanism of glial cell swelling in the postischemic retina. Mol Cell Neurosci 26(4):493–502. https://doi.org/10.1016/j.mcn.2004.04.005
Jaffe DB, Ross WN, Lisman JE, Lasser-Ross N, Miyakawa H, Johnston D (1994) A model for dendritic Ca2+ accumulation in hippocampal pyramidal neurons based on fluorescence imaging measurements. J Neurophysiol 71(3):1065–1077. https://doi.org/10.1152/jn.1994.71.3.1065
Schulien AJ, Justice JA, Di Maio R, Wills ZP, Shah NH, Aizenman E (2016) Zn2+-induced Ca2+ release via ryanodine receptors triggers calcineurin-dependent redistribution of cortical neuronal Kv2.1 K+ channels. J Physiol 594(10):2647–2659. https://doi.org/10.1113/jp272117
Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Ann Rev Genet 39:359–407. https://doi.org/10.1146/annurev.genet.39.110304.095751
Malaiyandi LM, Vergun O, Dineley KE, Reynolds IJ (2005) Direct visualization of mitochondrial zinc accumulation reveals uniporter-dependent and -independent transport mechanisms. J Neurochem 93(5):1242–1250. https://doi.org/10.1111/j.1471-4159.2005.03116.x
Sensi SL, Paoletti P, Bush AI, Sekler I (2009) Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci 10(11):780–791. https://doi.org/10.1038/nrn2734
Abramov AY, Canevari L, Duchen MR (2004) Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochem Biophys Acta 1742(1–3):81–87. https://doi.org/10.1016/j.bbamcr.2004.09.006
Angelova PR, Abramov AY (2014) Interaction of neurons and astrocytes underlies the mechanism of Aβ-induced neurotoxicity. Biochem Soc Trans 42(5):1286–1290. https://doi.org/10.1042/bst20140153
Görlach A, Bertram K, Hudecova S, Krizanova O (2015) Calcium and ROS: a mutual interplay. Redox Biol 6:260–271. https://doi.org/10.1016/j.redox.2015.08.010
Colacurcio DJ, Nixon RA (2016) Disorders of lysosomal acidification-the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev 32:75–88. https://doi.org/10.1016/j.arr.2016.05.004
Seo BR, Lee SJ, Cho KS, Yoon YH, Koh JY (2015) The zinc ionophore clioquinol reverses autophagy arrest in chloroquine-treated ARPE-19 cells and in APP/mutant presenilin-1-transfected Chinese hamster ovary cells. Neurobiol Aging 36(12):3228–3238. https://doi.org/10.1016/j.neurobiolaging.2015.09.006
Zheng L, Kågedal K, Dehvari N, Benedikz E, Cowburn R, Marcusson J, Terman A (2009) Oxidative stress induces macroautophagy of amyloid beta-protein and ensuing apoptosis. Free Radical Biol Med 46(3):422–429. https://doi.org/10.1016/j.freeradbiomed.2008.10.043
Funding
This work was supported by grants from the National Natural Science Foundation of China (31272317), the National Nature Science Youth Foundation of China (81801076), the Natural Science Foundation of Tianjin City (20JCYBJC01370), Natural Science Foundation of Tianjin City (15JCYBJC24500), the Tianjin Natural Science Youth Foundation (18JCQNJC11400), the Fundamental Research Funds for the Central Universities of Nankai University (BE123081), and the Natural Science Foundation of Hebei Province (C2020423055).
Author information
Authors and Affiliations
Contributions
W.B.C. designed and performed the experiments, analyzed the experimental data, prepared all figures, and wrote the manuscript. Y.Q.L. conceived the study, reviewed and revised the manuscript. The other authors help to perform the experiments, collect experimental data, review and revise the manuscript, and apply the funds. All the authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
All procedures were compliant with the approved protocol from the Animal Ethics Committee of Nankai University and the Chinese animal welfare act and the “Chinese code of practice and use of animals for scientific purposes.”
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, Wb., Wang, Yx., Wang, Hg. et al. Role of TPEN in Amyloid-β25–35-Induced Neuronal Damage Correlating with Recovery of Intracellular Zn2+ and Intracellular Ca2+ Overloading. Mol Neurobiol 60, 4232–4245 (2023). https://doi.org/10.1007/s12035-023-03322-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03322-x