
Abstract
Disruptions of brain energy and neurotransmitter metabolism are associated with several pathological conditions including neurodegenerative diseases such as Alzheimer’s disease. Transgenic rodent models, and in vitro preparations hereof, are often applied for studying pathological aspects of brain metabolism. However, despite the conserved cerebral development across mammalian species, distinct differences in cellular composition and structure may influence metabolism of the rodent and human brain. To address this, we investigated the metabolic function of acutely isolated brain slices and non-synaptic mitochondria obtained from the cerebral cortex of mice and neurosurgically resected neocortical tissue of humans. Utilizing dynamic isotope labeling with 13C-enriched metabolic substrates, we show that metabolism of glucose, acetate, β-hydroxybutyrate, and glutamine operates at lower rates in human cerebral cortical slices when compared to mouse slices. In contrast, human cerebral cortical slices display a higher capacity for converting exogenous glutamate into glutamine, which subsequently supports neuronal GABA synthesis, whereas mouse slices primarily convert glutamate into aspartate. In line with the reduced metabolic rate of the human brain slices, isolated non-synaptic mitochondria of the human cerebral cortex have a lower oxygen consumption rate when provided succinate as substrate. However, when provided pyruvate and malate, human mitochondria display a higher coupled respiration and lower proton leak, signifying a more efficient mitochondrial coupling compared to mouse mitochondria. This study reveals key differences between mouse and human brain metabolism concerning both neurons and astrocytes, which must be taken into account when applying in vitro rodent preparations as a model system of the human brain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The brain is a highly energy-demanding organ consuming up to 20% of the total body energy expenditure [1]. The majority of energy used by the brain is related to neurotransmission and restoration of ion gradients [1, 2], making a constant supply of energy essential to maintain cerebral function. Glucose is the primary energy substrate of the brain, but brain cells can utilize several other substrates for the production of energy [3]. As an example, the ketone body β-hydroxybutyrate (βHB), which is produced by the liver during fasting, can support up to 50–60% of the brain’s energy requirements [4]. Furthermore, amino acids, including neurotransmitters, also serve as cerebral metabolic fuels [5, 6]. Synthesis of glutamate, the principal excitatory neurotransmitter of the mammalian brain, is linked to cellular metabolism through the tricarboxylic acid (TCA) cycle intermediate α-ketoglutarate [7,8,9]. Astrocytes are the primary homeostatic glial cells of the brain and function in close metabolic collaboration with neurons. Astrocytic processes enclose most synapses and are responsible for uptake of the majority of the released neurotransmitter glutamate [10]. In the astrocytes, glutamate can either be utilized as a metabolic substrate in the TCA cycle or be converted into glutamine by the enzyme glutamine synthetase (GS) [11]. Glutamine is subsequently transferred from the astrocytes to neurons hereby aiding to replenish the neuronal glutamate pool [7, 12]. This shuttling of neuronal glutamate and astrocytic glutamine is collectively known as the glutamate-glutamine cycle and is crucial to sustain neurotransmission [13, 14].
Metabolic disturbances of the brain are linked to multiple neurological conditions, including neurodegenerative diseases. Alzheimer’s disease is strongly associated with cerebral hypometabolism of glucose [15], hampered neurotransmitter metabolism [16], and mitochondrial dysfunction [17]. Similarly, Huntington’s disease is also accompanied by cerebral alterations in glucose metabolism and impaired mitochondrial energetics [18, 19]. Transgenic rodent models are often applied in the attempt to explore potential metabolic mechanisms of neurodegeneration or effects of metabolic intervention [20, 21]. Animal models represent a valuable tool for studying human diseases and are commonly used for a large variety of experimental cerebral preparations including brain slices, cell cultures, and isolated mitochondria [22]. Although brain development appears to be conserved across mammalian species [23, 24], the human cerebral cortex displays unique features not found in rodents. Notable differences include the size and complexity of human astrocytes [25, 26] and the presence of specialized neuronal sub-types absent in the rodent brain [27]. Despite the generally conserved cellular architecture, comparative studies have revealed large differences between the transcriptome profiles of the human and mouse brain [23, 28, 29], which may profoundly affect brain signaling and metabolism. However, direct comparisons of functional metabolism of common in vitro preparations, with cellular and subcellular resolution, between the rodent and human brain remain scarce.
The overall aim of this study was therefore to assess and compare functional metabolic parameters of acutely isolated cerebral cortical tissue of mice and humans. To achieve this, we incubated acutely isolated slices prepared from cerebral cortical tissue of mice and humans, in the presence of energy substrates enriched with the stable carbon isotope 13C and assessed the intracellular isotopic enrichment of metabolites by gas chromatography-mass spectrometry (GC–MS) analysis. Moreover, we investigated the mitochondrial function of isolated non-synaptic mitochondria prepared from mouse and human cerebral cortical tissue. We report distinct differences in both energy and neurotransmitter metabolism between mouse and human brain slices, particularly of glucose and glutamate. Furthermore, we demonstrate substrate-dependent differences in mitochondrial function between isolated non-synaptic mitochondria from mouse and human brain tissue. Overall, our results provide new insights into functional brain metabolism, which will aid the use of rodents as metabolic model systems of the human brain in both health and disease.
Methods
Materials
The stable isotopes [U-13C]glucose (CLM-1396-PK, 99%), [U-13C]D-β-hydroxybutyrate (CLM-3853-PK, sodium salt, 97%), [U-13C]glutamate (CLM-1800-H-PK, 98%), and [U-13C]glutamine (CLM-1822-H-PK, 98%) were all from Cambridge Isotope Laboratories (Tewksbury, USA) and [1,2-13C]acetate (282,014, sodium salt, 99%) was from ISOTEC (St. Louis, MO, USA). Pyruvic acid (107360), succinic acid (S3674), malic acid (M1000), adenosine-5′-diphosphate (ADP, A5285), oligomycin A (75351), carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, C2920), antimycin A (A8674), and rotenone (R8875) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Percoll™ (17–0891-01) was from GE Healthcare, Uppsala, Sweden. All other reagents were of the purest grade available from regular commercial sources.
Preparation of Brain Tissue
Male NMRI mice (weight: 42.3 g ± 1.0 g) were purchased from Harlan (Horst, The Netherlands) and housed at the Department of Drug Design and Pharmacology, University of Copenhagen, in a specific pathogen-free, humidity, and temperature-controlled facility with 12-h light/dark cycle. Fluctuating levels of sex hormones can affect brain mitochondrial function, particularly in females [30]. To avoid this potential variation, only male mice were included in this study and sex-specific aspects of the work could therefore not be evaluated. The mice were acclimatized for 2 weeks before experiments and were single-housed in individually ventilated cages with free access to chow and water. The mice were used for experiments at 12–13 weeks of age (corresponding to early adulthood). Experiments were approved by the Danish National Ethics Committee and performed according to the European Convention (ETS 123 of 1986). Mice were euthanized without anesthesia by swift cervical dislocation and decapitated. The brain was quickly excised and transferred to ice-cold artificial cerebrospinal fluid (ACSF) containing in millimolar: NaCl, 128; NaHCO3, 25; D-glucose, 10; KCl, 3; CaCl2, 2; MgSO4, 1.2; KH2PO4, 0.4; pH = 7.4. The mouse cerebral cortex was isolated before slicing. Human neocortical tissue was obtained from patients undergoing hippocampal resections at Rigshospitalet (Copenhagen, Denmark). During surgery, patients were anesthetized by continuous infusion of the short-acting anesthetics propofol and remifentanil. The use of human neocortical tissue was approved by the local Ethics Committee in Copenhagen (H-2–2011-104) with written informed consent from all patients prior to surgery. In total, human neocortical tissue was obtained from 11 patients (7 female, 4 male) aged 24–59 years (39 years average). No significant correlation between patient age and metabolic outcomes was observed (Spearman correlation, p > 0.05 for all reported metabolic parameters, data not shown). The neocortical tissue of the temporal lobe was resected in order to facilitate access to the mesial temporal lobe, e.g., amygdala, hippocampus, and parahippocampal gyrus, involved in epileptic seizures. Histopathological examination of the neocortical tissue revealed no abnormal pathological features. Immediately after resection, the cortical tissue was transferred to ice-cold ACSF and transported on ice to the laboratory. To establish a baseline profile of the amino acid content, freshly isolated cerebral cortical tissue of both mouse and human origin was transferred to ice-cold 70% ethanol and tissue extracts were obtained as described for brain slices below.
Brain Slice Incubations
Incubation of acutely isolated brain slices was performed as previously reported for both mouse and human brain tissue [31, 32]. The tissue was sliced (350 µm) on a McIlwain tissue chopper (The Vibratome Company, O’Fallon, MO, USA). Two mouse slices or 1 human slice (grey matter only) were kept just below the surface of 10 mL 37 °C oxygenated (5% CO2/95% O2) ACSF and pre-incubated for 60 min. Subsequently, the media were exchanged for ACSF containing the 13C-enriched substrates: 5 mM [U-13C]glucose, 5 mM [1,2-13C]acetate, 5 mM [U-13C]β-hydroxybutyrate, 0.5 mM [U-13C]glutamate, or 1 mM [U-13C]glutamine and incubated for additional 60 min. All conditions, except [U-13C]glucose, were further supplemented with 5 mM unlabeled D-glucose. Incubations were terminated by quickly transferring the slices into ice-cold 70% ethanol. The tissue was subsequently sonicated and centrifuged (4000 g × 20 min) and the supernatant was removed and lyophilized before further analysis. Pellets were saved for protein determination (Pierce protein assay). Lactate released from brain slices to the medium during incubation was determined by an L-lactic acid kit (Boehringer Mannheim/R-Biopharm/Roche) according to the manufacturer’s instructions. Part of the metabolic data of human cerebral cortical slices (incubation with [1,2-13C]acetate) has previously been published as metabolic validation of the resected tissue [32], but has here been reanalyzed and compared to mouse brain slices.
Metabolic Mapping Using Gas Chromatography-Mass Spectrometry (GC–MS) Analysis
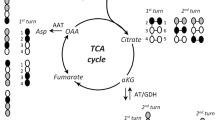
Brain slice extracts were analyzed by GC–MS for the determination of 13C enrichment of TCA cycle intermediates and connected amino acids as previously described in detail [33]. Briefly, slice extracts were reconstituted in water, acidified, extracted twice with ethanol, and derivatized using N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide. Samples were analyzed by GC (Agilent Technologies, 7820A, J&W GC column HP-5 MS) coupled to MS (Agilent Technologies, 5977E). The isotopic enrichment was corrected for the natural abundance of 13C by analyzing standards containing the unlabeled metabolites of interest. Data from the [U-13C]glucose (Figs. 1 and 2), [1,2-13C]acetate (Fig. 3), and [U-13C]β-hydroxybutyrate (Fig. 4) incubations is presented as M + X, where M is the molecular ion and X is the number of 13C atoms in the molecule. Furthermore, for these three substrates, the cycling ratio describing the rate of 13C accumulation and TCA cycle activity [34] is presented. Data from the [U-13C]glutamate (Fig. 5) and [U-13C]glutamine (Fig. 6) incubations is presented as M + X, showing the direct metabolism and first turn metabolism of the substrates [35].

Glycolytic activity in mouse and human cerebral cortical slices. Intracellular 13C enrichments of alanine and lactate and lactate amounts released to media of mouse and human cerebral cortical slices following metabolism of [U-13C]glucose. Metabolism of [U-13C]glucose via glycolysis results in M + 3 labeling of the glycolytic end-product pyruvate. As pyruvate is in equilibrium with alanine and lactate, through the enzymes alanine aminotransferase (ALAT) and lactate dehydrogenase (LDH), the 13C enrichment in alanine and lactate reflects the glycolytic activity. Mean ± SEM, n = 6–7, Welch t test with Benjamini–Hochberg correction, * < 0.05

Oxidative metabolism of [U-13C]glucose in mouse and human cortical slices. Intracellular 13C enrichment of TCA cycle intermediates and connected amino acids in mouse and human cerebral cortical brain slices following metabolism of [U-13C]glucose. [U-13C]Glucose is metabolized through glycolysis to yield pyruvate M + 3, which can enter the TCA cycle as acetylCoA M + 2, resulting in 13C enrichment in both TCA cycle intermediates and connected amino acids. First turn metabolism of [U-13C]glucose is reflected as M + 2 labeling, whereas second turn metabolism of [U-13C]glucose is represented as M + 3/M + 4 labeling. TCA cycling ratios, calculated from the 13C enrichments, reflect the overall rate of the TCA cycle. AAT, aspartate aminotransferase; GAD, glutamate decarboxylase; GDH, glutamate dehydrogenase; GS, glutamine synthetase; PDH, pyruvate dehydrogenase. Mean ± SEM, n = 6–7, Welch t test with Benjamini–Hochberg correction, * < 0.05

Astrocyte metabolism of [1,2-13C]acetate in mouse and human cortical slices. Intracellular 13C enrichment of TCA cycle intermediates and connected amino acids in mouse and human cortical brain slices following metabolism of [1,2-13C]acetate. [1,2-13C]Acetate is primarily metabolized in astrocytes and enters the TCA cycle as acetylCoA M + 2, giving rise to 13C enrichments in TCA cycle intermediates and amino acids. First and second turn metabolism of [1,2-13C]acetate is reflected as M + 2 and M + 3/M + 4 labeling, respectively. The cycling ratios calculated from 13C enrichments of [1,2-13C]acetate metabolism reflect the rate of the astrocyte TCA cycle. AAT, aspartate aminotransferase; GDH, glutamate dehydrogenase; GS, glutamine synthetase. Mean ± SEM, n = 6–7, Welch t test with Benjamini–Hochberg correction, * < 0.05

Metabolism of the ketone body [U-13C]β-hydroxybutyrate in mouse and human cortical slices. Intracellular 13C enrichment of TCA cycle intermediates and connected amino acids in mouse and human cortical brain slices following metabolism of [U-13C]β-hydroxybutyrate ([U-13C]βHB). Metabolism of [U-13C]βHB can primarily be attributed to neurons, and will enter the TCA cycle as acetylCoA M + 2, giving rise to 13C enrichments in TCA cycle intermediates and amino acids. First and second turn metabolism of [U-13C]βHB is reflected as M + 2 and M + 3/M + 4 labeling, respectively. The cycling ratios reflect the rate of the TCA cycle calculated from 13C enrichments. AAT, aspartate aminotransferase; GAD, glutamate decarboxylase; GDH, glutamate dehydrogenase; GS, glutamine synthetase. Mean ± SEM, n = 6–7, Welch t test with Benjamini–Hochberg correction, * < 0.05

Metabolism of [U-13C]glutamate in mouse and human cortical slices. Intracellular 13C enrichment of TCA cycle intermediates and connected amino acids in mouse and human cortical brain slices following metabolism of [U-13C]glutamate. Following uptake, [U-13C]glutamate can be converted to glutamine M + 5 or enter cellular metabolism as α-ketoglutarate M + 5. Direct and first turn metabolism of [U-13C]glutamate is reflected as M + 5/M + 4 and M + 3/M + 2 labeling, respectively. AAT, aspartate aminotransferase; GAD, glutamate decarboxylase; GDH, glutamate dehydrogenase; GS, glutamine synthetase. Mean ± SEM, n = 6–7, Welch t test with Benjamini–Hochberg correction, * < 0.05

Metabolism of [U-13C]glutamine in mouse and human cortical slices. Intracellular 13C enrichment of TCA cycle intermediates and connected amino acids in mouse and human cortical brain slices following metabolism of [U-13C]glutamine. Following uptake, [U-13C]glutamine can be converted to glutamate M + 5 by phosphate-activated glutaminase (PAG) and enter cellular metabolism as α-ketoglutarate M + 5. Direct and first turn metabolism of [U-13C]glutamine is reflected as M + 5/M + 4 and M + 3/M + 2 labeling, respectively. AAT, aspartate aminotransferase; GAD, glutamate decarboxylase; GDH, glutamate dehydrogenase; PAG, phosphate-activated glutaminase. Mean ± SEM, n = 6–7, Welch t test with Benjamini–Hochberg correction, * < 0.05
Determination of Amino Acid Amounts by High-Performance Liquid Chromatography (HPLC) Analysis
Amino acid content was determined by HPLC to assess the baseline amino acid levels of the cerebral cortical tissue and slice uptake of glutamate and glutamine. Tissue and slice extracts were reconstituted in water and analyzed by reverse-phase HPLC (Agilent Technologies, 1260 Inifinity, Agilent ZORBAX Eclipse Plus C18 column) to quantitatively determine the amounts of amino acids [36]. A pre-column derivatization with o-phthalaldehyde and fluorescent detection, λex = 338 nm, λem = 390 nm, was performed. Gradient elution with mobile phase A (10 mM NaH2PO4, 10 mM Na2B4O7, 0.5 mM NaN3, pH 8.2) and mobile phase B (acetonitrile 45%: methanol 45%: H2O 10%, V:V:V) was performed. The amounts of amino acids were determined from analysis of standards containing the amino acids of interest.
Isolation of Non-synaptic Mitochondria and Determination of Oxygen Consumption Rate
Isolation of non-synaptic mitochondria was performed using a Percoll gradient as previously described [37]. All procedures were performed on ice or at 4 °C. Acutely isolated brain tissue was submerged in isolation buffer (MSHE), containing in millimolar: mannitol, 210; sucrose, 70; HEPES, 5; EGTA, 1; and 0.5% BSA (fatty acid free); pH = 7.2, and gently homogenized using a Teflon on glass douncer at 500 revolutions/min for 7–8 strokes. The homogenate was centrifuged (500 g × 5 min) to pellet cell remnants. The supernatant was centrifuged (14,000 g × 10 min) and the pellet re-suspended in 12% Percoll MSHE solution. This suspension was gently layered on a 21% Percoll MSHE solution and centrifuged (18,000 g × 15 min). The supernatant was discarded and the pellet was washed with two subsequent centrifugations (18,000 g × 5 min and 14.000 g × 5 min) by suspending the pellet in MSHE buffer and discarding the supernatant after each centrifugation. The final pellet, containing the isolated mitochondria, was re-suspended and diluted in MSHE and protein amounts were determined using the Bradford protein assay with BSA as standard protein. The oxygen consumption rate (OCR) was investigated using a Seahorse XFe96 analyzer (Seahorse Biosciences, MA, USA). The isolated mitochondria were diluted in assay buffer (MAS), containing in millimolar: mannitol, 220; sucrose, 70; KH2PO4, 10; MgCl2, 5; HEPES, 2; EGTA, 1; and 0.2% BSA (fatty acid free); pH = 7.2. Twenty-five µL of mitochondrial suspension, containing 2 µg of protein for the succinate condition and 4 µg of protein for the pyruvate/malate condition, was added to a Seahorse 96-well plate and centrifuged (2000 g × 20 min × 4 °C). After centrifugation, 155 µL MAS containing pyruvate (10 mM) in combination with malate (2 mM) or succinate (10 mM) and rotenone (2 µM) (all final concentrations and pH 7.2) was added to the wells and the plate was analyzed immediately at 37 °C. In total, 7 measurement cycles (2 min mix, 1 min wait, 3 min measure) were performed (as outlined in Fig. 7B). After 3 initial baseline measurements, 4 injections were performed to stimulate or inhibit mitochondrial respiration. Firstly, ADP (4 mM) was injected, stimulating ATP synthesis and thus ATP synthase–driven respiration. Subsequently, oligomycin A, an inhibitor of the ATP synthase, was added to terminate ADP-mediated respiration. Then, the mitochondrial uncoupler FCCP, allowing protons from the intermembrane space to flow into the matrix hereby stimulating uncoupled respiration, was injected. Finally, antimycin A was injected to inhibit the flow of electrons from complex III to complex IV, thus leaving only residual non-mitochondrial oxygen consumption. Data was collected and extracted using the Wave software (Agilent Technologies, CA, USA). Non-mitochondrial OCR (i.e., OCR after antimycin A injection) was subtracted all values. The OCR is presented as pmol O2/min/µg protein. Basal OCR refers to the OCR before any compound addition. As a measure of general mitochondrial function, the respiratory control ratio (RCR) was calculated by dividing the OCR after ADP injection by the OCR after oligomycin A injection [38]. Mitochondrial function of non-synaptic mitochondria of the mouse cerebral cortex was previously determined [37] and is here compared to the isolated mitochondria of the human neocortex.

Metabolic function of isolated non-synaptic mitochondria of the mouse and human cerebral cortex. Oxygen consumption rates (OCRs) of non-synaptic isolated cortical mitochondria from human and mice. A The isolated mitochondria were provided with different metabolic substrates (1) pyruvate and malate (C and D; pink bars) or (2) succinate (E and F; green bars). B Schematic overview of the mitochondrial stress test; three baseline measurements were followed by applications of ADP stimulating coupled respiration; oligomycin A inhibiting the ATP synthase (complex V); FCCP inducing maximal uncoupled respiration, and finally antimycin A blocking mitochondrial respiration via complex III. The respiratory control ratio (RCR) was calculated as the coupled respiration (i.e., after ADP addition) divided by the proton leak (i.e., after oligomycin A addition) and is a general indicator of mitochondrial function. Mean ± SEM, n = 5–6, Welch t test with Benjamini–Hochberg correction, * < 0.05
Experimental Design and Statistical Analyses
Data is presented as mean ± standard error of the mean (SEM), with individual data point presented. Each data point (represented by a circle or a square in the graphs) represents biological replicates (i.e., obtained from one individual), which is denoted by “n” in the figure legends. Two independent groups were compared (mouse vs. human) and Welch unpaired t test was applied corrected for multiple comparisons using the Benjamini–Hochberg procedure with a critical value for false discovery of 0.05 [39]. The significance level was set at p < 0.05 and is indicated with a single asterisk.
Results
Intracellular Amino Acid Amounts
To establish a metabolic baseline, we first assessed a panel of central amino acids by high-performance liquid chromatography (HPLC) analysis of freshly isolated mouse and human cerebral cortical tissue. The intracellular taurine content of the mouse cerebral cortical tissue was higher when compared to that of human tissue (mouse vs. human: 158.2 ± 26.5 nmol/mg vs. 26.5 ± 5.2 nmol/mg) (Table 1). The levels of aspartate, glutamate, glutamine, and the inhibitory neurotransmitter GABA were all similar between mouse and human cerebral cortical tissue.
Glycolysis and Oxidative Glucose Metabolism
To functionally investigate cellular aspects of brain energy metabolism, we next incubated acutely isolated cerebral cortical brain slices with 13C-labeled metabolic substrates and determined the subsequent 13C enrichment of TCA cycle intermediates and connected amino acids by gas chromatography-mass spectrometry (GC–MS) analysis. Glucose is the primary energy substrate of the brain, and metabolism of [U-13C]glucose provides a general overview of metabolic function in brain slices [31]. Following uptake, [U-13C]glucose is metabolized via glycolysis to yield pyruvate M + 3, which can be transaminated to alanine M + 3 by alanine aminotransferase (ALAT) or reduced to lactate M + 3 by lactate dehydrogenase (LDH) (Fig. 1). The enrichments of both alanine M + 3 and lactate M + 3 were found to be lower in extracts of human slices when compared to mice, indicating a lower intracellular glycolytic activity in the human cortical slices. During incubation, brain slices release lactate into the incubation media [31]. No changes were found in the released amounts of lactate between the mouse and human slices, inferring that the differences in glycolysis may be limited to intracellular activity. In the mitochondria, pyruvate M + 3 can be converted into acetylCoA M + 2 by pyruvate dehydrogenase (PDH) and subsequently enter the TCA cycle (Fig. 2). AcetylCoA M + 2 will give rise to 13C enrichment in TCA cycle intermediates and connected amino acids (M + 2 in the first turn and M + 3/M + 4 in the second turn of the TCA cycle) [33]. The 13C enrichment from metabolism of [U-13C]glucose was overall lower in the human brain slices when compared to mice, observed as lower M + 2 labeling in malate, aspartate, glutamate, glutamine, and GABA following first turn metabolism of [U-13C]glucose (Fig. 2). No changes were observed in citrate M + 2, which may indicate that the flux of acetylCoA derived from PDH activity is similar between cerebral cortical slices of mouse and human origin. Despite this, the 13C labeling, following second turn metabolism of [U-13C]glucose, was lower in all measured metabolites (citrate, aspartate, malate, glutamate, glutamine, and GABA) in human cortical brain slices when compared to brain slices from mice. The overall reduction in 13C enrichment from [U-13C]glucose metabolism suggests a lower rate of glucose oxidation in the human brain slices. The intracellular 13C enrichment can furthermore be used to calculate the cycling ratio, which is a measurement of the overall rate of the TCA cycle [34]. In the human slices, the TCA cycling ratio was found to be lower in all measured metabolites, when compared to the cycling ratios obtained from the mouse slices. Collectively, these results show that oxidative glucose metabolism and TCA cycle activity operate at lower rates in human cerebral cortical slices.
Astrocyte Acetate Metabolism
Next, we provided the slices with [1,2-13C]acetate, which is a marker of astrocyte metabolism [40]. [1,2-13C]Acetate enters the astrocytic TCA cycle as acetylCoA M + 2 (Fig. 3). The overall highest 13C enrichment from metabolism of [1,2-13C]acetate, in both mouse and human brain slices, was observed in citrate and glutamine, which is in accordance with previous studies [31, 41, 42] and indicates active astrocyte metabolism in the slices. As observed for metabolism of [U-13C]glucose, the 13C enrichment of the measured metabolites was generally lower in the human brain slices following metabolism of [1,2-13C]acetate when compared to that in the mouse brain slices. This was the case for M + 2 labeling of citrate, malate, aspartate, glutamate, and GABA, but not glutamine M + 2. The M + 3/M + 4 labeling from [1,2-13C]acetate metabolism, reflecting the second turn of the TCA cycle, was likewise lower in the human slices, but only for glutamate, glutamine, and GABA, when compared to the mouse cortical slices. Similarly, the TCA cycling ratios from metabolism of [1,2-13C]acetate were lower for citrate, malate, glutamine, and GABA, in the human brain slices, suggesting a generally lower rate of astrocyte oxidative metabolism in human brain slices.
Metabolism of the Ketone Body β-Hydroxybutyrate
To further explore the metabolic function of the isolated brain slices, we next applied the ketone body [U-13C]β-hydroxybutyrate ([U-13C]βHB) as an energy substrate. Similar to [1,2-13C]acetate, [U-13C]βHB enters the TCA cycle directly as acetylCoA M + 2; however, βHB is preferentially metabolized in neurons rather than astrocytes [43, 44] (Fig. 4). The 13C enrichment from metabolism of [U-13C]βHB in human brain slices was lower in several of the measured TCA cycle metabolites and amino acids following first turn metabolism (M + 2 labeling), with the exception of glutamate M + 2 and citrate M + 2. Citrate M + 2 was significantly increased in the human slices which suggests an increased entry of acetylCoA from [U-13C]βHB metabolism when compared to mouse cortical slices. Following second turn metabolism of [U-13C]βHB, the M + 3/M + 4 labeling was lower in malate, glutamine, and GABA in the human brain slices. Only the TCA cycling ratios of glutamine and GABA were lower in the human brain slices when compared with mice. These results show that the rate of ketone body metabolism of human brain slices is lower than that of mouse slices, however, not as pronounced as observed for metabolism of [U-13C]glucose and [1,2-13C]acetate.
Uptake and Metabolism of Glutamate
Glutamate is the primary excitatory neurotransmitter in the mammalian brain. However, glutamate is also closely connected to cellular energy metabolism in the brain and serves as an energy substrate supporting oxidative metabolism of the TCA cycle [7,8,9]. Extracellular glutamate is rapidly removed from the synapse via high-affinity uptake systems present in both astrocytes and neurons [45]; however, astrocytes are the primary cell type responsible for glutamate uptake [10]. Cellular uptake of [U-13C]glutamate leads to M + 5 enrichment of the intracellular glutamate pool in brain slices [35]. The M + 5 labeling in glutamate was similar between mouse and human cortical brain slices (Fig. 5). This was also the case for the total amounts of intracellular glutamate (mouse vs. human: 358.7 ± 27.4 nmol/mg vs. 281.9 ± 42.6 nmol/mg), suggesting similar glutamate uptake capacities (Table 2). Once taken up, [U-13C]glutamate can enter the TCA cycle as α-ketoglutarate M + 5 and can hereby give rise to M + 5 or M + 4 labeling (direct metabolism) or M + 3/M + 2 labeling (first turn of TCA cycle) in the connected metabolites (Fig. 5). The M + 5 labeling of α-ketoglutarate was lower, whereas the 13C enrichment of malate, aspartate, and citrate was similar, between human and mouse slices, after direct metabolism of [U-13C]glutamate. Following a turn of the TCA cycle, the M + 3 and M + 2 labeling was found to be lower in α-ketoglutarate, malate, aspartate, and citrate in human cortical brain slices when compared to cortical brain slices from mice. Besides being a metabolic substrate of the TCA cycle, glutamate also serves as the precursor for glutamine and GABA synthesis. Interestingly, much higher labeling of glutamine M + 5 and GABA M + 4 from [U-13C]glutamate metabolism was observed in the human tissue when compared to cortical brain slices from mice. This observation suggests that human astrocytes have a larger capacity for glutamine synthesis supporting neuronal GABA synthesis. Moreover, we found that the total amounts of aspartate were higher in the mouse cortical slices compared to human brain slices (mouse vs. human: 117.8 ± 7.1 nmol/mg vs. 42.4 ± 7.0 nmol/mg) after [U-13C]glutamate incubation (Table 2). The elevated aspartate levels in the mouse slices may reflect a lower capacity of mouse astrocytes to convert glutamate to glutamine, instead directing glutamate towards aspartate synthesis via aspartate aminotransferase (AAT) activity to a higher extent than in human brain slices. Finally, as observed for the freshly prepared tissue, the amounts of taurine were higher in mouse cortical slices when compared to the human cortical slices following [U-13C]glutamate incubation (mouse vs. human: 56.7 ± 4.0 nmol/mg vs. 12.3 ± 2.3 nmol/mg), whereas the levels of glutamine and GABA were similar.
Uptake and Metabolism of Glutamine
Glutamine is selectively synthesized in astrocytes and released to the extracellular space for support of neuronal glutamate and GABA synthesis [12, 46]. No difference in the intracellular labeling of glutamine M + 5 was observed between human and mouse cortical brain slices (Fig. 6). However, the total amounts of glutamine were higher in the mouse tissue than in the human tissue (mouse vs. human: 89.3 ± 5.8 nmol/mg vs. 49.6 ± 5.2 nmol/mg), which may suggest an increased capacity for glutamine uptake in the mouse slices (Table 2). Intracellularly, glutamine can undergo hydrolysis to yield glutamate, a reaction catalyzed by the enzyme phosphate-activated glutaminase (PAG) being primarily expressed in neurons [47]. PAG converts [U-13C]glutamine into glutamate M + 5 which can enter the TCA cycle, as α-ketoglutarate M + 5, thus giving rise to the same labeling patterns as metabolism of [U-13C]glutamate (Fig. 6). The 13C enrichment of all measured TCA cycle intermediates and amino acids was lower in the human slices following both direct and first turn metabolism of [U-13C]glutamine, suggesting an overall lower rate of glutamine oxidation in the human cerebral cortex. Similar to the incubation with [U-13C]glutamate, the total amounts of aspartate were higher in the acutely isolated brain slices from mice following metabolism of [U-13C]glutamine (mouse vs. human: 79.2 ± 6.4 nmol/mg vs. 40.7 ± 8.7 nmol/mg) (Table 2), again suggesting an elevated aspartate production from glutamate in the mouse brain slices. In line with our previous observations, the amounts of taurine were again higher in mouse tissue when compared to the human tissue (mouse vs. human: 38.7 ± 3.2 nmol/mg vs. 11.2 ± 2.0 nmol/mg).
Mitochondrial Function of Isolated Non-synaptic Mitochondria
To compare the mitochondrial function between human and mouse cerebral cortex, we finally assessed the oxygen consumption rate (OCR) of isolated non-synaptic mitochondria. The mitochondria were provided two different substrate combinations: (1) pyruvate and malate or (2) succinate (Fig. 7A) and tested by sequential addition of mitochondrial modulators (Fig. 7B) [37]. The combination of pyruvate and malate stimulates the generation of NADH, whereas succinate is the direct substrate for complex II of the electron transport chain generating FADH2 [37]. In the presence of pyruvate and malate, no differences were observed in the basal OCR or in the OCR upon respiratory uncoupling by FCCP between isolated mitochondria from the mouse and human cerebral cortex (Fig. 7C). However, a higher coupled respiration in response to ADP stimulation was found in mitochondria from human cerebral cortex compared to mice, indicating a higher coupling efficiency of the human mitochondria. Furthermore, the human isolated mitochondria showed a lower proton leak. In accordance with the higher coupled OCR in the presence of ADP and lower proton leak, the respiratory control ratio (RCR), a general measure of mitochondrial function [38], was significantly higher in the human mitochondria when provided with pyruvate and malate (Fig. 7D). When the isolated mitochondria were provided with succinate as energy substrate, all OCR measurements (basal, coupled, proton leak, and uncoupled) were significantly lower in the isolated mitochondria from the human cerebral cortex when compared to mitochondria from mice (Fig. 7E). However, no changes in the RCR were observed between human and mouse mitochondria when provided with succinate (Fig. 7F), indicating that the coupling efficiency is comparable between mouse and human cerebral cortical mitochondria when using succinate as a mitochondrial substrate.
Discussion
Here, we demonstrate distinct differences in functional energy and neurotransmitter metabolism between cerebral cortical tissue of mice and humans. In particular, the rates of oxidative glucose metabolism and astrocyte acetate metabolism were considerably lower in human brain slices. In addition, we found that human brain slices have a larger capacity for conversion of exogenous glutamate into glutamine, and subsequently GABA synthesis, when compared to brain slices from mice. Finally, we show that isolated non-synaptic mitochondria from the human cerebral cortex display higher coupling efficiency when provided pyruvate and malate, whereas this was not the case with succinate as respiratory substrate.
Neuronal and Astrocyte Energy Metabolism
The mouse and human brains differ not only in size, but also in anatomical complexity and cellular composition. The size of rodent neurons correlates with the overall size of brain structures; thus, the size of a mouse neuron will be smaller than that of larger rodents, e.g., rats [48]. In contrast, neuronal size does not correlate with increasing brain mass of the primate brain. This has allowed the primate brain to maintain a higher neuronal density and more importantly to minimize the size necessary to accommodate the number of neurons [49, 50]. Glucose is the main neuronal energy substrate and is mandatory to maintain proper cerebral function. Based on in vivo 13C nuclear magnetic resonance (NMR) spectroscopy, it has been estimated that the total oxidative glucose consumption of the awake brain is over twice as high in rats (0.86 µmol/g/min) [51, 52] than in humans (0.41 µmol/g/min) [53,54,55,56]. Similar results were reached when comparing in vivo cerebral cortical glucose consumption in awake rats and humans, measured by [14C]2-deoxyglucose autoradiography and [18F]fluorodeoxyglucose positron emission tomography (18F-FDG-PET), respectively [57, 58]. Larger comparative studies across multiple mammalian species have demonstrated that the overall cerebral glucose consumption per gram of brain tissue is negatively correlated with brain size [50, 59]. Thus, of the investigated species, the mouse brain had the highest relative glucose consumption, whereas the human brain had the lowest [50, 59]. The in vivo observations described above agree well with our studies of [U-13C]glucose metabolism in cerebral cortical slices. Based on the cycling ratios derived from [U-13C]glucose metabolism, we find that the rate of TCA cycling in human brain slices was approximately half of that of mouse slices (Fig. 2). Importantly, these results demonstrate that acutely isolated brain slices maintain the metabolic profile of the in vivo brain. The species-specific differences in glucose metabolism have been suggested to be mediated by higher neuronal firing rate and synaptic density of the rodent brain [58], but could also be linked to differences in glial composition [59]. Indeed, astrocytes do not follow the same scaling rules as neurons. Human astrocytes are much larger in size, are more structurally complex, and modulate 20 times as many synapses than their rodent counterpart [26, 60]. In line with the total oxidative glucose metabolism, it has been estimated that the in vivo astrocytic contribution of glucose consumption is several fold lower in the human brain (0.07 µmol/g/min) [54, 56, 61, 62] when compared to the rat (0.18 µmol/g/min) [51]. Here, we applied [1,2-13C]acetate to functionally probe astrocyte metabolism in the brain slices [40, 41] and found that the rate of astrocyte TCA cycling indeed is lower in human cortical brain slices when compared to mice (Fig. 3). This observation supports our previous report of lower oxidative metabolism of branched-chain amino acids, primarily reflecting astrocyte metabolism, in human brain slices when compared to mice [63]. Furthermore, cultured human astrocytes display a lower basal and ATP-linked mitochondrial oxygen consumption when compared to mouse astrocytes [64]. In addition to the lower mitochondrial respiration, it was suggested that human astrocytes produce more lactate than mouse astrocytes [64]. This observation, may be in agreement with our results of similar amounts of released lactate from human and mouse slices, despite lower 13C enrichment of the intracellular lactate pool in human brain slices (Fig. 1). This could suggest a larger lactate export, relative to oxidative glucose metabolism, from the human slices. In contrast, to the pronounced differences in [U-13C]glucose metabolism between human and mouse slices, metabolism of the ketone body βHB was more similar (Fig. 4). βHB has been found to be primarily metabolized in neurons in vivo [43, 44], but βHB metabolism is glucose sparing in both cultured neurons [65] and astrocytes [66]. Interestingly, we observed higher labeling in citrate M + 2 following metabolism of [U-13C]βHB in the human brain slices. As ketone bodies can compete with glucose for acetylCoA generation, the higher M + 2 labeling in citrate likely reflects a higher entry of βHB-derived acetylCoA into the TCA cycle [67, 68]. Furthermore, metabolism of [U-13C]βHB was not reduced to the same extent as [U-13C]glucose in the human slices, indicating that the human brain retains a larger basal capacity for ketone body metabolism and that ketones may serve as an important neuronal substrate in the human cerebral cortex. It could be argued that the mouse cohort used in this study (12–13 weeks, corresponding to early adulthood) was young compared to the human patient group (39 years on average). However, the mouse brain metabolome is nearly conserved from early adulthood (16 weeks) to middle adulthood (60 weeks), including all cerebral cortical metabolites investigated in this study [69]. Furthermore, despite an age span of 35 years in the human patient group, no age-related metabolic correlations were observed, supporting the notion that cerebral cortical metabolic function is maintained throughout adulthood. Finally, it should be noted that, although the surgically resected human neocortical tissue was not taken from the epileptic focus, recurrent seizures may affect the surrounding neocortical tissue. However, no abnormal histopathological features were present in the tissue and glial morphology and organization have likewise been found to be maintained in similar resected human tissue [60].
Glutamate and Glutamine Metabolism and Recycling
Besides being the primary excitatory neurotransmitter, glutamate is also an integral part of cellular metabolism, connecting amino acid, and glucose metabolism [7, 9]. Glutamate is extensively recycled between neurons and astrocytes, as part of the glutamate-glutamine cycle [12] and neuronal glucose consumption is linearly correlated with glutamate recycling [54, 57, 58]. We found significantly higher labeling in glutamine M + 5 and GABA M + 4 following direct metabolism of [U-13C]glutamate in human slices (Fig. 5), whereas the intracellular levels of glutamate, glutamine, and GABA were similar in human and mouse brain slices (Table 2). We have previously demonstrated functional glutamine transfer from astrocytes to neurons in our slice incubation setup, particularly supporting neuronal GABA synthesis [70]. The elevated 13C enrichment in GABA from metabolism of [U-13C]glutamate therefore likely reflects increased astrocytic glutamine synthesis and transfer. As the slices were incubated with elevated concentrations of exogenous glutamate (0.5 mM), the augmented transfer of glutamine does not faithfully reflect neurotransmitter recycling. However, our results demonstrate that human astrocytes have an increased capacity for glutamine synthesis for neuronal replenishment of GABA. Indeed, in vivo NMR studies have suggested a lower rate of glutamine synthesis in the rat brain [71] than in the human brain [53]. The 13C enrichment in TCA cycle intermediates after direct metabolism of [U-13C]glutamate was overall similar in the human and mouse slices. This is in line with our recent report of functional GABA metabolism, showing similar rates of oxidative metabolism of [U-13C]GABA between mouse and human cerebral cortical slices [32]. In contrast, the 13C enrichment from direct [U-13C]glutamine metabolism was lower in all measured metabolites in the human slices when compared to mouse (Fig. 6). This overall reduction may reflect either reduced capacity for the conversion of glutamate to glutamine, or a general lower metabolic rate. The specific activity of PAG has been found to be lower in mouse brain [72] as well as cultured neurons [73], when compared to human brain [74], suggesting that the enzymatic capacity of PAG does not limit glutamine metabolism in the human slices. However, as glutamine is primarily metabolized in the neuronal compartment [47], the reduced 13C enrichment from metabolism of [U-13C]glutamine is in accordance with the lower metabolic rate of [U-13C]glucose. Interestingly, we found higher intracellular amounts of aspartate in mouse brain slices following incubations with [U-13C]glutamate and [U-13C]glutamine (Table 2). In contrast, the human brain slices appeared to be more efficient in converting exogenous glutamate into glutamine (Fig. 5). Glutamate primarily enters the TCA cycle via activity of two enzymes: glutamate dehydrogenase (GDH) and aspartate aminotransferase (AAT) [7, 75]. Indeed, the expression levels of AAT (genes: Got1 and Got2) are several fold greater in the mouse cerebral cortex than in humans [29], supporting that aspartate synthesis is the primary metabolic pathway handling elevated glutamate concentrations in the mouse brain. GDH is particularly enriched in astrocytes [76] and astrocytic deletion of GDH leads to decreased glutamate oxidation in the TCA cycle and instead directs glutamate towards aspartate via AAT activity [77, 78]. Interestingly, human astrocytes express an additional isoform of GDH, GDH2, which is absent in rodents [28, 29, 79]. Introduction of GDH2 in mice increases the astrocytic capacity of both uptake and oxidative metabolism of glutamate [80]. As GDH acts as a metabolic safeguard elevating oxidative glutamate metabolism during intensive glutamatergic signaling [7, 8], the results of this study further suggest that the combination of high GS and GDH activity of human astrocytes collectively serves as a metabolic defense against excess glutamatergic signaling and potential excitotoxicity.
Non-synaptic Mitochondrial Function
Finally, to obtain a better understanding of the metabolic differences between the human and the mouse brain, we investigated the functionality of isolated non-synaptic mitochondria (Fig. 7). It has previously been shown that isolated non-synaptic mitochondria display higher respiration with succinate as metabolic substrate when compared to the combination of pyruvate and malate [37, 81,82,83]. The results presented here are in line with these previous reports, as the non-synaptic mitochondria from both mouse and human cerebral cortex had higher OCRs with succinate as a substrate when compared to pyruvate/malate. Succinate provides electrons directly to the electron transport chain via complex II, i.e., succinate dehydrogenase [37, 84]. The non-synaptic mitochondria from humans had an overall lower OCR with succinate as substrate, when compared to mouse mitochondria. Since mitochondrial respiration on succinate primarily reflects activity of complex II, this observation agrees well with the general lower metabolic rate of the human cerebral cortical slices (Figs. 2, 3, 4). Comparative studies have found a negative correlation between cerebral oxygen consumption and brain size [50, 59] agreeing well with these results. In contrast to the overall lower respiration with succinate, we observed similar basal and uncoupled OCRs, and even a higher coupled OCR, of the human mitochondria when provided pyruvate/malate as respiratory substrates. Oxidative metabolism of pyruvate relies on multiple metabolic pathways including functional TCA cycling and reflects NADH-mediated, and hence complex I–driven, respiration [37]. In addition, we also observed a lower proton leak in the human non-synaptic mitochondria. The proton leak is the flow of protons across the inner mitochondrial membrane not related to energy production, which constitutes a significant fraction of the resting mitochondrial metabolic rate [85, 86]. The lower proton leak, in combination with the higher coupled respiration, leads to much higher RCR values for the human mitochondria. This suggests that the human mitochondria are more efficiently coupled and are better equipped to handle complex I respiration than brain mitochondria of mice. The preference for pyruvate metabolism of human mitochondria may be explained by the high enzyme activity of complex I of the electron transport chain in human brain [87, 88], which is several fold higher than what has been observed in mice [89]. As a higher mitochondrial coupling efficiency will lead to more efficient energy production, it may be speculated that the enhanced coupling of the human mitochondria is linked to the overall lower TCA cycling ratios observed in the human brain slices. Finally, it should be noted that isolated non-synaptic mitochondria are derived from multiple different cell types and that depriving mitochondria of their cellular environment may influence metabolic outcomes. Further comparative studies of mitochondrial function in situ or in vivo are therefore highly encouraged.
Conclusions
Here, we report distinct metabolic differences between cerebral cortical slices and isolated mitochondria from humans and mice. Overall, the neuronal and astrocytic metabolism was lower in the human brain slices, whereas human astrocytes displayed an increased capacity for glutamine synthesis when faced with elevated glutamate concentrations. Complex I–driven respiration was more efficient in human mitochondria when compared to mice, whereas respiration mediated by complex II was lower in the human mitochondria. The reported metabolic differences between the human and mouse cerebral cortex are in line with the metabolic profile of the in vivo brain and have to be considered when applying rodents as model systems. The overall higher metabolic rate of the rodent brain could lead to overestimations of the metabolic consequences when using transgenic rodent models of neurodegenerative diseases and may not accurately reflect similar outcomes in humans.
Data Availability
All data of this study is available from the corresponding author upon request.
Abbreviations
- AAT:
-
Aspartate aminotransferase
- ADP:
-
Adenosine-5'-diphosphate
- ALAT:
-
Alanine aminotransferase
- βHB:
-
β-Hydroxybutyrate
- FCCP:
-
Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- GC-MS:
-
Gas chromatography-mass spectrometry
- GAD:
-
Glutamate decarboxylase
- GDH:
-
Glutamate dehydrogenase
- GS:
-
Glutamine synthetase
- HPLC:
-
High-performance liquid chromatography
- LDH:
-
Lactate dehydrogenase
- M:
-
Molecular ion
- OCR:
-
Oxygen consumption rate
- PAG:
-
Phosphate-activated glutaminase
- PDH:
-
Pyruvate dehydrogenase
- RCR:
-
Respiratory control ratio
- TCA:
-
Tricarboxylic acid (cycle)
References
Attwell D, Laughlin SB (2001) An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21(10):1133–1145. https://doi.org/10.1097/00004647-200110000-00001
Harris JJ, Jolivet R, Attwell D (2012) Synaptic energy use and supply. Neuron 75(5):762–777. https://doi.org/10.1016/j.neuron.2012.08.019
Zhang S, Lachance BB, Mattson MP, Jia X (2021) Glucose metabolic crosstalk and regulation in brain function and diseases. Prog Neurobiol 204:102089. https://doi.org/10.1016/j.pneurobio.2021.102089
Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF Jr (1967) Brain metabolism during fasting. J Clin Invest 46(10):1589–1595. https://doi.org/10.1172/jci105650
Schousboe A, Bak LK, Waagepetersen HS (2013) Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front Endocrinol (Lausanne) 4:102. https://doi.org/10.3389/fendo.2013.00102
Waagepetersen HS, Sonnewald U, Schousboe A (2007) Glutamine, glutamate, and GABA: metabolic aspects. In: Lajtha A, Oja SS, Schousboe A, Saransaari P (eds) Handbook of Neurochemistry and Molecular Neurobiology: Amino Acids and Peptides in the Nervous System. Springer US, Boston, MA 1–21. https://doi.org/10.1007/978-0-387-30373-4_1
Andersen JV, Markussen KH, Jakobsen E, Schousboe A, Waagepetersen HS, Rosenberg PA, Aldana BI (2021) Glutamate metabolism and recycling at the excitatory synapse in health and neurodegeneration. Neuropharmacol 196:108719. https://doi.org/10.1016/j.neuropharm.2021.108719
McKenna MC (2013) Glutamate pays its own way in astrocytes. Front Endocrinol (Lausanne) 4:191. https://doi.org/10.3389/fendo.2013.00191
Schousboe A, Scafidi S, Bak LK, Waagepetersen HS, McKenna MC (2014) Glutamate metabolism in the brain focusing on astrocytes. Adv Neurobiol 11:13–30. https://doi.org/10.1007/978-3-319-08894-5_2
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65(1):1–105
Martinez-Hernandez A, Bell KP, Norenberg MD (1977) Glutamine synthetase: glial localization in brain. Science 195(4284):1356–1358. https://doi.org/10.1126/science.14400
Bak LK, Schousboe A, Waagepetersen HS (2006) The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 98(3):641–653. https://doi.org/10.1111/j.1471-4159.2006.03913.x
Tani H, Dulla CG, Farzampour Z, Taylor-Weiner A, Huguenard JR, Reimer RJ (2014) A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron 81(4):888–900. https://doi.org/10.1016/j.neuron.2013.12.026
Cheung G, Bataveljic D, Visser J, Kumar N, Moulard J, Dallérac G, Mozheiko D, Rollenhagen A et al (2022) Physiological synaptic activity and recognition memory require astroglial glutamine. Nat Commun 13(1):753. https://doi.org/10.1038/s41467-022-28331-7
Gordon BA, Blazey TM, Su Y, Hari-Raj A, Dincer A, Flores S, Christensen J, McDade E et al (2018) Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol 17(3):241–250. https://doi.org/10.1016/s1474-4422(18)30028-0
Andersen JV, Schousboe A, Verkhratsky A (2022) Astrocyte energy and neurotransmitter metabolism in Alzheimer's disease: integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol:102331. https://doi.org/10.1016/j.pneurobio.2022.102331
Moreira PI, Carvalho C, Zhu X, Smith MA (1802) Perry G (2010) Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim Biophys Acta 1:2–10. https://doi.org/10.1016/j.bbadis.2009.10.006
Mochel F, Haller RG (2011) Energy deficit in Huntington disease: why it matters. J Clin Invest 121(2):493–499. https://doi.org/10.1172/jci45691
Kim J, Moody JP, Edgerly CK, Bordiuk OL, Cormier K, Smith K, Beal MF, Ferrante RJ (2010) Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet 19(20):3919–3935. https://doi.org/10.1093/hmg/ddq306
Rosenthal N, Brown S (2007) The mouse ascending: perspectives for human-disease models. Nat Cell Biol 9(9):993–999. https://doi.org/10.1038/ncb437
Scearce-Levie K, Sanchez PE, Lewcock JW (2020) Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat Rev Drug Discov 19(7):447–462. https://doi.org/10.1038/s41573-020-0065-9
Blanchard JW, Victor MB, Tsai LH (2022) Dissecting the complexities of Alzheimer disease with in vitro models of the human brain. Nat Rev Neurol 18(1):25–39. https://doi.org/10.1038/s41582-021-00578-6
Hodge RD, Bakken TE, Miller JA, Smith KA, Barkan ER, Graybuck LT, Close JL, Long B et al (2019) Conserved cell types with divergent features in human versus mouse cortex. Nature 573(7772):61–68. https://doi.org/10.1038/s41586-019-1506-7
Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ (2013) Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 106–107:1–16. https://doi.org/10.1016/j.pneurobio.2013.04.001
de Majo M, Koontz M, Rowitch D, Ullian EM (2020) An update on human astrocytes and their role in development and disease. Glia 68(4):685–704. https://doi.org/10.1002/glia.23771
Oberheim NA, Wang X, Goldman S, Nedergaard M (2006) Astrocytic complexity distinguishes the human brain. Trends Neurosci 29(10):547–553. https://doi.org/10.1016/j.tins.2006.08.004
Boldog E, Bakken TE, Hodge RD, Novotny M, Aevermann BD, Baka J, Bordé S, Close JL et al (2018) Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat Neurosci 21(9):1185–1195. https://doi.org/10.1038/s41593-018-0205-2
Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK et al (2016) Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89(1):37–53. https://doi.org/10.1016/j.neuron.2015.11.013
Sjöstedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, Adori C, Oksvold P, Edfors F et al (2020) An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367 (6482)https://doi.org/10.1126/science.aay5947
Torrens-Mas M, Pons DG, Sastre-Serra J, Oliver J, Roca P (2020) Sexual hormones regulate the redox status and mitochondrial function in the brain. Pathological implications Redox Biol 31:101505. https://doi.org/10.1016/j.redox.2020.101505
McNair LF, Kornfelt R, Walls AB, Andersen JV, Aldana BI, Nissen JD, Schousboe A, Waagepetersen HS (2017) Metabolic characterization of acutely isolated hippocampal and cerebral cortical slices using [U-13C]glucose and [1,2–13C]acetate as substrates. Neurochem Res 42(3):810–826. https://doi.org/10.1007/s11064-016-2116-5
Andersen JV, Jakobsen E, Westi EW, Lie MEK, Voss CM, Aldana BI, Schousboe A, Wellendorph P et al (2020) Extensive astrocyte metabolism of γ-aminobutyric acid (GABA) sustains glutamine synthesis in the mammalian cerebral cortex. Glia 68(12):2601–2612. https://doi.org/10.1002/glia.23872
Walls AB, Bak LK, Sonnewald U, Schousboe A, Waagepetersen HS (2014) Metabolic mapping of astrocytes and neurons in culture using stable isotopes and gas chromatography-mass spectrometry (GC-MS). In: Hirrlinger J, Waagepetersen HS (eds) Brain Energy Metabolism. Neuromethods, vol 90. Humana Press, New York, NY,
Andersen JV, Skotte NH, Christensen SK, Polli FS, Shabani M, Markussen KH, Haukedal H, Westi EW et al (2021) Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer’s disease. Cell Death Dis 12(11):954. https://doi.org/10.1038/s41419-021-04237-y
Andersen JV, Nissen JD, Christensen SK, Markussen KH, Waagepetersen HS (2017) Impaired hippocampal glutamate and glutamine metabolism in the db/db mouse model of type 2 diabetes mellitus. Neural Plast 2017:2107084. https://doi.org/10.1155/2017/2107084
Andersen JV, Westi EW, Jakobsen E, Urruticoechea N, Borges K, Aldana BI (2021) Astrocyte metabolism of the medium-chain fatty acids octanoic acid and decanoic acid promotes GABA synthesis in neurons via elevated glutamine supply. Mol Brain 14(1):132. https://doi.org/10.1186/s13041-021-00842-2
Andersen JV, Jakobsen E, Waagepetersen HS, Aldana BI (2019) Distinct differences in rates of oxygen consumption and ATP synthesis of regionally isolated non-synaptic mouse brain mitochondria. J Neurosci Res 97(8):961–974. https://doi.org/10.1002/jnr.24371
Brand MD, Nicholls DG (2011) Assessing mitochondrial dysfunction in cells. Biochem J 435(2):297–312. https://doi.org/10.1042/bj20110162
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B (Methodol) 57(1):289–300. https://doi.org/10.1111/j.2517-6161.1995.tb02031.x
Wyss MT, Magistretti PJ, Buck A, Weber B (2011) Labeled acetate as a marker of astrocytic metabolism. J Cereb Blood Flow Metab 31(8):1668–1674. https://doi.org/10.1038/jcbfm.2011.84
Sonnewald U, Westergaard N, Schousboe A, Svendsen JS, Unsgard G, Petersen SB (1993) Direct demonstration by [13C]NMR spectroscopy that glutamine from astrocytes is a precursor for GABA synthesis in neurons. Neurochem Int 22(1):19–29
Andersen JV, Christensen SK, Nissen JD, Waagepetersen HS (2017) Improved cerebral energetics and ketone body metabolism in db/db mice. J Cereb Blood Flow Metab 37(3):1137–1147. https://doi.org/10.1177/0271678x16684154
Edmond J, Robbins RA, Bergstrom JD, Cole RA, de Vellis J (1987) Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J Neurosci Res 18(4):551–561. https://doi.org/10.1002/jnr.490180407
Pan JW, de Graaf RA, Petersen KF, Shulman GI, Hetherington HP, Rothman DL (2002) [2,4–13 C2 ]-beta-Hydroxybutyrate metabolism in human brain. J Cereb Blood Flow Metab 22(7):890–898. https://doi.org/10.1097/00004647-200207000-00014
Olivares-Bañuelos TN, Chí-Castañeda D, Ortega A (2019) Glutamate transporters: gene expression regulation and signaling properties. Neuropharmacol 161:107550. https://doi.org/10.1016/j.neuropharm.2019.02.032
Walls AB, Waagepetersen HS, Bak LK, Schousboe A, Sonnewald U (2015) The glutamine-glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res 40(2):402–409. https://doi.org/10.1007/s11064-014-1473-1
Kvamme E, Torgner IA, Roberg B (2001) Kinetics and localization of brain phosphate activated glutaminase. J Neurosci Res 66(5):951–958. https://doi.org/10.1002/jnr.10041
Herculano-Houzel S, Mota B, Lent R (2006) Cellular scaling rules for rodent brains. Proc Natl Acad Sci U S A 103(32):12138–12143. https://doi.org/10.1073/pnas.0604911103
Herculano-Houzel S, Collins CE, Wong P, Kaas JH (2007) Cellular scaling rules for primate brains. Proc Natl Acad Sci U S A 104(9):3562–3567. https://doi.org/10.1073/pnas.0611396104
Herculano-Houzel S (2011) Scaling of brain metabolism with a fixed energy budget per neuron: implications for neuronal activity, plasticity and evolution. PLoS One 6(3):e17514. https://doi.org/10.1371/journal.pone.0017514
Öz G, Berkich DA, Henry PG, Xu Y, LaNoue K, Hutson SM, Gruetter R (2004) Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J Neurosci 24(50):11273–11279. https://doi.org/10.1523/jneurosci.3564-04.2004
McNair LM, Mason GF, Chowdhury GM, Jiang L, Ma X, Rothman DL, Waagepetersen HS, Behar KL (2022) Rates of pyruvate carboxylase, glutamate and GABA neurotransmitter cycling, and glucose oxidation in multiple brain regions of the awake rat using a combination of [2-(13)C]/[1-(13)C]glucose infusion and (1)H-[(13)C]NMR ex vivo. J Cereb Blood Flow Metab 271678x221074211. https://doi.org/10.1177/0271678x221074211
Mason GF, Gruetter R, Rothman DL, Behar KL, Shulman RG, Novotny EJ (1995) Simultaneous determination of the rates of the TCA cycle, glucose utilization, alpha-ketoglutarate/glutamate exchange, and glutamine synthesis in human brain by NMR. J Cereb Blood Flow Metab 15(1):12–25. https://doi.org/10.1038/jcbfm.1995.2
Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF, Petroff OA, Shulman GI et al (1999) Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc Natl Acad Sci U S A 96(14):8235–8240. https://doi.org/10.1073/pnas.96.14.8235
Pan JW, Stein DT, Telang F, Lee JH, Shen J, Brown P, Cline G, Mason GF et al (2000) Spectroscopic imaging of glutamate C4 turnover in human brain. Magn Reson Med 44(5):673–679. https://doi.org/10.1002/1522-2594(200011)44:5%3c673::aid-mrm3%3e3.0.co;2-l
Gruetter R, Seaquist ER, Ugurbil K (2001) A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am J Physiol Endocrinol Metab 281(1):E100-112. https://doi.org/10.1152/ajpendo.2001.281.1.E100
Hyder F, Rothman DL, Bennett MR (2013) Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc Natl Acad Sci U S A 110(9):3549–3554. https://doi.org/10.1073/pnas.1214912110
Yu Y, Herman P, Rothman DL, Agarwal D, Hyder F (2018) Evaluating the gray and white matter energy budgets of human brain function. J Cereb Blood Flow Metab 38(8):1339–1353. https://doi.org/10.1177/0271678x17708691
Karbowski J (2007) Global and regional brain metabolic scaling and its functional consequences. BMC Biol 5:18. https://doi.org/10.1186/1741-7007-5-18
Oberheim NA, Takano T, Han X, He W, Lin JH, Wang F, Xu Q, Wyatt JD et al (2009) Uniquely hominid features of adult human astrocytes. J Neurosci 29(10):3276–3287. https://doi.org/10.1523/jneurosci.4707-08.2009
Blüml S, Moreno-Torres A, Shic F, Nguy CH, Ross BD (2002) Tricarboxylic acid cycle of glia in the in vivo human brain. NMR Biomed 15(1):1–5. https://doi.org/10.1002/nbm.725
Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI et al (2002) Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci 22(5):1523–1531. https://doi.org/10.1523/jneurosci.22-05-01523.2002
Salcedo C, Andersen JV, Vinten KT, Pinborg LH, Waagepetersen HS, Freude KK, Aldana BI (2021) Functional metabolic mapping reveals highly active branched-chain amino acid metabolism in human astrocytes, which is impaired in iPSC-derived astrocytes in Alzheimer’s disease. Front Aging Neurosci 13:736580. https://doi.org/10.3389/fnagi.2021.736580
Li J, Pan L, Pembroke WG, Rexach JE, Godoy MI, Condro MC, Alvarado AG, Harteni M et al (2021) Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat Commun 12(1):3958. https://doi.org/10.1038/s41467-021-24232-3
Lund TM, Ploug KB, Iversen A, Jensen AA, Jansen-Olesen I (2015) The metabolic impact of β-hydroxybutyrate on neurotransmission: reduced glycolysis mediates changes in calcium responses and KATP channel receptor sensitivity. J Neurochem 132(5):520–531. https://doi.org/10.1111/jnc.12975
Valdebenito R, Ruminot I, Garrido-Gerter P, Fernández-Moncada I, Forero-Quintero L, Alegría K, Becker HM, Deitmer JW et al (2016) Targeting of astrocytic glucose metabolism by beta-hydroxybutyrate. J Cereb Blood Flow Metab 36(10):1813–1822. https://doi.org/10.1177/0271678x15613955
Cox PJ, Kirk T, Ashmore T, Willerton K, Evans R, Smith A, Murray AJ, Stubbs B et al (2016) Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab 24(2):256–268. https://doi.org/10.1016/j.cmet.2016.07.010
Valente-Silva P, Lemos C, Köfalvi A, Cunha RA, Jones JG (2015) Ketone bodies effectively compete with glucose for neuronal acetyl-CoA generation in rat hippocampal slices. NMR Biomed 28(9):1111–1116. https://doi.org/10.1002/nbm.3355
Ding J, Ji J, Rabow Z, Shen T, Folz J, Brydges CR, Fan S, Lu X et al (2021) A metabolome atlas of the aging mouse brain. Nat Commun 12(1):6021. https://doi.org/10.1038/s41467-021-26310-y
Andersen JV, McNair LF, Schousboe A, Waagepetersen HS (2017) Specificity of exogenous acetate and glutamate as astrocyte substrates examined in acute brain slices from female mice using methionine sulfoximine (MSO) to inhibit glutamine synthesis. J Neurosci Res 95(11):2207–2216. https://doi.org/10.1002/jnr.24038
Sibson NR, Dhankhar A, Mason GF, Behar KL, Rothman DL, Shulman RG (1997) In vivo 13C NMR measurements of cerebral glutamine synthesis as evidence for glutamate-glutamine cycling. Proc Natl Acad Sci U S A 94(6):2699–2704. https://doi.org/10.1073/pnas.94.6.2699
Gaisler-Salomon I, Miller GM, Chuhma N, Lee S, Zhang H, Ghoddoussi F, Lewandowski N, Fairhurst S et al (2009) Glutaminase-deficient mice display hippocampal hypoactivity, insensitivity to pro-psychotic drugs and potentiated latent inhibition: relevance to schizophrenia. Neuropsychopharmacol 34(10):2305–2322. https://doi.org/10.1038/npp.2009.58
Hogstad S, Svenneby G, Torgner IA, Kvamme E, Hertz L, Schousboe A (1988) Glutaminase in neurons and astrocytes cultured from mouse brain: kinetic properties and effects of phosphate, glutamate, and ammonia. Neurochem Res 13(4):383–388. https://doi.org/10.1007/bf00972489
Butterworth J, Yates CM, Reynolds GP (1985) Distribution of phosphate-activated glutaminase, succinic dehydrogenase, pyruvate dehydrogenase and gamma-glutamyl transpeptidase in post-mortem brain from Huntington’s disease and agonal cases. J Neurol Sci 67(2):161–171. https://doi.org/10.1016/0022-510x(85)90112-1
McKenna MC, Stridh MH, McNair LF, Sonnewald U, Waagepetersen HS, Schousboe A (2016) Glutamate oxidation in astrocytes: roles of glutamate dehydrogenase and aminotransferases. J Neurosci Res 94(12):1561–1571. https://doi.org/10.1002/jnr.23908
Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T et al (2007) The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci 27(45):12255–12266. https://doi.org/10.1523/jneurosci.3404-07.2007
Skytt DM, Klawonn AM, Stridh MH, Pajęcka K, Patruss Y, Quintana-Cabrera R, Bolaños JP, Schousboe A et al (2012) siRNA knock down of glutamate dehydrogenase in astrocytes affects glutamate metabolism leading to extensive accumulation of the neuroactive amino acids glutamate and aspartate. Neurochem Int 61(4):490–497. https://doi.org/10.1016/j.neuint.2012.04.014
Nissen JD, Pajecka K, Stridh MH, Skytt DM, Waagepetersen HS (2015) Dysfunctional TCA-cycle metabolism in glutamate dehydrogenase deficient astrocytes. Glia 63(12):2313–2326. https://doi.org/10.1002/glia.22895
Spanaki C, Zaganas I, Kleopa KA, Plaitakis A (2010) Human GLUD2 glutamate dehydrogenase is expressed in neural and testicular supporting cells. J Biol Chem 285(22):16748–16756. https://doi.org/10.1074/jbc.M109.092999
Nissen JD, Lykke K, Bryk J, Stridh MH, Zaganas I, Skytt DM, Schousboe A, Bak LK et al (2017) Expression of the human isoform of glutamate dehydrogenase, hGDH2, augments TCA cycle capacity and oxidative metabolism of glutamate during glucose deprivation in astrocytes. Glia 65(3):474–488. https://doi.org/10.1002/glia.23105
Clark JB, Nicklas WJ (1970) The metabolism of rat brain mitochondria Preparation and characterization. J Biol Chem 245(18):4724–4731
Iuso A, Repp B, Biagosch C, Terrile C, Prokisch H (2017) Assessing mitochondrial bioenergetics in isolated mitochondria from various mouse tissues using Seahorse XF96 analyzer. Methods Mol Biol 1567:217–230. https://doi.org/10.1007/978-1-4939-6824-4_13
Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN (2011) High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One 6(7):e21746. https://doi.org/10.1371/journal.pone.0021746
Lores-Arnaiz S, Lombardi P, Karadayian AG, Orgambide F, Cicerchia D, Bustamante J (2016) Brain cortex mitochondrial bioenergetics in synaptosomes and non-synaptic mitochondria during aging. Neurochem Res 41(1–2):353–363. https://doi.org/10.1007/s11064-015-1817-5
Brand MD, Chien LF, Ainscow EK, Rolfe DF, Porter RK (1994) The causes and functions of mitochondrial proton leak. Biochim Biophys Acta 1187(2):132–139
Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD (2010) Mitochondrial proton and electron leaks. Essays Biochem 47:53–67. https://doi.org/10.1042/bse0470053
Ojaimi J, Masters CL, Opeskin K, McKelvie P, Byrne E (1999) Mitochondrial respiratory chain activity in the human brain as a function of age. Mech Ageing Dev 111(1):39–47. https://doi.org/10.1016/s0047-6374(99)00071-8
Ronsoni MF, Remor AP, Lopes MW, Hohl A, Troncoso IH, Leal RB, Boos GL, Kondageski C et al (2016) Mitochondrial respiration chain enzymatic activities in the human brain: methodological implications for tissue sampling and storage. Neurochem Res 41(4):880–891. https://doi.org/10.1007/s11064-015-1769-9
Pollard AK, Craig EL, Chakrabarti L (2016) Mitochondrial complex 1 activity measured by spectrophotometry is reduced across all brain regions in ageing and more specifically in neurodegeneration. PLoS One 11(6):e0157405. https://doi.org/10.1371/journal.pone.0157405
Funding
The Scholarship of Peter and Emma Thomsen is gratefully acknowledged for financial support to EWW and JVA.
Author information
Authors and Affiliations
Contributions
Emil W. Westi, Jens V. Andersen, and Caroline M. Voss performed slice incubations. Emil Jakobsen and Jens V. Andersen performed mitochondrial analysis. Jens V. Andersen and Blanca I. Aldana conceived the overall project. Emil W. Westi and Jens V. Andersen wrote the original manuscript. All authors contributed to the article and approved the final version.
Corresponding author
Ethics declarations
Ethics Approval
The use of human neocortical tissue was approved by the local Ethics Committee in Copenhagen (H-2–2011-104) and performed in line with the principles of the Declaration of Helsinki. All animal experiments were approved by the Danish National Ethics Committee and performed in accordance with the European Convention (ETS 123 of 1986).
Consent to Participate
The use of human neocortical tissue was performed with written informed consent from all patients prior to surgery.
Consent for Publication
Written informed consent for publication was obtained from all individuals included in the study.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Blanca I. Aldana and Jens V. Andersen shared last authorship.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Westi, E.W., Jakobsen, E., Voss, C.M. et al. Divergent Cellular Energetics, Glutamate Metabolism, and Mitochondrial Function Between Human and Mouse Cerebral Cortex. Mol Neurobiol 59, 7495–7512 (2022). https://doi.org/10.1007/s12035-022-03053-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-03053-5