
Abstract
Glial cell-line derived neurotrophic factor (GDNF) is a powerful astroglioma (AG) proliferation and migration factor that is highly expressed in AG cells derived from astrocytes. However, it is still unclear whether high levels of GDNF promote AG occurrence or if they are secondary to AG formation. We previously reported that high concentrations of GDNF (200 and 500 ng/mL) can inhibit DNA damage-induced rat primary astrocytes (RA) apoptosis, suggesting that high concentrations of GDNF may be involved in the malignant transformation of astrocytes to AG cells. Here we show that 200 ng/mL GDNF significantly increased the proliferation and migration ability of RA cells and human primary astrocytes (HA). This treatment also induced RA cells to highly express Pgf, Itgb2, Ibsp, Loxl2, Lif, Cxcl10, Serpine1, and other genes that enhance AG proliferation and migration. LOXL2 is an important AG occurrence and development promotion factor and was highly expressed in AG tissues and cells. High concentrations of GDNF promote LOXL2 expression and secretion in RA cells through GDNF family receptor alpha-1(GFRα1)/rearranged during transfection proto-oncogene (RET)/mitogen-activated protein kinase (MAPK)/phosphorylated cyclic AMP response element binding protein (pCREB) signaling. GDNF-induced LOXL2 significantly promotes RA and HA cell proliferation and migration, and increases the expression of Ccl2, Gbp5, MMP11, TNN, and other genes that regulate the extracellular microenvironment in RA cells. Our results demonstrate that high concentrations of GDNF activate LOXL2 expression and secretion via the GFRα1/RET/MAPK/pCREB signal axis, which leads to remodeling of the astrocyte extracellular microenvironment through molecules such as Ccl2, Gbp5, MMP11, TNN. This ultimately results in abnormal astrocyte proliferation and migration. Collectively, these findings suggest that high GDNF concentrations may promote the malignant transformation of astrocytes to AG cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Astroglioma (AG) originating from astrocytes is the most common malignant tumor in the central nervous system, but effective therapies are limited due to its migratory and invasive growth capacities [1, 2]. Recent evidence indicates that AG occurrence and development are closely related to multiple cytokines and trophic factors including glial cell line-derived neurotrophic factor (GDNF) [3,4,5]. A member of the transforming growth factor beta super-family, GDNF was first isolated and purified from the rat B49 glioma cell line in 1993 [6]. It widely expressed in different brain regions and nourishes and protects many types of neurons including midbrain dopaminergic, sensory, and motor neurons [7]. Until recently, GDNF was known only as a specific physiological nutrient and pro-differentiation factor. However, studies have reported abnormally increased GDNF expression and secretion in human AG tissues and AG cell lines, and these events positively correlated with pathological grade [8, 9]. Excessive GDNF is chiefly secreted from AG cells, and it effectively promotes AG cell proliferation, invasion, and migration via specific signal pathways such as mitogen-activated protein kinase (MAPK) [10,11,12]. Moreover, reducing the expression of GDNF and its receptor GFRα1 significantly inhibits AG progression [13], indicating that GDNF and its downstream signaling pathways play important roles in maintaining abnormal AG cell proliferation and migration.
GDNF is mainly secreted by astrocytes and neurons and is maintained at a low level in adult brain tissues [14, 15]. Recent studies have shown that some neurons secrete high levels of GDNF following oxidative stress stimuli [16,17,18], indicating that high concentrations can exist in local brain regions. Researchers have confirmed that low concentrations of GDNF nourish astrocytes and neurons [19], while cells treated with high concentrations can resist DNA damage-induced astrocyte apoptosis [20], suggesting that physiologically expressed GDNF may be tumorigenic at abnormally high concentrations. Malignant proliferation and migration are the most significant characteristics of tumor cells [21], and GDNF can promote both events in AG cells [12, 22, 23]. However, it is still unclear whether and how high concentrations of GDNF can induce astrocytes to gradually acquire malignant proliferation and migration abilities.
To address these issues, we investigated the effects of a high concentration of GDNF (200 ng/mL) on the proliferation and migration of rat primary astrocytes (RA) and human primary astrocytes (HA) using 5-ethynyl-2′-deoxyuridine (EdU) staining, flow cytometry, wound healing and transwell assays. The results show that 200 ng/mL GDNF significantly enhanced astrocyte proliferation and migration. Subsequently, the expression of global mRNAs in phosphate-buffered saline (PBS)- or GDNF (200 ng/mL)-treated RA cells and rat C6 astroglioma cells were determined by RNA-sequencing (RNA-seq). We found that GDNF-treated RA cells and C6 cells shared a variety of pro-proliferation and pro-migration molecules including lysyl oxidase-like protein 2 (LOXL2). This copper and lysine tyrosyl-quinone (LTQ)-dependent amine oxidase catalyzes covalent cross-linking of extracellular matrix (ECM) proteins including collagens and elastin [24]. Recent studies have shown that LOXL2 is highly expressed in multiple cancers including glioma, especially glioma stem cells [25], and it is associated poor prognosis, epithelial-mesenchymal transition, metastasis, and tumor progression [26]. LOXL2 promotes glioma cell proliferation and migration by regulating key proteins such as E-cadherin, Snail1, Src, and FAK [27, 28], and it induces oncogenic transformation of normal human mammary epithelial cells into breast cancer cells [29]. Moreover, LOXL2 expression can be regulated through MAPK signaling that is a classic pathway activated by the GDNF receptors GFRα1/RET [30]. Hence, we speculated that high concentrations of GDNF may promote astrocyte proliferation and migration via GFRα1/RET/MAPK-induced LOXL2 activity.
To test this hypothesis, LOXL2 knockdown was performed in RA cells by RNA interference (RNAi) to determine whether LOXL2 is involved in GDNF-induced proliferation and migration. Suppression of GFRα1/RET receptor or MAPK signaling in RA cells by neutralizing antibodies or specific inhibitors were performed to determine if GDNF increases LOXL2 expression via the GFRα1/RET/MAPK signaling pathway. Finally, the molecular mechanisms of LOXL2-mediated GDNF-promoting astrocyte proliferation and migration were investigated by microarray, real-time polymerase chain reaction (RT-PCR) and bioinformatics analyses. Our results clarify how GDNF promotes astrocyte proliferation and migration and suggest that high concentrations of GDNF may contribute to AG occurrence, providing new ideas for AG prevention and treatment.
Materials and Methods
Animals and Tissue Samples
Newborn Sprague Dawley (SD) rats were purchased from the Xuzhou Medical University Experimental Animal Center (Xuzhou, China) and reared in pathogen-free conditions. All experiments were carried out in compliance with the Provisions and General Recommendations of the Chinese Experimental Animal Administration Regulations, as well as institutional consent from the Xuzhou Medical University Animal Ethics Committee. Three human glioblastoma (GBM) tissue samples were obtained from the surgical specimen archives of Xuzhou Medical University’s affiliated hospitals with written informed consent from the patients. Tissue samples from the normal brain (NB) were collected from three patients who had suffered acute brain damage and underwent intracranial decompression.
Cell Culture and Growth Curves
Cryopreserved rat C6 astrocytoma cells, normal human astrocytes (HA, ScienCell, Carlsbad, CA, USA), and human U251, U87, and A172 astrocytoma cells were cultured as previously described [31, 32]. Rat astrocytes (RA) were isolated from postnatal (days 1–3) SD rats as discussed previously [20]. The purity of the RA cells was evaluated by immunostaining for glial fibrillary acidic protein (GFAP, astrocyte marker), 2′, 3′-cyclic nucleotide phosphodiesterase (CNPase, oligodendrocyte marker), and Iba1 (microglial marker). Image-Pro Plus 6.0 software was then used to count the number of cells. Using five randomly selected fields, the percentages of GFAP-, CNPase-, and Iba1-positive cells were determined. The percentage of GFAP-positive cells represents RA cell purity. The RA and C6 cells’ growth curves were plotted using CCK8 assay results. The primary astrocytes were treated with rat GDNF (enQuire Bio, Denver, CO, USA, #QP5504), human GDNF (Gibco, Grand Island, NY, USA#RP-8602), neutralizing GFRα1 antibody (Bio-techne, Minneapolis, MN, USA, #AF560-SP), and inhibitors of particular receptors or kinases.
Enzyme-Linked Immunosorbent Assay (ELISA)
The concentration of GDNF in the cell-conditioned media of RA and C6 cells was determined using a commercial GDNF ELISA kit (Abnova, Taipei, Taiwan) and the manufacturer’s instructions. The secretion of LOXL2 from RA and C6 cells in response to PBS or GDNF treatments was measured using Rat LOXL2 ELISA kits (Meimian Biotechnology, Yancheng, China, #MM-70467R1). Three biological replicates were performed for each treatment and assay.
Cell Proliferation and Cell Cycle Assays
The CCK8, EdU, and flow cytometry for cell cycle assays were carried out in accordance with the protocols published in earlier publications [33]. In brief, cell vitality was measured using a cell counting kit-8 (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. The Cell Light EdU Apollo®488 In Vitro Kit (RiboBio, Guangzhou, China, #C10310-3) was used to assess the cell proliferation of astrocytes. PI/RNase Staining Buffer (Becton Dickinson Biosciences, Franklin Lakes, NJ, USA, #550,825) was used to assess RA cell cycle by flow cytometry. DNA content was monitored with a flow cytometer (FACScan; Becton Dickinson Biosciences). Flow cytometry data were analyzed using ModFit LTTM software (Verity Software House, Topsham, ME, USA). For the plate clone formation test, 600 RA and HA cells were seeded in 6-well plates with 10% FBS. Different concentrations of GDNF were added into the culture medium every 3 days. The developed colonies were stained with crystal violet (Beyotime, Shanghai, China, #C0121) after 12–15 days, and the number was counted using ImageJ software (National Institutes of Health, Bethesda, MD, USA). All of the above experiments were carried out in accordance with the manufacturers’ instructions.
Cell Migration Assays
The wound-healing and the transwell assay results were analyzed as previously described [33]. The dynamic wound healing assay was used to further investigate the migratory potential of astrocytes using the live-cell workstation (OLYMPUS advanced live-cell imaging system XCELLENCE). The variations in cell mobility near the scratch’s edge were continuously collected by the live-cell workstation for 24 h. Each point was captured for 5 min and looped once per hour.
RNA Extraction and RNA-Sequencing
Total RNA was isolated from cells using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA, #15,596–018), purified using the RNeasy micro kit (QIAGEN, GmBH, Hilden, Germany), and the RNase-Free DNase Set (QIAGEN, GmBH), and sequenced. To confirm the insert size and determine the mole concentration, purified libraries were measured using a Qubit®2.0 fluorometer (Invitrogen) and validated using an Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Clusters were generated by cBot using a diluted library of 10 pM and sequenced on an Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) using a two-terminal sequencing model (Shanghai Biotechnology Corporation, Shanghai, China). Tophat 2.0.9 was used to align high-quality reads that passed the Illumina quality filters to the rat reference genome Rnor 6.0 [34]. EdgeR [35] was used to perform the differentially expressed gene (DEG) analysis (P ≤ value 0.05, fold change ≥ 2). R3.6.3 software, including the ggplot2 and cluster profile packages, was used to perform the volcano plot, gene ontology (GO) enrichment, and Kyoto Encyclopedia Genes and Genomes (KEGG) pathway analysis and visualization [36].
RT-PCR
Total RNA was extracted in the manner described in the previous section. The PrimeScript® RT reagent kit (TaKaRa, Tokyo, Japan) was used to generate first-strand cDNA according to the manufacturer's instructions. In a 20-μL reaction volume, RT-PCR was done using SYBR® qPCR Mix (Toyobo, Tokyo, Japan) on a Roche LightCycler® 480 real-time PCR detection system. The 2−ΔΔCt method was used to calculate the relative expression level of each gene, which was then normalized to the housekeeping gene β-actin or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). RT-PCR was performed using three biological replicates per group. Supplementary Table S1 displays the mRNA primer pairs.
Bioinformatics Analysis of DEGs
To explore the expression levels of the same gene in glioma tissues and normal brain tissues, we used RNA-seq data from the TCGA and GTEx databases, which were uniformly processed by the Toil process in UCSC XENA (https://xenabrowser.net/datapages/) [37]. The normal brain data set includes 1152 normal brain tissue samples from GTEx and 5 paracancerous samples from the TCGA database. The glioma data comes from the TCGA database, which contains 523 LGG and 166 GBM cases. To compare the expression levels of different samples, the RNA-seq data in TPM format was converted by log2. Statistical significance was determined using the Wilcoxon rank-sum test. R3.6.3 software, comprising pROC (1.17.0.1 version), ggplot2 (3.3.3, for visualization), survival (3.2–10), and survminer (0.4.9, for visualization), was used to draw the receiver operating characteristic (ROC) and Kaplan–Meier curves.
Western Blot
To isolate total protein, cells or tissues were homogenized ultrasonically in RIPA buffer. The nuclear and cytoplasmic proteins were extracted from RA and C6 cells by using a nuclear and cytoplasmic protein extraction kit (Beyotime, #P0027) according to the manufacturer’s instructions. A western blot analysis was performed on the protein in each group using antibodies against LOXL2 (Abcam, Cambridge, UK, ab96233 and ab179810), ERK1/2 (Proteintech, Rosemont, IL, USA #67,170–1-Ig), phospho-ERK1/2 (Thr202/Tyr204) (Affinity, Cincinnati, OH, USA, #AF1015), JNK1/2/3 (Proteintech, #66,210–1-Ig), phospho-JNK1/2/3 (Thr183 + Tyr185) (Affinity, #AF3318), p38 MAPK(Affinity, #AF6456), phospho-p38 MAPK (Thr180/Tyr182) (Affinity, #AF4001), β-actin (Proteintech, #66,009–1-Ig), GAPDH (Proteintech, #60,004–1-Ig), Lamin B1 (Proteintech, #66,095–1-Ig), and β-tubulin (Proteintech, #66,240–1-Ig) as described previously [31]. β-actin, GAPDH, Lamin B1, and β-tubulin were used as internal references.
Immunofluorescence Staining
Immunofluorescence staining was carried out exactly as stated previously [38]. Cells were fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.5% (w/v) Triton X-100 in PBS. The cells were incubated with GFAP antibody (Abcam, #ab7260), CNPase antibody (Abcam, #ab6319), Iba1 antibody (Wako Chemicals, Ann Arbor, MI, USA, #NCNP24), RET antibody (Abcam, # ab134100), and LOXL2 antibody (Abcam, #ab96233). Following secondary antibody incubation, the nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI, KeyGEN BioTECH, Nanjing, China). Fluorescence images were captured using a laser scanning confocal microscope (Leica, Wetzlar, Germany, STELLARIS 5), and analyzed using Leica Application Suite X software.
Chromatin immunoprecipitation (ChIP)-PCR
ChIP-PCR assays were performed essentially as described elsewhere [31]. CREB monoclonal antibody (Cell Signaling Technology, Danvers, MA, USA, #9197) and normal rabbit IgG (SCBT, Dallas, TX, USA #sc-2027X) were used for ChIP, and RT-PCR was employed to identify DNA from immunoprecipitated target antibodies. The results were calculated as %Input = 2(CtInput−CtChIP) × Input dilution factor × 100%. Supplementary Table S2 shows the primer sequences.
Lentivirus Construction and Infection
Three siRNAs targeting the Rat Loxl2 mRNA as well as NC-siRNA were generated and cloned into the GV248 lentiviral vector (hU6-MCS-Ubiquitin-EGFP-IRES- puromycin) (Supplementary Table S3). Sequence alignment ensured the validity of the above constructions’ sequences. HEK 293 T cells were employed to create recombinant lentiviruses, and the virus of NC-siRNA was used as a negative control (NC). RA cells were grown on a 24-well plate (Corning, Corning, NY, USA) at a density of 2 × 104 cells/well for 24 h before being infected with 1 µL concentrated lentivirus in the presence of polybrene (8 µg/mL). Cells were extracted 72 h after infection to be used in the subsequent investigations.
Microarray Analysis of mRNA Expression Profiling
Microarray-based mRNA expression profiling was conducted at Genechem (Shanghai, China). Briefly, total RNAs were extracted and quantified using the Thermo Nanodrop 2000 and Agilent 2100 Bioanalyzers, purified using the Gene Chip WT PLUS Reagent Kit (Thermo Fisher Scientific, Waltham, MA, USA), and hybridized to the Affymetrix Gene Chip Clariom™ D (Rat) Assay using the Gene Chip Hybridization Wash and Stain Kit according to the manufacturer’s instructions. Chips were scanned using the Affymetrix Gene Chip Scanner 3000 (Affymetrix, Santa Clara, CA, USA) and analyzed with the gene matrix.
Statistical Analysis
All data were processed with SPSS 25.0 (IBM Corp. Armonk, NY, USA). Data from at least three distinct experiments are provided as mean ± SD. Independent sample t-tests were employed to discover significant differences in mean values between the two groups. The mean values of multiple groups were compared using one-way analyses of variance and Bonferroni post hoc tests. For all tests, the statistical significance criteria were set at P < 0.05.
Results
GDNF Secretion is Higher in AG Cells Compared to Astrocytes
To identify the purity of primary cultured RA cells, the numbers of third-pass cells positive for GFAP (labeling astrocytes), CNPase (labeling oligodendrocytes), and Iba1 (labeling microglia) were detected by immunofluorescence staining. Primary cultured oligodendrocytes and microglia were used as positive controls for CNPase and Iba1 labeling (Supplementary Fig. S1). The results showed that GFAP-, CNPase-, and Iba1-positive cells accounted for 96.77 ± 0.58%, 2.92 ± 1.64%, and 1.39 ± 0.50% of cells, respectively (Fig. 1a), indicating that primary cultured astrocytes were appropriate for the subsequent experiments.

GDNF secretion is higher in AG cells compared to astrocytes. a Immunofluorescence was used to determine the purity of rat primary astrocytes. CNP and Iba1 in green represent oligodendrocytes and microglia, respectively, whereas GFAP in red denotes astrocytes. The GFAP positive cell rate is calculated by comparing the number of GFAP stained cells to the number of DAPI stained cells. Bar = 50 µm. b CCK8 assays were used to analyze the growth curves of RA and C6 cells (n = 3). c ELISA was used to identify GDNF secretion from RA and C6 cells throughout the logarithmic growth period (n = 4). All data are presented in mean ± SD. ## P < 0.01; *** P < 0.001
The growth characteristics of RA and C6 cells were tested by CCK-8 assays. The results showed that 24–72 h was the exponential growth phase for both types of cells, and the proliferation activity of C6 cells was significantly higher than that of RA cells during this period (P < 0.01, Fig. 1b). GDNF secretion of RA and C6 cells was then determined by ELISA during the exponential growth period. The results showed that GDNF secretion of C6 cells was significantly higher than that of RA cells (C6, 0.87 ± 0.22 pg/10,000 cells; RA, 0.10 ± 0.03 pg/10,000 cells after 24 h; C6, 1.65 ± 0.28 pg/10,000 cells; RA, 0.30 ± 0.09 pg/10,000 cells after 48 h; P < 0.001), and GDNF secretion increased over time (P < 0.01, Fig. 1c). GDNF was undetectable in DMEM-F12 medium and fetal calf serum in this experimental system.
High-Concentration GDNF Promotes Astrocyte Proliferation
We previously reported that a high concentration of GDNF (200 ng/mL) enables astrocytes to resist DNA damage-induced apoptosis [20], suggesting that physiologically expressed GDNF may be oncogenic in the context of abnormally high secretion. To investigate the effect of different concentrations of GDNF on RA cell proliferation, we applied low (10 ng/mL), medium (50 ng/mL) and high concentrations (100, 200, and 500 ng/mL) to treat RA cells for 24 h before measuring viability with CCK8 assays. The results showed that medium and high concentrations of GDNF significantly enhanced RA cell viability in a concentration-dependent manner (P < 0.05, Fig. 2a). The effects of medium and high concentrations of GDNF on RA cell proliferation, cell cycle, and clone formation were further explored by EdU staining, flow cytometry, and plate cloning. The results showed that only high concentrations of GDNF promoted RA cell proliferation in a dose-dependent manner after 24-h treatment (P < 0.01, Fig. 2b, c). The 100 and 200 ng/mL GDNF-treated groups had a significantly reduced proportion of cells in the G0/G1 phase, while the percentages of S phase cells increased (P < 0.05, Fig. 2d and e). Long-term 15 d-treatment with 100 and 200 ng/mL GDNF significantly promoted RA cell clonogenic ability in a dose-dependent manner (P < 0.01, Fig. 2f). Since 200 ng/mL GDNF had the most significant effect on proliferation and clonogenic abilities, we selected this concentration for subsequent experiments. To verify whether the above phenomenon occurs in human primary astrocytes, the effects of 200 ng/mL GDNF on HA cell viability, proliferation, and clone formation were detected by CCK8, EdU, and plate cloning assays. The results showed that HA cell activity and proliferation were significantly increased after 24-h 200 ng/mL GDNF treatment (P < 0.05, Fig. 2g and h), and the clonogenic ability also significantly enhanced following 12-day treatment with 200 ng/mL GDNF (P < 0.05, Fig. 2i–k), which was consistent with the RA cell results.

High-concentration GDNF promotes astrocyte proliferation significantly. a CCK-8 assay was used to determine cell viability in RA cells after 24 h of treatment with various doses of GDNF (n = 3). b and c EdU analysis of the impact of various GDNF concentrations on RA cell proliferation (n = 3). Bar = 50 µm. d and e Flow cytometry analysis of the cell cycle of RA cells treated with different concentrations of GDNF over 24 h (n = 3). f A plate cloning formation assay was used to assess colony formation of RA cells after 15 days of treatment with different doses of GDNF; the number of clones containing > 50 cells were counted (n = 3). g CCK8 was used to test the cell viability of HA cells treated with 200 ng/mL GDNF for 24 h (n = 3). h EdU was used to detect cell proliferation in HA cells after 24 h of treatment with 200 ng/mL GDNF (n = 3). i–k Plate cloning assay was used to assess cloning formation ability in GDNF-treated HA cells after 12 days (n = 3). All data are presented as mean ± SD. * and # P < 0.05; ** and ## P < 0.01
High-Concentration GDNF Promotes Astrocyte Migration
RA and HA cell migration was determined by wound healing and transwell assays after 200 ng/mL GDNF treatment for 24, 48, and 72 h. PBS was used as a negative control. Dynamic scratch tests were performed to further verify the migration ability of RA cells treated with GDNF. Compared with the PBS group, 200 ng/mL GDNF significantly increased the migration of RA cells (Fig. 3a, b) and HA cells (Fig. 3e and f) at each time point (P < 0.05), which was consistent with the results of the dynamic scratch experiments in RA cells (Supplementary Video 1). Transwell assay results further confirmed that 200 ng/mL GDNF significantly (P < 0.05) increased the migration levels of RA cells (Fig. 3c and d) and HA cells (Fig. 3g and h) at each time point.

High-concentration GDNF promotes astrocyte migration drastically. a and b GDNF at 200 ng/mL enhanced RA cell wound repair at 24, 48, and 72 h (n = 3). c and d Transwell migration experiment detected RA cell migration following GDNF exposure at 24, 48, and 72 h (n = 3). The number of migrating cells was enumerated in seven microscopic fields. e and f At 24, 48, and 72 h, 200 ng/mL GDNF also promoted HA cell wound healing (n = 3). g and h Transwell migration assay was used to detect cell migration in GDNF-treated HA cells at 24, 48, and 72 h (n = 3). Bar = 50 µm. All data are presented as mean ± SD. * P < 0.05; ** P < 0.01
High-Concentration GDNF Induces Astrocytes to Express Genes that Promote Glioma Proliferation and Migration
To investigate the molecular mechanism by which high-concentration GDNF promotes astrocyte proliferation and migration, the transcriptomes of RA cells with or without 200 ng/mL GDNF treatment were assessed with RNA-seq. DEGs were screened using following criteria: |log2 (fold change)|≥ 1 and P < 0.05, and 253 upregulated, and 92 downregulated genes (Supplementary Table S4) are shown in a volcano plot (Fig. 4a). The upregulated DEGs were selected to perform GO and KEGG pathway analyses, which showed that these genes were enriched in the GO terms of in response to tumor necrosis factor (TNF), positive regulation of cell–cell adhesion, response to hypoxia, regulation of angiogenesis, epithelial cell proliferation, cytokine activity, and cell adhesion molecule binding. Similarly, KEGG pathway analysis showed that these upregulated genes were mainly involved in TNF signaling pathway, cell adhesion molecules, and cytokine-cytokine receptor interaction (Fig. 4b, Supplementary Table S5). Subsequently, the global RNA expression levels of RA cells and C6 cells were examined by RNA-seq; the results revealed that 2828 genes were upregulated and 3436 genes were downregulated in C6 cells (Fig. 4c, Supplementary Table S6). The upregulated genes were mainly enriched in GO terms of nuclear division, positive regulation of cell cycle, ECM organization, angiogenesis, and cell-substrate adhesion and mainly involved in the signaling pathways of cell cycle, DNA replication, focal adhesion, MAPK signaling pathway, and glioma (Fig. 4d, Supplementary Table S7). Venn diagrams showed that there were 39 co-upregulated genes and 37 co-downregulated genes in GDNF-treated RA cells and C6 cells (Fig. 4e, Supplementary Table S8). The co-upregulated DEGs were mainly enriched in the GO terms of angiogenesis, epithelial cell migration, regulation of fibroblast proliferation, ECM, and cell adhesion molecules and the pathways of TNF signaling pathway, cell adhesion molecules, and focal adhesion (Fig. 4f, Supplementary Table S9).

High-concentration GDNF drives astrocytes to express genes that promote glioma proliferation and migration. a Volcano plots of DEGs in PBS-treated RA cells versus GDNF-treated RA cells (n = 1). The red color denotes up-regulated genes, whereas the blue color shows downregulated genes. b GO annotations and KEGG signaling analyses of the GDNF-treated group’s elevated genes. The Y-axis shows the GO terms that are significantly enhanced. The enrichment levels are shown by the Gene ratio on the X-axis. The dot in different colors represents the p-adjusted value, which is the p-value corrected by the BH (Benjamini & Hochberg) approach, and its size shows the number of enriched genes. BP stands for biological process, CC stands for cellular component, and MF stands for molecular function. c Volcano plots of C6 and RA cells (n = 1). d A signaling analysis of the elevated genes in C6 cells using GO and KEGG. e Venn diagrams of co-upregulated and co-downregulated genes in GDNF-treated RA cells and C6 cells, compared with RA cells. f GO and KEGG signaling analyses of co-upregulated genes in GDNF-treated RA and C6 cells. g RT-PCR was used to confirm the expression of seven genes involved in proliferation and migration (n = 3). h Human PGF, ITGB2, IBSP, LOXL2, LIF, CXCL10, and SERPINE1 mRNA levels (log2 (TPM + 1)) were compared in normal brain samples (n = 1157) and glioma samples (n = 689) from the TCGA and GTEx datasets. The statistical significance was determined using the Wilcoxon rank-sum test. i ROC curve of seven overlapping genes. The data source is the same as described previously. * P < 0.05; ** P < 0.01; *** P < 0.001
To verify the sequencing results, co-upregulated genes associated with proliferation and migration such as Pgf, Itgb2, Ibsp, Loxl2, Lif, Cxcl10, and Serpine1 were detected by RT-PCR. These genes were significantly increased in GDNF-treated RA cells and C6 cells (P < 0.05), which was consistent with the sequencing results (Fig. 4g), indicating that the above genes may be involved in GDNF-induced abnormal proliferation and migration of astrocytes. In addition, the TCGA and GTEx datasets showed that the expression levels of these seven genes in glioma tissues were significantly higher than in normal brain tissues (P < 0.001, Fig. 4h); the ROC curves showed that the above seven genes can be used as predictive variables for glioma; PGF (area under the curve [AUC] = 0.925, confidence interval [CI] = 0.911–0.938) and ITGB2 (AUC = 0.937, CI = 0.927–0.947) had higher accuracy; LOXL2 (AUC = 0.843, CI = 0.825–0.861), LIF (AUC = 0.772, CI = 0.751–0.793), CXCL10 (AUC = 0.751, CI = 0.729–0.774), and SERPINE1 (AUC = 0.847, CI = 0.828–0.865) had moderate accuracy, and IBSP (AUC = 0.656, CI = 0.630–0.683) had the lowest accuracy (Fig. 4i).
High Concentration of GDNF Increases Glioma Potential Prognostic Indicator LOXL2 Expression and Secretion in Astrocytes
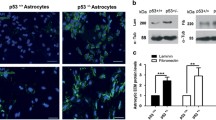
LOXL2 is a new target for cancer treatment that is involved in glioma development [26, 28]. In order to clarify the relationships among LOXL2, GDNF, and AG occurrence, we detected LOXL2 expression in normal brain tissues (NB) and glioblastoma tissues derived from astrocytes (GBM) and in HA, U251, U87, and A172 AG cell lines using RT-PCR and western blot. The results showed that LOXL2 mRNA and protein in GBM tissues and AG cells were significantly increased (P < 0.05, Fig. 5a–d), and 24-h treatment with 200 ng/mL GDNF significantly increased LOXL2 mRNA and protein levels in HA cells (P < 0.05, Fig. 5c and d), similar to the results of RA cells (Fig. 5e and f). The effect of GDNF on subcellular LOXL2 localization was tested by immunofluorescence in RA cells treated with 200 ng/mL GDNF for 24 h and C6 cells. The results showed that LOXL2 was present in both the cytoplasm and nuclei of RA cells and C6 cells, but it was mainly distributed in the nuclei (especially around the nucleoli) (Fig. 5g). LOXL2 protein was highly expressed in the cytoplasm and nuclei of GDNF-treated RA cells and C6 cells (Fig. 5g-i), and both cell types secreted higher levels of LOXL2 (Fig. 5j, P < 0.05). Subsequently, the effect of LOXL2 expression (from the TCGA database) on glioma patient survival (from Liu’s data [39]) was analyzed by Cox regression. Kaplan–Meier survival curves showed that high LOXL2 mRNA expression was significantly associated with worse overall survival (hazard ratio [HR] 3.92 [2.97–5.16]), disease-specific survival (HR 3.82 [2.87–5.09]) and progress free interval (HR 2.58 [2.06–3.21]) for glioma patients (P < 0.001, Supplementary Fig. S2a–c), confirming the correlation between LOXL2 and glioma progression.

High-concentration GDNF increases LOXL2 expression and release in RA cells, which is a possible prognostic indicator for glioma. a and b Analysis of LOXL2 mRNA and protein expression in glioblastoma (GBM) and normal brain (NB) tissue (n = 3). c and d LOXL2 mRNA and protein expression in HA cells treated with PBS or 200 ng/mL GDNF and different astroglioma cells (n = 3). e and f LOXL2 mRNA and protein expression in RA cells treated with PBS or 200 ng/mL GDNF and C6 cells (n = 3). g Immunofluorescence was used to examine the subcellular location and expression of LOXL2 in the three cell types mentioned above (n = 3). The cytoplasm of a cell is shown in red by GFAP, while the nucleus is shown in blue by DAPI. Bar = 50 µm. h–j LOXL2 protein levels in the nucleus and cytoplasm, and LOXL2 secretion levels in RA, GDNF-treated RA, and C6 cells measured by western blot and ELISA (n = 3). *P < 0.05, **P < 0.01
High-Concentration GDNF Increases LOXL2 Expression in Astrocytes Through the GFRα1/RET/MAPK/pCREB Signaling Axis
MAPK signaling is a classic downstream pathway of GDNF [11, 40, 41]. To clarify whether high concentrations of GDNF promote LOXL2 expression by activating this pathway in astrocytes, the phosphorylation levels of Erk, JNK, and P38 and LOXL2 protein levels were measured by western blot in RA cells after treatment with 200 ng/mL GDNF with or without the MAPK inhibitors PD98059, SP600125, and SB203580. The results showed that after 200 ng/mL GDNF treatment for 0.5 h and 1 h, phosphorylated Erk, JNK, and P38 were significantly increased (P < 0.05) in RA cells; PD98059, SP600125, and SB203580 significantly reduced the GDNF-induced increases in phosphorylated Erk, JNK, and P38, respectively (P < 0.05, Fig. 6a–c). Elevated LOXL2 protein expression induced by GDNF was significantly decreased after inhibition of Erk, JNK, and P38 signaling pathways (P < 0.05, Fig. 6d).

The GFRα1/RET/MAPK/pCREB signaling pathway was activated by a high concentration of GDNF in astrocytes. a–c The total and phosphorylated Erk, JNK, and P38 proteins, d as well as LOXL2 protein, were assessed by western blot after pretreatment of RA cells with 50 µM PD98059, 20 µM SP600125, and 20 µM SB203580 for 2 h, and 200 ng/mL GDNF treatment for further 0.5 h, 1 h, and 3 h (n = 3). Following that, the RA cells were pretreated for 3 h with GFRα1 neutralizing antibody (1:200 dilution) or 50 µM RET inhibitor selpercatinib, followed by another 1 h with 200 ng/mL GDNF. e and g Western blot identified phosphorylated Erk, f and h P38, and i and j LOXL2 proteins (n = 3). k RET mRNA was tested by real-time PCR after transfected siRETs in RA cells (n = 3). RET-siRNA2 was used in the following experiments. l LOXL2 mRNA was examined by real-time PCR in RA cells with different treatments (n = 3). m A conserved CREB binding motif (MA0018.2) from the JASPAR database in humans, mice, and rats, as well as a probable CRE site within the rat Loxl2 promoter. The primers for ChIP-PCR are indicated by arrows. n ChIP-PCR was used to examine CREB binding to the Loxl2 promoter in RA cells treated with 200 ng/mL GDNF for 24 h. o For 2 h, the RA cells were pretreated with 50 µM PD98059, 10 µM SP600125, and 20 µM SB203580, followed by 1 h of treatment with 200 ng/mL GDNF. The phosphorylated CREB was examined using a western blot (n = 3). * and # P < 0.05, ** and ## P < 0.01
The classic GFRα1/RET receptor were expressed in rat, mouse and human primary astrocyte [42,43,44], which was consistent with our findings (Supplementary Fig. S3a and b). To further clarify whether the GFRα1/RET receptor is involved in GDNF-induced MAPK signal activation and LOXL2 expression, the optimal dilution and concentration for GFRα1 neutralizing antibody (1:200 dilution) and the RET inhibitor selpercatinib (50 µM, LOXO-292; 2,152,628–33-4, InvivoChem, Libertyville, IL, USA) were screened by western blot (Supplementary Fig. S3c and d). Subsequently, RA cells pretreated with anti-GFRα1 antibody (1:200 dilution) and 50 µM selpercatinib for 3 h were then treated 200 ng/mL GDNF for another 1 h, and phosphorylated Erk, JNK, and P38 and LOXL2 expression were detected by western blot and RT-PCR. The results showed that neutralizing GFRα1 significantly decreased GDNF-induced phosphorylated Erk and P38 (P < 0.05, Fig. 6e and f) and LOXL2 mRNA (Supplementary Fig. S3e) and protein (Fig. 6i) levels, but it did not significantly alter JNK signal activation (P > 0.05, Supplementary Figure S3f). Blocking RET also reduced GDNF-induced phosphorylated Erk and P38 (Fig. 6g and h) and LOXL2 protein levels (Fig. 6j), which was similar to neutralizing GFRα1. Notably, neutralizing GFRα1 receptors or blocking RET with selpercatinib was sufficient to inhibit Erk1/2 and p38MAPK activation and LOXL2 expression in a GDNF-independent manner (Fig. 6e–j). Moreover, the RET knockdown assay was performed to further confirm if GDNF-induced LOXL2 expression was mediated by RET. The results showed that knockdown RET significantly decreased GDNF-induced LOXL2 mRNA expression in RA cells (P < 0.01, Fig. 6k and l). Additionally, knockdown RET was sufficient to inhibit LOXL2 expression in a GDNF-independent manner in RA cells, but 200 ng/mL GDNF can still increase LOXL2 mRNA level in RETi-RA cells (P < 0.01, Fig. 6l). Bioinformatics analysis revealed a potential CRE binding site for CREB in the rat Loxl2 promoter (Fig. 6m). CREB can bind to this CRE of Loxl2 promoter, and 200 ng/mL GDNF significantly increased the binding of CREB to CRE in Loxl2 promoter (P < 0.05, Fig. 6n). Furthermore, 200 ng/mL GDNF significantly increased phosphorylated CREB levels in RA cells (P < 0.05); total CREB protein also showed an increasing tendency, but the difference was not significant (P > 0.05, Supplementary Fig. S3g). Inhibiting Erk, JNK, and P38 signaling pathways significantly attenuated GDNF-induced phosphorylated CREB (P < 0.05, Fig. 6o).
LOXL2 Knockdown Significantly Inhibits GDNF-Induced Astrocyte Proliferation and Migration
To investigate whether LOXL2 is involved in GDNF-induced astrocyte proliferation and migration, RNAi was used to generate an RA cell model with knocked down LOXL2 (Fig. 7a and b). The effects of 200 ng/mL GDNF on LOXL2i-RA cell activity and proliferation were examined by CCK8 and EdU assays, and the effects of 200 ng/mL GDNF on LOXL2i-RA cell migration were determined by scratch and transwell assays. The results showed that LOXL2-KD2 and LOXL2-KD3 significantly reduced LOXL2 mRNA and protein levels in RA cells (P < 0.01), and LOXL2-KD2 was selected for subsequent experiments (Fig. 7c and d). Compared with the NC group, LOXL2 knockdown significantly reduced RA cell activity at 24, 48, and 72 h and proliferation at 24 h (P < 0.01), and the addition of 200 ng/mL GDNF significantly increased RA cell activity and proliferation (P < 0.01). Compared with the NC plus GDNF group, knocking down LOXL2 significantly reduced the pro-proliferative effect of GDNF on RA cells (P < 0.05, Fig. 7e and f). Additionally, compared with the NC group, RA cell migration was significantly reduced after LOXL2 knockdown for 24, 48, and 72 h (P < 0.05), while it was significantly increased after treatment with 200 ng/mL GDNF (P < 0.05). Compared with the NC plus GDNF group, knocking down LOXL2 significantly decreased the pro-migration effect of GDNF on RA cells (P < 0.05, Fig. 7g and h), suggesting that high concentrations of GDNF can promote astrocyte proliferation and migration via LOXL2.

LOXL2 knockdown dramatically reduces GDNF-induced RA cell proliferation and migration. a and b Fluorescence microscopy (100 ×) indicated that after 72 h of infection, > 90% of RA cells continued to proliferate and emit green fluorescence. c, d RT-PCR and western blot were used to confirm the knockdown efficiency of LOXL2 at the mRNA and protein levels in RA cells (n = 3). e CCK8 was used to assess the effects of 200 ng/mL GDNF on LOXL2i-RA cell viability after 24, 48, and 72 h (n = 3). f EdU detects the influence of 200 ng/mL GDNF on LOXL2i-RA cell proliferation for 24 h (n = 3). Bar = 50 µm. g and h Wound healing assay was used to test the migratory ability of LOXL2i-RA cells following treatment with 200 ng/mL GDNF for 24, 48, and 72 h (n = 3). Bar = 50 µm. i A transwell was utilized to test the migratory ability of LOXL2i-RA cells in the presence of 200 ng/mL GDNF for 24 h (n = 3). Bar = 50 µm. * P < 0.05, ** and ## P < 0.01
High-Concentration GDNF Remodels the Extracellular Microenvironment via LOXL2 to Promote Astrocyte Proliferation and Migration
To elucidate the molecular mechanism by which high-concentration GDNF promotes astrocyte proliferation and migration via LOXL2, we assessed the effects of LOXL2 knockdown on GDNF-induced gene expression profiles by performing microarrays in RA cells. The R package from SAM was used to screen the differential expression of mRNAs using the following criteria: |fold change|≥ 1.5 and a false discovery rate < 0.05. A total of 720 DEGs were identified in LOXL2i-RA cells (Fig. 8a); 334 genes were upregulated and 386 genes were downregulated after 24-h GDNF treatment for 24 h (Supplementary Table S10). This is clearly visualized in the scatter plot and volcano plot (Fig. 8b, c). To validate array data reliability, we randomly selected 26 mRNAs (4 upregulated and 22 downregulated mRNAs) and confirmed their expression in GDNF-treated LOXL2i-RA cells using conventional PCR (Fig. 8d) and RT-PCR (Fig. 8e). The DEGs were mainly enriched in the GO terms of extracellular space, ECM, cell junction, cellular response to organic substance, regulation of cell differentiation, sulfur compound binding, receptor binding, cell adhesion molecular binding and the signaling pathways of ECM receptor interaction, focal adhesion, pathways in cancer, and cytokine receptor interaction (Fig. 8g). These results suggest that high GDNF concentrations may promote astrocyte proliferation and migration by remodeling the extracellular microenvironment through LOXL2. Seven genes (Ccl2, Ccl7, Gbp2, Gbp5, Slfn3, C1s, and Cxcl1) were screened out according to the following conditions: significantly increased expression after GDNF treatment and significantly decreased expression after further LOXL2 knockdown in RA cells (Fig. 8h). The TCGA and GTEx datasets showed that compared with normal brain tissues, the expression levels of Ccl2, Gbp2, Gbp5, and C1s were significantly increased in glioma tissues (P < 0.001), while Ccl7 expression was significantly decreased (P < 0.001, Fig. 8i). An Oncomine database search revealed significantly increased Ccl7 and Cxcl1 expression in glioma tissues (P < 0.05, Fig. 8j). As tumor malignant transformation factors, Ccl2 and GBP5 play a pivotal role in driving GBM malignancy [45,46,47]. Moreover, 200 ng/mL GDNF significantly increased Ccl2 and Gbp5 expression (P < 0.05), and further knocking down LOXL2 significantly decreased their expression (P < 0.05, Fig. 8k). Collectively, these results indicate that GDNF can regulate the extracellular microenvironment through LOXL2.

Effect of LOXL2 knockdown on global mRNA expression profiles in RA cells treated with GDNF (200 ng/mL). a Heatmap of global mRNAs in NC- or LOXL2i- RA cells following GDNF exposure (n = 3). b and c Scatter plot and Volcano plot comparing the global mRNA expression profiles of NC plus GDNF and LOXL2i plus GDNF groups. d Conventional PCR was used to determine the primer specificity. e RT-PCR analysis of the expression pattern of selected mRNAs associated with extracellular components (n = 3). f GO analysis of DEGs in LOXL2i-RA cells treated with GDNF. Based on the P-value, the top 30 significant GO terms were chosen. g Analysis of DEGs using the KEGG pathway. h A Venn diagram depicts the collection of up-regulated genes in GDNF-treated RA cells and down-regulated genes in GDNF-treated LOXL2i-RA cells. i mRNA levels [log2 (TPM + 1)] of human Ccl2, Ccl7, Gbp2, Gbp5, C1s, and Cxcl1 in normal brain samples (n = 1157) and glioma samples (n = 689) from the TCGA and GTEx datasets. j Oncomine® Platform analysis of mRNA levels (log2 median-centered ratio) of human Cxcl1 and Ccl7 in normal brain samples (n = 4, 3, 23) and glioblastoma samples (n = 10, 27, 81) from the Pomeroy, Bredel, and Sun Brain datasets. k Ccl2 and Gbp5 expression levels in RA cells with different treatments were determined by RT-PCR (n = 3). * P < 0.05, ** P < 0.01
Discussion
GDNF is an important factor that promotes AG development, but the relationship between GDNF and AG occurrence is unclear. To clarify this issue, we used primary astrocytes as a model to test the effects of different concentrations of GDNF on proliferation, migration, and apoptosis. The results showed that high concentrations of GDNF significantly promoted the proliferation and migration of RA and HA cells, and this treatment also enabled RA cells to significantly resist DNA damage-induced apoptosis [20]. Collectively, these findings suggest that high concentrations of GDNF may explain the malignant transformation of astrocytes to AG cells.
To clarify how high-concentration GDNF promotes astrocyte proliferation and migration, the transcriptomes of PBS-treated and GDNF-treated RA cells and C6 AG cells were detected by RNA-seq, and the accuracy of the results was verified using RT-PCR. The results showed that GDNF-treated RA cells and C6 cells shared a variety of proliferation and migration mechanisms. The co-upregulated genes such as Serpine1, Itgb2, Ibsp, Loxl2, and Has2 were mainly enriched in the GO terms of epithelial cell migration, regulation of fibroblast proliferation, ECM, and cell adhesion molecules and the signaling pathways such as cell adhesion molecules and focal adhesion. This suggests that high concentrations of GDNF may promote the malignant transformation of astrocyte to AG by affecting the ECM and cell adhesion. Moreover, the expression levels of Serpine1, Itgb2, Loxl2, and Has2 genes are also significantly increased in human glioma tissues, and their expression levels are negatively correlated with patient prognosis [28–, 48,49,50], again indicating that the above molecules may be involved in AG occurrence and development. Our next question was if high-concentration GDNF promoted astrocytes to gradually acquire the ability of malignant proliferation and migration through the above molecules.
To answer this, we screened 39 genes that were co-upregulated in GDNF-treated RA cells and C6 cells and identified LOXL2 as a potential candidate molecule. This member of the LOX family can catalyze the covalent cross-linking of collagen and elastin in the ECM, thus regulating ECM stability [51]. Recent studies reported that LOXL2 is an oncogene; it is highly expressed in a variety of cancers to promote tumor occurrence, metastasis, chemotherapy resistance, and angiogenesis, making it a potential target for tumor treatment [26, 52]. Our study found significantly increased LOXL2 expression in AG tissues and cells, which is consistent with the results of Du et al. [27]. Furthermore, highly expressed LOXL2 can promote glioma cell proliferation and migration by regulating proteins such as E-cadherin, Snail1, Src, and FAK [27, 28]. High GDNF concentrations also significantly increased LOXL2 expression and secretion in RA and HA cells, suggesting that elevated GDNF may promote astrocyte proliferation and migration by increasing the expression and secretion of LOXL2. Next, we performed more experiments to determine if and how GDNF regulates Loxl2 gene expression.
MAPK signaling (Erk/JNK/P38) is a classic pathway downstream of GDNF [11, 40, 41]; it is involved in promoting the proliferation and migration of glioma cells [11, 33]. Moreover, LOXL2 expression is regulated by the MAPK pathway (Erk/JNK/P38) in breast cancer, esophageal squamous cell carcinoma, and liver cancer [30, 53, 54], suggesting that GDNF may induce LOXL2 expression via the MAPK signaling pathway. Our results confirmed its involvement in AG, in that high concentrations of GDNF significantly increased the phosphorylation levels of Erk, JNK, and P38. Notably, after treatment with PD98059, SP600125, and SB203580 inhibitors, the increased phosphorylation of Erk, JNK, and P38 and higher LOXL2 expression induced by GDNF was significantly lower. GFRα1 and RET are classic GDNF receptors expressed in astrocytes [42,43,44, 55]. To further clarify whether a high concentration of GDNF activates MAPK signaling to regulate LOXL2 expression via GFRα1/RET receptors, the GFRα1-neutralizing antibody and selpercatinib were used to block these receptors in RA cells. After inhibiting GFRα1 or RET, GDNF-induced phosphorylated Erk, phosphorylated P38, and LOXL2 protein were significantly decreased, but these treatments did not significantly affect GDNF-induced JNK activation, indicating that high concentrations of GDNF activate the Erk and P38 pathways via the GFRα1/RET receptor. Wu et al. reported that integrin β1/α5/JNK/c-JUN signaling increased LOXL2 expression in hepatocellular carcinoma cells [30]. GDNF can increase integrin β1 expression to activate downstream signaling to promote human pancreatic cancer cell invasion [56], suggesting that GDNF may activate JNK signaling through integrin β1 to increase LOXL2 expression, which is an issue we will examine in future studies. In addition, neutralizing GFRα1 or blocking RET with selpercatinib was sufficient to inhibit Erk and p38MAPK activation and LOXL2 expression in a GDNF-independent manner, suggesting that astrocyte-derived GDNF can activate Erk and p38MAPK signaling to regulate LOXL2 levels. Interestingly, selpercatinib reduced total Erk1/2 independently of the addition of GDNF. Jiang et al. reported that application of a RET inhibitor downregulated the expression and activation of c-Myc [57], which are involved in ERK expression regulation in GBM cells [58]. Collectively, the evidence suggests that selpercatinib may decrease the total Erk1/2 levels through inhibition of c-Myc activation. CREB is an important downstream nuclear transcription factor of the MAPK pathway [59]. It binds to the CRE domains in gene promoter regions to activate the expression of specific genes and plays an important role in tumor cell immortalization and transformation [60]. CREB expression and phosphorylation level in AG tissues and cells are significantly increased, and this can promote glioma cell proliferation and migration by increasing GDNF expression [32, 61]. We showed that CREB can bind to the potential CRE site on the Loxl2 promoter in RA cells, and high concentrations of GDNF increased CREB binding to this CRE. As a key modification for CREB binding to CRE, the phosphorylation of CREB at Ser133 was significantly increased by the Erk, JNK, and P38 signaling pathways after the treatment with high concentrations of GDNF, suggesting that high GDNF concentrations increase CREB binding to the Loxl2 promoter to upregulate LOXL2 expression. Additionally, pCREB may further increase GDNF expression to form a positive feedback loop to maintain the high expression level of LOXL2 in RA cells.
To further clarify whether high concentrations of GDNF promote astrocyte proliferation and migration via LOXL2, we tested the effects of GDNF on LOXL2i-RA cell proliferation and migration. The results showed that knocking down LOXL2 significantly inhibited GDNF-induced RA cell proliferation and migration, indicating that high concentrations of GDNF can promote astrocyte proliferation and migration via LOXL2. To explore the molecular mechanism by which GDNF induces LOXL2 to promote astrocyte proliferation and migration, we performed gene expression profiling in GDNF-treated NC and LOXL2i RA cells using microarray analyses. The results showed that 720 DEGs including Csf1, Ccl2, Mmp11, Tnn, Flrt2, and Itga1, which were mainly enriched in GO terms such as extracellular space, ECM, cell junction, cellular response to organic substance, cell adhesion, and molecular binding and signaling pathways such as ECM receptor interaction, focal adhesion, pathways in cancer, and cytokine receptor interaction. These results suggest that extracellular microenvironment remodeling may be involved in GDNF-induced astrocyte proliferation and migration via LOXL2.
Many studies have reported that secreted LOXL2 participates in ECM remodeling through its prototypical lysyl oxidase activity to enhance tumor cell proliferation and migration [26], and it also mediates crosstalk between tumor cells and fibroblasts to promote tumor progression [62]. We found that high-concentration GDNF increased the amount of LOXL2 secreted by RA cells; ECM-related or microenvironment-related genes were differentially expressed in LOXL2i RA cells after GDNF treatment, indicating that enhanced LOXL2 secretion promotes astrocyte proliferation and migration by changing its ECM or microenvironment. LOXL2 is distributed in the cytoplasm and nucleus, and high concentrations of GDNF increase LOXL2 protein levels in both cellular compartments. Recent studies demonstrated that non-secreted LOXL2 can regulate cytoskeletal reorganization, oxidation, and de-acetylation activities in the nucleus to promote tumor progression [26, 63]. In addition, high concentrations of GDNF promote LOXL2 entry into the nucleus, which can increase the expression of oncogenes such as Ccl2 and Gbp5. Ccl2 and GBP5 are important factors driving GBM malignancy [45,46,47]. It is speculated that high concentrations of GDNF may increase LOXL2 expression, upregulating the expression of Ccl2 and Gbp5 and other genes related to the extracellular microenvironment, which then promotes RA cell proliferation and migration.
In summary, this study provides the first evidence that high concentrations of GDNF promote abnormal astrocyte proliferation and migration via the GFRα1/RET/MAPK/pCREB/LOXL2 axis. Our results suggest that high concentrations of GDNF may be involved in the malignant transformation of astrocytes to AG cells. These findings provide new ideas for AG prevention and treatment. Our future study will focus on investigating how LOXL2 remodels the astrocyte microenvironment.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
References
Errico A (2014) CNS cancer: new options for glioblastoma. Nat Rev Clin Oncol 11(3):124. https://doi.org/10.1038/nrclinonc.2014.27
Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C et al (2019) Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 565(7738):240–245. https://doi.org/10.1038/s41586-018-0810-y
Zhang B, Han X, Gao Q, Liu J, Li S, Zha W et al (2021) Enhancer II-targeted dsRNA decreases GDNF expression via histone H3K9 trimethylation to inhibit glioblastoma progression. Brain Res Bull 167:22–32. https://doi.org/10.1016/j.brainresbull.2020.11.022
Fielder GC, Yang TW, Razdan M, Li Y, Lu J, Perry JK et al (2018) The GDNF family: a role in cancer? Neoplasia 20(1):99–117. https://doi.org/10.1016/j.neo.2017.10.010
Ayanlaja AA, Zhang B, Ji G, Gao Y, Wang J, Kanwore K et al (2018) The reversible effects of glial cell line-derived neurotrophic factor (GDNF) in the human brain. Semin Cancer Biol 53:212–222. https://doi.org/10.1016/j.semcancer.2018.07.005
Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F (1993) GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260(5111):1130–1132. https://doi.org/10.1126/science.8493557
Chermenina M, Schouten P, Nevalainen N, Johansson F, Oradd G, Stromberg I (2014) GDNF is important for striatal organization and maintenance of dopamine neurons grown in the presence of the striatum. Neuroscience 270:1–11. https://doi.org/10.1016/j.neuroscience.2014.04.008
Yu ZQ, Zhang BL, Ren QX, Wang JC, Yu RT, Qu DW et al (2013) Changes in transcriptional factor binding capacity resulting from promoter region methylation induce aberrantly high GDNF expression in human glioma. Mol Neurobiol 48(3):571–580. https://doi.org/10.1007/s12035-013-8443-5
Wiesenhofer B, Stockhammer G, Kostron H, Maier H, Hinterhuber H, Humpel C (2000) Glial cell line-derived neurotrophic factor (GDNF) and its receptor (GFR-alpha 1) are strongly expressed in human gliomas. Acta Neuropathol 99(2):131–137. https://doi.org/10.1007/pl00007416
Huang Y, Zhang B, Haneke H, Haage V, Lubas M, Yuan Y et al (2021) Glial cell line-derived neurotrophic factor increases matrix metallopeptidase 9 and 14 expression in microglia and promotes microglia-mediated glioma progression. J Neurosci Res 99(4):1048–1063. https://doi.org/10.1002/jnr.24768
Song H, Moon A (2006) Glial cell-derived neurotrophic factor (GDNF) promotes low-grade Hs683 glioma cell migration through JNK, ERK-1/2 and p38 MAPK signaling pathways. Neurosci Res 56(1):29–38. https://doi.org/10.1016/j.neures.2006.04.019
Shabtay-Orbach A, Amit M, Binenbaum Y, Na’ara S, Gil Z (2015) Paracrine regulation of glioma cells invasion by astrocytes is mediated by glial-derived neurotrophic factor. Int J Cancer 137(5):1012–1020. https://doi.org/10.1002/ijc.29380
Wiesenhofer B, Weis C, Humpel C (2000) Glial cell line-derived neurotrophic factor (GDNF) is a proliferation factor for rat C6 glioma cells: evidence from antisense experiments. Antisense Nucleic Acid Drug Dev 10(5):311–321. https://doi.org/10.1089/oli.1.2000.10.311
Duarte Azevedo M, Sander S, Tenenbaum L (2020) GDNF, A neuron-derived factor upregulated in glial cells during disease. J Clin Med 9 (2). https://doi.org/10.3390/jcm9020456
Schaar DG, Sieber BA, Sherwood AC, Dean D, Mendoza G, Ramakrishnan L et al (1994) Multiple astrocyte transcripts encode nigral trophic factors in rat and human. Exp Neurol 130(2):387–393. https://doi.org/10.1006/exnr.1994.1218
Saavedra A, Baltazar G, Santos P, Carvalho CM, Duarte EP (2006) Selective injury to dopaminergic neurons up-regulates GDNF in substantia nigra postnatal cell cultures: role of neuron-glia crosstalk. Neurobiol Dis 23(3):533–542. https://doi.org/10.1016/j.nbd.2006.04.008
Gao J, Kang XY, Sun S, Li L, Gao DS (2020) MES23.5 DA immortalized neuroblastoma cells self-protect against early injury by overexpressing glial cell-derived neurotrophic factor via Akt1/Eya1/Six2 signaling. J Molec Neurosci 70(3):328–339. https://doi.org/10.1007/s12031-019-01416-7
Nakajima H, Itakura M, Sato K, Nakamura S, Azuma YT, Takeuchi T (2017) Extracellular poly(ADP-ribose) is a neurotrophic signal that upregulates glial cell line-derived neurotrophic factor (GDNF) levels in vitro and in vivo. Biochem Biophys Res Commun 484(2):385–389. https://doi.org/10.1016/j.bbrc.2017.01.129
Bilak MM, Kuncl RW (2001) Delayed application of IGF-I and GDNF can rescue already injured postnatal motor neurons. NeuroReport 12(11):2531–2535. https://doi.org/10.1097/00001756-200108080-00048
Wang Y, Qin Y, Guo T, Tang C, Liu L, Gao D (2018) High concentration of glial cell line-derived neurotrophic factor protects primary astrocytes from apoptosis. Dev Neurosci 40(2):134–144. https://doi.org/10.1159/000487853
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. https://doi.org/10.1016/j.cell.2011.02.013
Xiong Y, Liu L, Zhu S, Zhang B, Qin Y, Yao R et al. (2017) Precursor N-cadherin mediates glial cell line-derived neurotrophic factor-promoted human malignant glioma. Oncotarget 8(15):24902–24914. https://doi.org/10.18632/oncotarget.15302
Qu DW, Liu Y, Wang L, Xiong Y, Zhang CL, Gao DS (2015) Glial cell line-derived neurotrophic factor promotes proliferation of neuroglioma cells by up-regulation of cyclins PCNA and Ki-67. Eur Rev Med Pharmacol Sci 19(11):2070–2075
Ferreira S, Saraiva N, Rijo P, Fernandes AS (2021) LOXL2 inhibitors and breast cancer progression. Antioxidants (Basel) 10 (2). https://doi.org/10.3390/antiox10020312
Shevchenko V, Arnotskaya N, Pak O, Sharma A, Sharma HS, Khotimchenko Y et al (2020) Molecular determinants of the interaction between glioblastoma CD133(+) cancer stem cells and the extracellular matrix. Int Rev Neurobiol 151:155–169. https://doi.org/10.1016/bs.irn.2020.03.005
Wen B, Xu LY (1874) Li EM (2020) LOXL2 in cancer: regulation, downstream effectors and novel roles. Biochim Biophys Acta Rev Cancer 2:188435. https://doi.org/10.1016/j.bbcan.2020.188435
Du XG, Zhu MJ (2018) Clinical relevance of lysyl oxidase-like 2 and functional mechanisms in glioma. Onco Targets Ther 11:2699–2708. https://doi.org/10.2147/OTT.S164056
Zhang Q, Yang L, Guan G, Cheng P, Cheng W, Wu A (2020) LOXL2 upregulation in gliomas drives tumorigenicity by activating autophagy to promote TMZ resistance and trigger EMT. Front Oncol 10:569584. https://doi.org/10.3389/fonc.2020.569584
Chang J, Nicolau MM, Cox TR, Wetterskog D, Martens JW, Barker HE et al (2013) LOXL2 induces aberrant acinar morphogenesis via ErbB2 signaling. Breast Cancer Res 15(4):R67. https://doi.org/10.1186/bcr3461
Wu S, Zheng Q, Xing X, Dong Y, Wang Y, You Y et al (2018) Matrix stiffness-upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre-metastatic niche formation. J Exp Clin Cancer Res 37(1):99. https://doi.org/10.1186/s13046-018-0761-z
Zhang BL, Ni HB, Liu J, Lei Y, Li H, Xiong Y et al (1839) (2014) Egr-1 participates in abnormally high gdnf gene transcription mediated by histone hyperacetylation in glioma cells. Biochem Biophys Acta 11:1161–1169. https://doi.org/10.1016/j.bbagrm.2014.08.014
Zhang B, Gu X, Han X, Gao Q, Liu J, Guo T et al (2020) Crosstalk between DNA methylation and histone acetylation triggers GDNF high transcription in glioblastoma cells. Clin Epigenetics 12(1):47. https://doi.org/10.1186/s13148-020-00835-3
Zhang BL, Dong FL, Guo TW, Gu XH, Huang LY, Gao DS (2017) MiRNAs mediate GDNF-induced proliferation and migration of glioma cells. Cell Physiol Biochem 44(5):1923–1938. https://doi.org/10.1159/000485883
Trapnell C, Pachter L, Salzberg SL (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25(9):1105–1111. https://doi.org/10.1093/bioinformatics/btp120
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–140. https://doi.org/10.1093/bioinformatics/btp616
Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16(5):284–287. https://doi.org/10.1089/omi.2011.0118
Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A et al (2017) Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 35(4):314–316. https://doi.org/10.1038/nbt.3772
Zhou N, Wang L, Fu P, Cui Z, Ge Y, Jiang F et al (2021) Conditioned medium-preconditioned EPCs enhanced the ability in oligovascular repair in cerebral ischemia neonatal rats. Stem Cell Res Ther 12(1):118. https://doi.org/10.1186/s13287-021-02157-4
Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD et al (2018) An integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality survival outcome analytics. Cell 173(2):400-416 e411. https://doi.org/10.1016/j.cell.2018.02.052
Villegas SN, Njaine B, Linden R, Carri NG (2006) Glial-derived neurotrophic factor (GDNF) prevents ethanol (EtOH) induced B92 glial cell death by both PI3K/AKT and MEK/ERK signaling pathways. Brain Res Bull 71(1–3):116–126. https://doi.org/10.1016/j.brainresbull.2006.08.014
Sawai H, Okada Y, Kazanjian K, Kim J, Hasan S, Hines OJ et al (2005) The G691S RET polymorphism increases glial cell line-derived neurotrophic factor-induced pancreatic cancer cell invasion by amplifying mitogen-activated protein kinase signaling. Can Res 65(24):11536–11544. https://doi.org/10.1158/0008-5472.CAN-05-2843
Remy S, Naveilhan P, Brachet P, Neveu I (2001) Differential regulation of GDNF, neurturin, and their receptors in primary cultures of rat glial cells. J Neurosci Res 64(3):242–251. https://doi.org/10.1002/jnr.1072
Ku MC, Wolf SA, Respondek D, Matyash V, Pohlmann A, Waiczies S et al (2013) GDNF mediates glioblastoma-induced microglia attraction but not astrogliosis. Acta Neuropathol 125(4):609–620. https://doi.org/10.1007/s00401-013-1079-8
Nakamura T, Ishizaka Y, Nagao M, Hara M, Ishikawa T (1994) Expression of the ret proto-oncogene product in human normal and neoplastic tissues of neural crest origin. J Pathol 172(3):255–260. https://doi.org/10.1002/path.1711720305
Yu X, Jin J, Zheng Y, Zhu H, Xu H, Ma J et al (2021) GBP5 drives malignancy of glioblastoma via the Src/ERK1/2/MMP3 pathway. Cell Death Dis 12(2):203. https://doi.org/10.1038/s41419-021-03492-3
Soria G, Ben-Baruch A (2008) The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett 267(2):271–285. https://doi.org/10.1016/j.canlet.2008.03.018
Yang C, Zhan H, Zhao Y, Wu Y, Li L, Wang H (2021) MEX3A contributes to development and progression of glioma through regulating cell proliferation and cell migration and targeting CCL2. Cell Death Dis 12(1):14. https://doi.org/10.1038/s41419-020-03307-x
Vachher M, Arora K, Burman A, Kumar B (2020) NAMPT, GRN, and SERPINE1 signature as predictor of disease progression and survival in gliomas. J Cell Biochem 121(4):3010–3023. https://doi.org/10.1002/jcb.29560
Xu H, Zhang A, Han X, Li Y, Zhang Z, Song L et al (2021) ITGB2 as a prognostic indicator and a predictive marker for immunotherapy in gliomas. Cancer Immunol Immunother. https://doi.org/10.1007/s00262-021-03022-2
Valkonen M, Haapasalo H, Rilla K, Tyynela-Korhonen K, Soini Y, Pasonen-Seppanen S (2018) Elevated expression of hyaluronan synthase 2 associates with decreased survival in diffusely infiltrating astrocytomas. BMC Cancer 18(1):664. https://doi.org/10.1186/s12885-018-4569-1
Smith-Mungo LI, Kagan HM (1998) Lysyl oxidase: properties, regulation and multiple functions in biology. Matrix Biol 16(7):387–398. https://doi.org/10.1016/s0945-053x(98)90012-9
Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M et al (2010) Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med 16(9):1009–1017. https://doi.org/10.1038/nm.2208
Lv GQ, Zou HY, Liao LD, Cao HH, Zeng FM, Wu BL et al (2014) Identification of a novel lysyl oxidase-like 2 alternative splicing isoform, LOXL2 Deltae13, in esophageal squamous cell carcinoma. Biochem Cell Biol 92(5):379–389. https://doi.org/10.1139/bcb-2014-0046
Brekhman V, Lugassie J, Zaffryar-Eilot S, Sabo E, Kessler O, Smith V et al (2011) Receptor activity modifying protein-3 mediates the protumorigenic activity of lysyl oxidase-like protein-2. FASEB J 25(1):55–65. https://doi.org/10.1096/fj.10-162677
Takasaki I, Takarada S, Fukuchi M, Yasuda M, Tsuda M, Tabuchi Y (2007) Identification of genetic networks involved in the cell growth arrest and differentiation of a rat astrocyte cell line RCG-12. J Cell Biochem 102(6):1472–1485. https://doi.org/10.1002/jcb.21369
Funahashi H, Takeyama H, Sawai H, Furuta A, Sato M, Okada Y et al (2003) Alteration of integrin expression by glial cell line-derived neurotrophic factor (GDNF) in human pancreatic cancer cells. Pancreas 27(2):190–196. https://doi.org/10.1097/00006676-200308000-00013
Jiang Y, Peng X, Ji Y, Dai Y, Fang Y, Xiong B et al (2021) The novel RET inhibitor SYHA1815 Inhibits RET-driven cancers and overcomes gatekeeper mutations by inducing G1 cell-cycle arrest through c-Myc downregulation. Mol Cancer Ther 20(11):2198–2206. https://doi.org/10.1158/1535-7163.MCT-21-0127
Velpula KK, Dasari VR, Tsung AJ, Dinh DH, Rao JS (2012) Transcriptional repression of Mad-Max complex by human umbilical cord blood stem cells downregulates extracellular signal-regulated kinase in glioblastoma. Stem Cells Dev 21(10):1779–1793. https://doi.org/10.1089/scd.2011.0424
Srinivasan S, Totiger T, Shi C, Castellanos J, Lamichhane P, Dosch AR et al (2018) Tobacco carcinogen-induced production of GM-CSF activates CREB to promote pancreatic cancer. Can Res 78(21):6146–6158. https://doi.org/10.1158/0008-5472.CAN-18-0579
Conkright MD, Montminy M (2005) CREB: the unindicted cancer co-conspirator. Trends Cell Biol 15(9):457–459. https://doi.org/10.1016/j.tcb.2005.07.007
Liu XF, Tang CX, Zhang L, Tong SY, Wang Y, Abdulrahman AA et al (2020) Down-regulated CUEDC2 increases GDNF expression by stabilizing CREB through reducing its ubiquitination in glioma. Neurochem Res 45(12):2915–2925. https://doi.org/10.1007/s11064-020-03140-w
Torres S, Garcia-Palmero I, Herrera M, Bartolome RA, Pena C, Fernandez-Acenero MJ et al (2015) LOXL2 is highly expressed in cancer-associated fibroblasts and associates to poor colon cancer survival. Clin Cancer Res 21(21):4892–4902. https://doi.org/10.1158/1078-0432.CCR-14-3096
Zhan XH, Jiao JW, Zhang HF, Xu XE, He JZ, Li RL et al (2019) LOXL2 Upregulates phosphorylation of ezrin to promote cytoskeletal reorganization and tumor cell invasion. Can Res 79(19):4951–4964. https://doi.org/10.1158/0008-5472.CAN-19-0860
Acknowledgements
We thank Zhonglin Li (the affiliated hospitals of Xuzhou Medical University, China) for providing clinical human glioma samples and Piniel Alphayo Kambey for revising this manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (nos. 81602464 to B. L. Zhang, and 81772688 to D. S. Gao), Six Talent Peaks in Jiangsu Province Jiangsu (SWYY-088 to B. L. Zhang), the 333 Project of Jiangsu Province (to B. L. Zhang), Qing Lan Project in Jiangsu Province (2017 to B. L. Zhang), Xuzhou Science and Technology Plan Project (KC20145 to B. L. Zhang), and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD to D. S. Gao). Special thanks to the staff of the Public Experimental Research Center of Xuzhou Medical University. We acknowledge the use of their laboratories to complete some of our research works.
Author information
Authors and Affiliations
Contributions
BLZ and DSG designed the study and oversaw the project. MMW, XH, WZ, XYW, LYL, ZML, YFS, XGK, GW, and BLZ performed the experiment and collected the data. MMW, XH, WZ, XYW, LYL, GW, and BLZ performed data analyses and produced the manuscript and the figures. All authors reviewed the manuscript and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics Approval
All experiments were carried out in compliance with the Provisions and General Recommendations of the Chinese Experimental Animal Administration Regulations, as well as institutional consent from the Xuzhou Medical University Animal Ethics Committee. Human GBM and NB tissue samples were obtained from the surgical specimen archives of Xuzhou Medical University’s affiliated hospitals with written informed consent from the patients.
Consent to Participate
Informed consent was obtained from all individual participants included in the study.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file2 (MP4 5307 KB)
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, M., Han, X., Zha, W. et al. GDNF Promotes Astrocyte Abnormal Proliferation and Migration Through the GFRα1/RET/MAPK/pCREB/LOXL2 Signaling Axis. Mol Neurobiol 59, 6321–6340 (2022). https://doi.org/10.1007/s12035-022-02978-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02978-1