
Abstract
Current evidence suggests that mild cerebrovascular changes could induce neurodegeneration and contribute to HIV-associated neurocognitive disease (HAND) in HIV patients. We investigated both the quantitative and qualitative impact of HIV infection on brain microvessels, especially on hippocampal microvessels, which are crucial for optimal O2 supply, and thus for maintaining memory and cognitive abilities. The results obtained using cultured human brain microvascular endothelial cells (HBMEC) were reproduced using a suitable mouse model and autopsied human HIV hippocampus. In HBMEC, we found significantly higher oxidative stress-dependent apoptotic cell loss following 5 h of treatment of GST-Tat (1 µg/ml) compared to GST (1 µg/ml) control. We noticed complete recovery of HBMEC cells after 24 h of GST-Tat treatment, due to temporal degradation or inactivation of GST-Tat. Interestingly, we found a sustained increase in mitochondrial oxidative DNA damage marker 8-OHdG, as well as an increase in hypoxia-inducible factor hypoxia-inducible factor-1α (HIF-1α). In our mouse studies, upon short-term injection of GST-Tat, we found the loss of small microvessels (mostly capillaries) and vascular endothelial growth factor (VEGF), but not large microvessels (arterioles and venules) in the hippocampus. In addition to capillary loss, in the post-mortem HIV-infected human hippocampus, we observed large microvessels with increased wall cells and perivascular tissue degeneration. Together, our data show a crucial role of Tat in inducing HIF-1α-dependent inhibition of mitochondrial transcriptional factor A (TFAM) and dilated perivascular space. Thus, our results further define the underlying molecular mechanism promoting mild cerebrovascular disease, neuropathy, and HAND pathogenesis in HIV patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
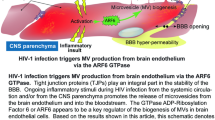
Neuropathy and mild HIV-associated neurological disorder (HAND) remain a problem that is frequently found in the brains of long-term-treated people living with HIV/AIDS (PLWHA) [1]. There is increased recognition that cardiovascular disease and stroke are becoming a complex health problem in PLWHA [2, 3]. Current data suggest that stroke may be a coincidence of long-term-treated PLWHA [3,4,5]. The incidence of cerebrovascular change is also increased in both age and young PLWHA [4, 6, 7]. Certain anti-HIV drugs can also result in vascular damage and confer additional pathogenesis [8, 9]. Mild cerebrovascular disease or cerebral small vascular disease (e.g., microinfarct, microbleed) in the brain was found to contribute more significantly to HAND than was previously believed [4]. However, the underlying mechanisms are not clearly defined [6].
HIV infection to the brain is associated with capillary rarefaction (reduced blood flow and oxygen delivery [10]. In general, cerebral rarefaction of capillaries and terminal arterioles and microvascular (MV) damage (blood–brain barrier [BBB] malfunction, arteriolar change, perivascular space dilation, etc.) result in reduced microcirculation and hypoxia, leading to microinfarcts in the brain, and are often attributed to pathologies such as neurodegeneration, hypertension, and aging [11,12,13,14]. Cerebral MV rarefaction and damage are also associated with aging and Alzheimer’s disease [15,16,17]. In addition, current data suggest that perivascular space dilation is another marker of increased risk for cognitive decline and dementia [18, 19].
The hippocampus, one of the most well-studied brain structures, is widely believed to be involved in learning and memory. Increasing evidence indicates that HIV-1-infected individuals with cognitive deficits show impairment of structure and function within the hippocampus as well as the circuitry between the hippocampus and prefrontal cortex, which plays a central role in cognitive control [20, 21]. Hippocampal microvascular dysfunction and hypoperfusion, e.g., by hypertension and aging, can induce hippocampal impairment and neurocognitive disorders [13, 22]. However, to our knowledge, the effect of HIV on hippocampal microvessels and function has never been studied.
HIV-1 Tat (trans-activator of transcription) is a regulatory protein encoded by the tat gene that enhances viral transcription [23,24,25,26,27]. Tat is secreted into the extracellular micro-environment by HIV-infected cells [28,29,30,31]. In the circulation, Tat is suggested to act as a proto cytokine, modulating the functions of several cells including endothelial cells [32]. Circulating Tat alters BBB permeability in the capillaries [33, 34]. Tat exhibits a dual function regarding survival regulation, exhibiting either endothelial cell proliferation or apoptosis depending on the micro-environment conditions [32, 35,36,37].
Normal O2 level (normoxia) activates prolyl hydroxylases (PHD) that promote pVHL-dependent hypoxia-inducible factor-1α (HIF-1α) degradation [38, 39]. Under hypoxia, the activity of PHDs is inhibited by low O2 levels and HIF1-α is upregulated and stabilized [40,41,42]. Although HIF-1 is the major transcription factor responsible for the specific induction of genes in hypoxia, HIF-1 can also be activated by cytokines, hormones, and nitric oxide (NO) [43]. The HIF-1α forms a heterodimer with HIF-1β to function as a transcription factor resulting in the upregulation of VEGF and VEGFR2 [44]. Activation of VEGF induces the downstream tyrosine kinase signal pathway, Src/Akt, the serine/threonine kinase signal through protein kinase C (PKC), and extracellular signal-regulated kinase (ERK1/2) pathways. ERK1/2 subsequently phosphorylates and activates a coactivator P300/CREB binding protein (CBP), resulting in vascular cell proliferation [45, 46].
The embryonic lethal, abnormal vision, Drosophila (ELAV)–like or human antigen (Hu) family proteins can specifically bind with AU-rich (ARE) sequence in the 3′-untranslated region (3′-UTR) of mRNA. PKC activates the mRNA stabilizing protein, the Hu family proteins, which results in the nuclear export of mRNA and Hu to the cytoplasm [47,48,49]. HIF-1α, MnSOD, and VEGF mRNAs have multiple ARE sequences. The HuR protein binds and prevents HIF-1α, MnSOD, and VEGF mRNA degradation, thus enhancing protein synthesis in vascular endothelial cells and their proliferation [50,51,52,53,54,55].
The hippocampus is crucial for memory, executive function, and learning in the neurocognitive network. Due to continuous synaptic loss, the volume of the hippocampus reduces with age [56]. The deterioration of hippocampal function plays a crucial role during certain neurocognitive impairments. Insufficient oxygen supply is one of the factors that contributes to hippocampal atrophy, which further accelerates under certain pathologies, including upon HIV infection [57]. The brain microvascular network is comprised of capillaries, arterioles, and venules, all of which retain, although to a different extent, BBB properties [58]. The effect of HIV on the microvascular network and on the brain oxygen supply and function has not been clearly understood. Moreover, the loss of microvessels in the hippocampus due to HIV infection has never or rarely been studied. The present study, therefore, investigated the effect of HIV on micro vessels in the hippocampus.
Materials and Methods
Cell Culture
Human brain microvascular endothelial (HBMEC) cells (Neuromics, Edina, MN, USA or ScienCell Research Laboratories, Carlsbad, CA, USA) at subculture passage 2–5 were grown on fibronectin-coated glass slides or wells plated. Cultured cells were maintained in endothelial medium (ScienCell) supplemented with the endothelial cell growth supplement (ECGS, ScienCell) and 5% fetal calf serum. After subculture, the cultured medium was used at < 0.5 cm above the apical surface of cultured cells without fresh culture medium change to allow cultured cells to initiate proliferation and subsequently activate HBMEC cell mature (single layer of squamous cells) because of the depletion of ECGS and fetal calf serum. Whenever necessary, a culture medium was added to cultured cells in small amounts each time only to prevent cell death (due to the low level of essential nutrients and growth factor) but not to keep the cells in the proliferation phase (multiple layers of spindle-shaped cells).
Measurement of Superoxide in Cultured Endothelial Cells
The specific probe hydroethidine (Thermo Fisher Scientific, Grand Island, NY, USA) was used to measure changes in cellular superoxide (O2•−) production[59]. The reaction between O2•− and non-fluorescent hydroethidine generates a highly specific red fluorescent product 2-hydroxyethidium. In biological systems, another red fluorescent product ethidium is also formed, usually at a much higher concentration than 2-hydroxyethidium[60]. Cultured cells, grown on glass slides (4 independent experiments per group), were incubated with hydroethidine (2 µg/ml) at room temperature for 15–20 min and then fixed with 4% paraformaldehyde. Increased O2•− production was quantified at 488 nm/ > 510 nm (excitation/emission). For confocal microscopy studies, the cultured cells were fixed with 4% paraformaldehyde at room temperature for 15–20 min and stored in phosphate buffer saline at 4
Cell Proliferation Assay
Cells were seeded at a concentration of 1 × 104 or 1 × 105 cells per well in 96-well plates with a total media volume of 100 µl. Cells were treated with different doses of GST or GST-Tat for 72 h. Later, 20 µl per well of CellTiter 96® Aqueous One Solution Reagent was added to the plate and incubated at 37 °C for 4 h in a humidified, 5% CO2 incubator. Absorbance was recorded at 490 nm using a 96-well plate reader.
PKCε Knockdown in Cultured Human Neurons
PKCε knockdown was performed using 100 nM of three target-specific 19–25 nucleotide PKCε si-RNA constructs (sc-36251), purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). For negative control, a scrambled si-RNA (sc-37007) was used. Transfection was performed using Lipofectamine RNAimax, per the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA). After 6 h, the medium was changed, and fresh media was added. PKC downregulation was assessed 72 h after transfection [59, 61].
Western Blot Analysis
Cells were harvested in homogenizing buffer (HB) containing 10 mM Tris–Cl (pH 7.4), 1 mM PMSF (phenylmethylsulfonylfluoride), 1 mM EGTA, 1 mM EDTA, 50 mM NaF, and 20 µM leupeptin and were lysed by sonication. The homogenate was centrifuged at 100,000 Å ~ g for 15 min at 4 °C to obtain the cytosolic fraction (soluble) and membrane (particulate). The pellet was resuspended in the HB by sonication. HB used for whole-cell protein isolation from cultured cells containing 1% Triton X-100. Protein concentration was measured using the Bradford Protein Assay kit (Pierce, Rockford, IL, USA). Following quantification, 20 µg of protein from each sample was subjected to SDS-PAGE analysis in a 4–20% gradient Tris–glycine polyacrylamide gel (Invitrogen, Carlsbad, CA, USA). The separated protein was then transferred to a nitrocellulose membrane. The membrane was blocked with 3% BSA and incubated with primary antibodies overnight at 4 °C. Primary antibodies were anti-HIF-1α (rabbit polyclonal IgG; 1:250; Novus Biological, Centennial, CO); anti-TFAM (rabbit polyclonal IgG, 1:500; Invitrogen, Carlsbad, CA); anti-VEGF (rabbit polyclonal IgG; 1:500; Invitrogen); anti-PKCε (rabbit polyclonal IgG; 1:100; Novus Biological); anti-MnSOD (rabbit polyclonal IgG; 1:4,000; MilliporeSigma); and anti-β-Actin IgG (mouse monoclonal IgG; 1:25,000; MilliporeSigma or rabbit polyclonal IgG; 1:25,000; Novus Biological). After incubation, the blot was washed 3 × with TBS-T (Tris-buffered saline-0.1% Tween 20) and further incubated with alkaline phosphatase-conjugated secondary antibody at 1:10,000 dilution for 45 min. The membrane was finally washed 3 × with TBS-T and developed using the 1-step NBTBCIP substrate (Pierce, Rockford, IL, USA). The blot was imaged in ImageQuant RTECL (GE Life Sciences, Piscataway, NJ), and densitometric quantification was performed using ImageJ software. For quantifying the expression of a protein, the densitometric value for the protein of interest was normalized against β-actin (loading control). The results were reproduced at least three times.
Generation of VSV-G Pseudotyped Lentiviral Vector Generation and Infection
Human Embryonic Kidney 293 cells (HEK 293 or 293 T) were cultured with Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 4.5 g/l glucose and L-glutamine (Lonza, Walkersville, MD, USA); 10% fetal bovine serum (Gibco, Waltham, MA, USA); and 1 U/mL penicillin/streptavidin (Gibco, Waltham, MA, USA). Cells were grown to 70–80% confluency and washed with Opti-MEMs GlutaMAX reduced serum media (Gibco, Waltham, MA, USA) before transfection. Transfection with Lipofectamine 3000 (Invitrogen, Waltham, MA, USA) was carried out according to the manufacturer’s protocol. Briefly, 35 µl of Lipofectamine 3000 reagent was diluted in 500 µl Opti-MEM. In a separate tube, 18 µg of plasmid DNA mixture (4 µg pMD.G, 3 µg pCMVΔ8.9.1, 3 µg pMDL-g/p-RRE, 1 µg pRSV-Rev, and 7 µg of either pHR’P-H13LTat or pHR’P-WtTat, to generate pseudotyped HIV viruses either expressing mutated H13L Tat or Wild Tat, respectively) and 35 µl of P3000 reagent were diluted in 500 µl Opti-MEM. The two separated dilutions were mixed and allowed to incubate at room temperature for 10 min to form the lipid-DNA complex, which was then added to the cells. Three to 5 h following the addition of the transfection cocktail, a DMEM culture medium was added to the cells. The virus-containing cell supernatant was collected at 48 and 72 h post-transfection. For infecting the HBMEC cell with lentivirus (4 independent experiments per group), the cells were incubated with supernatant for 1 h, and later the same volume of cell culture medium was added to the cells. The infectivity was confirmed after 48 h by performing western for Tat expression.
Animal Brain Tissue Preparation
Nine mice were deeply anesthetized with sodium pentobarbital (120 mg/kg body weight, i.p.). Animals were perfused through the heart with cold phosphate-buffered saline (PBS) for less than 4 min to wash out the blood and subsequently with 4% paraformaldehyde in PBS. Brains were then removed, postfixed for 20 min, and stored in PBS at 4 °C. The hippocampi were sectioned with a cryostat, and 4 hippocampal Sects. (10 µm thickness) were selected every 400–600 µm (for each mouse) and 600–900 µm (for each rat) and processed free-floating for immunohistochemistry and cytochemistry.
Human Brain Tissue Preparation
All patients (or relatives/representatives who had the power of attorney) signed informed consent forms. The present work was carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans [62]. Autopsied brains (4 control and 2 HIV subjects) were fixed in 10% formalin. Human hippocampal samples were processed for paraffin embedding. Paraffin-embedded Sects. (10 µm) were mounted on glass slides and deparaffinized with xylene (2 times; 5 min each) and 100% ethanol for 5 min. Tissue sections were then rehydrated with graded alcohol. For antigen retrieval of human autopsies, deparaffinized hippocampal sections were incubated in 10 mM citrate buffer pH 6.0 added with 0.05% Tween at 95 °C for 30 min.
Immunohistochemistry, Cytochemistry, and Confocal Microscopy
Tissue specimens were treated with PBS containing 5% normal goat serum and 0.5% Triton X-100 for 30 min at room temperature to block non-specific protein binding sites. Tissue samples were then incubated with primary antibodies diluted in PBS containing 2.5% normal goat serum and 0.5% Triton X-100 overnight at 4 °C, but human tissue was incubated at 4 °C for 2–3 days and at room temperature for 24 h. Primary antibodies were anti-8-OHdG (mouse monoclonal IgG; 1:100; Genox, Torrance, CA); anti-cleaved caspase 3 (rabbit polyclonal IgG; 1:100; Cell Signaling Tech, Danvers, MA); anti-HIF-1α (rabbit polyclonal IgG; 1:500; Novus Biological); anti-TFAM (rabbit polyclonal IgG, 1:500; Invitrogen); anti-PKCε (rabbit polyclonal IgG; 1:100; MilliporeSigma); anti-MnSOD (rabbit polyclonal IgG; 1:100; MilliporeSigma); anti-VEGF (rabbit polyclonal IgG; 1:50; MilliporeSigma); anti-ERK1/2 (mouse monoclonal IgG; 1:500; Invitrogen); anti-HuR (mouse monoclonal IgG; 1:500; Invitrogen); and/or anti-OGG1 (rabbit polyclonal IgG; 1:250; Protein Tech, Rosemont, IL). Specimens were then incubated with Alexa Fluor 568 horse anti-mouse or Alexa 488 anti-rabbit (1:1,000; Thermo Fisher Scientific) in PBS + 0.5% Triton X-100 for 1 h at room temperature. For blood vessel staining, rodent tissue sections were incubated with DyLight fluor 594-conjugated Lycopersicon esculentum (tomato) lectin (Vector Laboratories, Burlingame, CA, 1:50) at room temperature for 3 h. After each lectin and/or antibody incubation, the samples were washed with PBS + 0.5% Triton X-100 3 times (each time for a period of 5 min). Sections were mounted with VECTASHIELD mounting medium containing DAPI (Vector Laboratories) or with Polong Glass antifade mountant with NucBlue stain (Thermo Fisher Scientific). For negative controls, primary or secondary antibody was replaced with protein with no antigenicity against the tested antibody.
Hippocampal slices were oriented with a Zeiss Axio Observer Z1 microscope equipped with a 710 confocal scanning system using the 10 × objective lens in the DAPI channel (for staining nuclei). The random CA1 area that appeared immediately after switching to the higher magnification lens, either 63X or 100 × Plan-APO Chromat oil immersion objectives (1.4 NA), was imaged for appropriate fluorescence (e.g., Alexa 488 and/or 568). Confocal images of hippocampal sections were acquired in line scan mode with a pinhole of approximately 1.00 Airy unit, and averaged data from several images were reported. At least 5 images were randomly collected from each sample and were quantified with the ImageJ program (http://rsb.info.nih.gov/ij/). For fluorescence intensity, data were normalized to DAPI. Control data were set at 100%, and all other experiment data were defined as % of their controls.
Statistical Analyses
Data with two experimental groups were analyzed with Student’s t-test. For experiments with three or more groups, data with a significant overall difference among the groups as demonstrated by ANOVA were further analyzed by Tukey’s multiple comparisons. The confidence level was set at 95% (α = 0.05).
Results
Tat Enhances HBMEC Cell Death by Stimulating Superoxide Production
The HBMEC cells, at 95% confluence, were treated with either glutathione S-transferase (GST) or GST-Tat. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) and counted from the confocal micrographs (Fig. 1A). The time-dependent study of GST or GST-Tat at 1 µg/ml (Fig. 1B) showed that while GST had no effect, GST-Tat induced an early decrease in cell density at 2.5 (p < 0.05) and 5 (p < 0.001) h. However, HBMEC cell density was recovered after 24 h once GST-Tat became inactive due to either degradation or oxidization. Accordingly, after 72 h of GST-Tat treatment, we observed over-proliferation of HBMEC cells (Fig. 1B). Interestingly, the treatment of GST-Tat for 72 h induced an over-proliferation of HBMEC cells compared to GST treatment (p < 0.001), indicating the triggering of a compensatory mechanism to revive the system. However, dose-dependent effects of Tat on cell density and proliferation were not proportional. At 72 h, we noted profound over-proliferation of HBMEC cells at 1 µg/ml, but relatively less at 4 µg/ml and 8 µg/ml (Fig. 1C, D). These results suggest a significant depletion of cells due to higher Tat amount and resultant slower recovery. Fluorescence imaging of ethidium was used to determine O2•− levels (Fig. 1D). Intracellular O2•− level was gradually increased (p < 0.001) at 2.5 and 5 h and decreased at 24 h (p < 0.001, compared to at 5 h) but still higher (p < 0.01) than the unstimulated control at 0 h (Fig. 1E). Both GST and GST-Tat at 1 µg/ml were therefore used in later experiments.

Tat induces early strong oxidative stress and cell loss, followed by a gradual decrease in oxidative stress and over-proliferation of human brain microvascular endothelial cell (HBMEC). Cultured cells were treated with GST or GST-Tat both at 0, 1, 4, or 8 µg/ml before harvested at 4 or 24 or 72 h. A For cell density, nuclei were stained with DAPI, imaged with confocal microscopy, and counted in 100 µm2 areas. B Time-dependent effect of GST and GST-tat, both at 1 µg/ml, on cell density. Concentration-dependent effect of GST-Tat on C cell density and D cell proliferation assay after 72-h treatment. E To evaluate oxidative stress, cells were loaded with non-fluorescent dihydroethidium (2 µg/ml for 10 min), which was oxidized with superoxide (O2•−) to fluorescent oxyethidium and imaged with a confocal fluorescence microscope. F A slow increase in O2•− was maximal at 5 h before recovery. Data are % of according to control cells that were treated parallel to treated cells without GST or GST-Tat for 24 or 48 h (sham treatment) and represented at 100% at 0 h. Data are mean ± SE (n = 22–30 random areas or n = 79–216 random cells from 3–6 independent experiments per group). Asterisks indicate significant differences compared with controls. Asterisks over the lines indicate significant differences between groups. *, p < .05; **, p < .01; ***, p < .001; ANOVA and Tukey multiple comparison
Tat Stimulates HBMEC Apoptosis by Inducing Oxidative Stress and DNA Damage
The interaction of hydroxyl radical (HO), the most toxic reactive oxygen species (ROS), with the nucleoside, such as deoxyguanosine, leads to the formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG). The 8-OHdG undergoes keto-enol tautomerism, which favors the oxidized product 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG). In the scientific literature, 8-OHdG and 8-oxodG are used for the same compound [63]. Using double immunohistochemistry of the oxidative DNA damage marker 8-OHdG and the mitochondria-specific protein mitofilin (Fig. 2A), GST-Tat increased 8-OHdG in the mitochondria at 5 h (p < 0.05) and 24 h (p < 0.001) and in nuclei only at 5 h (p < 0.001) (Fig. 2B, C), suggesting local oxidative stress within mitochondria that are the main source of O2•− is sustained up to 24 h. The result further supports that the Tat-dependent increase in O2•− level is sufficient to induce strong oxidative stress and damage mitochondrial and nuclear DNAs.

Tat induces early apoptosis associated with oxidative DNA damage in mitochondria and nuclei of HBMEC cells. Cells cultured (> 95% confluence) were treated with GST or GST-Tat at 1 µg/ml in the presence or absence of the superoxide scavenger Nac (3 mM with 15 h pretreated before incubation together with GST-Tat) or the membrane-permeable superoxide dismutase mimetic MnTMPyP (25 µM with 45 min pretreatment) compared to untreated control cells. A Immunohistochemistry of the oxidative DNA damage marker 8-OHdG in B mitochondria, colocalized with the mitochondria marker mitofilin, and C nuclei. N, nucleus. D, E Immunohistochemistry of the apoptotic marker cleaved caspase 3. F, G Western blot analysis of the apoptotic marker cleaved caspase 3, the active fragments of caspase 3. H HBMEC cell density after 72-h treatment. Nac and/or MnTMPyP inhibited an increase oxidative DNA damage and apoptosis at 5 h and prevented the over-proliferation of HBMEC cells at 72 h after the GST-Tat treatment. B, C, E, G Data are % of according control cells that were treated parallel to treated cells without GST or GST-Tat for 5 or 24 h (sham treatment) and represented at 100% at 0 h. Data are mean ± SE; asterisks indicate significant differences compared with controls, and asterisks over the lines indicate significant differences between groups. *, p < .05; **, p < .01; ***, p < .001; ANOVA and Tukey multiple comparison (n = 79–216 random cells or 30 random areas from 3–4 independent experiments per group) or paired T-test (n = 3 independent experiments per group for western blot)
Caspases, or cysteine-dependent aspartate specific proteases, are a family of enzymes crucial for initiating and executing apoptosis within the cell. In endothelial cells during ischemic stroke, caspase 3 (32 kDa molecular weight) is cleaved into several small fragments and thus activated upon the initiation of apoptosis [64]. Western blot and/or immunohistochemistry of the apoptosis marker cleaved caspase 3 was used with an antibody that selectively detects cleaved fragments of caspase 3 (10-21 kDa). Immunohistochemistry revealed the GST-tat-dependent increase (p < 0.001) in cleaved caspase 3 was seen in both cytoplasm and nuclei of HBMEC cells after GST-Tat treatment for 5 h (Fig. 2D, E). Western blot analysis showed that GST-Tat incubation increased (p < 0.01) large subunits of cleaved caspase 3 at 5 h (Fig. 2F, G) that returned to the basal level at 24 h (Fig. 2G).
Antioxidants Prevent the Tat-Dependent Apoptosis and Subsequent Compensatory Over-proliferation of Surviving HBMEC Cell
In order to validate the direct impact of Tat-induced superoxide in promoting HBMEC cell death/apoptosis, we assessed the impact of the superoxide scavenger N-acetyl cysteine (Nac) and a cell-permeable superoxide dismutase mimetic Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin (MnTMPyP). Using immunocytochemistry, we found that at 5 h after treatment, Nac prevented the increase in the oxidative DNA damage marker in the mitochondria (p < 0.05) and nucleus of an HBMEC cell (p < 0.001, Fig. 2B, C). Nac also protected the presence of apoptotic marker cleaved caspase 3 in both the cytoplasm and nucleus (p < 0.001, Fig. 2E). In Fig. 2G, western blot studies showed that at 5 h, the cleaved caspase 3 level in cells treated with GST-Tat in the presence of Nac or MnTMPyP for 5 h was not significantly different from that in the unstimulated cells. At 72 h after GST-Tat treatment, Nac and MnTMPyP prevented (p < 0.001) the Tat-dependent increase in cell density (Fig. 2H). Thus, the results confirm that Tat induces HBMEC cells apoptosis by promoting oxidative stress, which leads to DNA damage and cell loss. However, once Tat is degraded/oxidized, HBMEC cells recover and achieve cell proliferation rate.
Tat Increases Hypoxia-Inducible Factor-1α (HIF-1α) Stability that Inhibits Mitochondrial Transcription Factor A (TFAM) Expression
GST-Tat, but not GST, treatment for 24 h increased the total HIF-1α level in cultured HBMEC cells, determined by the western blots (Fig. 3A, B). Immunohistochemistry showed that HIF-1α increased in both cytoplasms (p < 0.001) and nuclei (p < 0.05) of HBMEC cells (Fig. 3C, D). We further studied the effect of Tat on the downstream HIF-1α cascade on mitochondrial transcription factor A (TFAM) that is inhibited by HIF-1α [42]. The western blot analysis showed that the total TFAM protein in GST-Tat-treated cell cultures was not statistically different from the GST-treated and control cell cultures (Fig. 3E, F). GST-Tat increased (p < 0.01) nuclear TFAM compared to GST (Fig. 3G, H). To study the effect of HIF-1α on TFAM, we used the PHD inhibitor dimethyloxalylglysine (DMOG) that increases the stability of HIF-1α [65]. Accordingly, we found rapid degradation of TFAM upon DMOG treatment in both nucleus and cytoplasm (p < 0.001; Fig. 3G, H). These data indicate that although a mild increase in HIF-1α has no effect on TFAM, the DMOG-activated increase in HIF-1α stability (presumably high HIF-1α level) decreases the TFAM expression in HBMEC cells.

Tat increases hypoxia-inducible factor-1α (HIF-1α) but decreases mitochondrial transcription factor A (TFAM) in surviving HBMEC cells. Cultured cells were treated with GST or GST-Tat both at 1 µg/ml for 24 h in the presence or absence of the PHD inhibitor dimethyloxalylglysine (DMOG, 0.3 mM with 4 h pretreatment), compared to untreated control. DMOG was used to increase the stability of HIF-1α (Heikal et al., 2018). A, B Western blot analysis and C, D immunohistochemistry imaged with confocal microscopy of HIF-1α. E, F Western blot analysis and G, H immunohistochemistry of TFAM. Data are mean ± SE; asterisks over bars indicate significant differences compared with controls, and asterisks over the lines indicate significant differences between groups. *, p < .05; ***, p < .001; ANOVA and Tukey multiple comparison (n = 72–195 cells from 3 independent experiments per group for immunohistochemistry) or paired T-test (n = 3 independent experiments per group for western blot)
Tat Activates VEGF and VEGF Receptor 2 (VEGFR-2) Related Proliferation of Surviving Cells
VEGF is involved in angiogenesis (the growth of existing blood vessels). Although VEGF binds to both VEGF receptors (VEGFR)-1 and -2, only VEGFR-2 appears to mediate almost all the known cellular responses to VEGF [66]. After 24 h of treatment, western blots showed that GST-Tat significantly increased VEGF (p < 0.05) compared to the GST-treated cells, although not different from the control cells (Fig. 4A, B). Immunohistochemistry (Fig. 4C) also demonstrated that GST-Tat increased (p < 0.05) cytoplasmic and nuclear VEGF (Fig. 4D). Cytoplasmic and nuclear VEGF in cells treated with both Tat and DMOG was lower (p < 0.001) than those activated with Tat alone (Fig. 4D). Taken together, data suggest that the proliferation of surviving cells after the Tat-induced apoptosis of HBMEC cells is associated with the upregulation of VEGF. The DMOG-dependent increase in HIF-1α stability, presumably resulting in higher HIF-1α, can decrease TFAM (Fig. 3G, H) and VEGF (Fig. 4C, D) expression.

Tat-dependent over-proliferation of surviving HBMEC cells associated with an increase in VEGF expression and inhibited by the VEGF receptor (VEGFR-2) blocker. Cultured cells were treated with GST or GST-Tat at 1 µg/ml in the presence or absence of the PHD inhibitor dimethyloxalylglysine (DMOG, 0.3 mM with 4 h pretreatment) or the VEGFR-2 blocker SU-1498 (20 µM with 1 h pretreatment), compared to untreated control. A, B Western blot analysis and C, D immunohistochemistry imaged with confocal microscopy of VEGF after 24-h treatment. E HBMEC cell density at 72 h after treatment. GST-Tat prevented a GST-dependent decrease in VEGF, indicating that Tat increased VEGF expression. The VEGFR-2 blocker SU-1498 abolished the Tat-dependent cell over-proliferation. Data are mean ± SE; asterisks over bars indicate significant differences as compared with controls, and asterisks over the lines indicate significant differences between groups. *, p < .05; **, p < .01; ***, p < .001; ANOVA and Tukey multiple comparison (n = 3 129–222 cells or 30 random areas from 4 independent experiments per group) or paired T-test (n = 3 independent experiments per group for western blot)
We then studied the physiological effect of VEGF on HBMEC cell density after the 72-h treatment of GST-Tat (Fig. 4E). The VEGFR-2 inhibitor SU-1498 significantly suppressed (p < 0.001) the Tat-dependent increase in HBMEC cell density (Fig. 4E). The results indicate that transient Tat treatment induces cell apoptosis; however, a compensatory mechanism takes over during prolonged Tat treatment and increases VEGF expression and cell proliferation through stimulation of the VEGFR2 receptor.
Tat Increases Protein Kinase C Epsilon (PKCε), Extracellular Signal–Regulated Kinase 1/2 (ERK1/2), and Cell Proliferation Pathway
Subsequently, we investigated Tat effect on the downstream VEGFR-2 pathway, involving phospholipase C (PLC)/diacylglycerol (DAG)/PKC/ERK1/2 and on cell proliferation [45, 46, 67]. We found a significant (p < 0.05) increase in PKCε levels with GST-Tat treatment after 24 h compared to the unstimulated control and GST-treated cells (Fig. 5A, B). In support of these findings, we also noted upregulation of ERK1/2 expression in both cytoplasm and nuclei (Fig. 5C, D). However, as expected upon PKCε knockdown (KD) by transfection of small interfering (si), RNA reduced (p < 0.001) ERK1/2 to the basal level in the cytoplasm (Fig. 5E). At 72 h after GST-Tat treatment, the over-proliferation of HBMEC cells was suppressed (p < 0.001) with PKCε KD or with the ERK inhibitor FR180204 (Fig. 5I). The results indicate that the Tat-dependent VEGF induction is associated with the stimulation of VEGFR-2 receptor molecular cascade, involving PKCε and ERK1/2 and resultant cell proliferation. Incubation of the ERK inhibitor FR180204 alone (without GST-Tat) increased the number of HBMEC cells. The result suggests that FR180204 inhibits cytosolic ERK1/2 that can potentiate the catalytic activity of some pro-apoptotic protein, such as dead associated protein (DAP) kinase under normal cell aging[68]. By contrast, under sudden apoptotic attack by Tat, FR180204 may inhibit the nuclear ERK1/2-dependent cell over-proliferation [68].

Tat-induced HBMEC cell overgrowth is associated with PKCε-dependent activation of ERK1/2 and the mRNA-stabilizing protein. Cultured cells were treated with GST or GST-Tat both at 1 µg/ml for 24 or 72 h in the presence or absence of the HuR blockers dihydrotanshinone-I (DHTS, 10 µM with 30 min pretreatment); CMLD-2 (35 µM with 30 min pretreatment); or the ERK inhibitor (5 µM with 30 min pretreatment), compared to untreated control. In other experiments, the effect of PKCε knockdown (KD) was studied in cells transfected with and without si-RNA for 5–6 h and cultured for 72 h (in the absence of GST and GST-Tat at all stages). A Western blot analysis showed that B Tat significantly increased PKCε after 24-h treatment. C Immunohistochemistry was used to demonstrate D an increase in ERK1/2 in the cytoplasm and nuclei after 24-h treatment and E an effect of PKCε KD on ERK1/2 expression. F Immunohistochemistry was used to study effect of G GST-Tat and H PKCε KD on the nuclear export of the mRNA-stabilizing protein HuR. I Effect of PKCε KD and the ERK1/2 inhibitor FR 180,204 and J effect of DHTS and CMLD-2 to interfere with the binding of HuR to the ARE-binding site on mRNA on Tat-dependent cell over-proliferation at 72 h, using DAPI staining. Data are mean ± SE; asterisks over bars indicate significant differences compared with controls, and asterisks over the lines indicate significant differences between groups. *, p < .05; **, p < .001; ***, p < .001; ANOVA and Tukey multiple comparison (n = 165–220 cells or 20–30 random areas from 3 independent experiments per group) or paired T-test (n = 3 independent experiments per group for western blot)
An Increase in PKCε-Dependent mRNA Stabilizing HuR Protein During HBMEC Cell Proliferation After Tat Treatment
PKC activates the mRNA stabilizing protein ELAV-like protein Hu, which can bind to AU-rich elements in 3′-untranslated regions of HIF-1α, MnSOD, and VEGF mRNAs and enhances their mRNA translation [52, 53, 55]. PKCε has also been shown to stimulate cardiovascular endothelial cell proliferation and angiogenesis [69, 70]. To investigate whether PKCε can activate HuR that can bind and chaperone the mRNA from the nucleus to the cytoplasm, immunohistochemistry was used to study the export of HuR from the nucleus to the cytoplasm (Fig. 5F). At 24 h of treatment, GST-Tat decreased (p < 0.001) the nuclear HuR level, indicating the nuclear export of HuR to the cytoplasm (Fig. 5F, G). PKCε KD reduced the basal PKCε-dependent nuclear export HuR, resulting in an increased nuclear HuR level (Fig. 5H). At 72-h incubation, the Tat-dependent over-proliferation of HBMEC cells was suppressed (p < 0.001) with dihydrotanshinone-I (DHTS) and CMLD-2 that interfered with the binding of HuR to the ARE-binding site on mRNA (Fig. 5J). The results suggest that Tat-induced PKCε activation plays a crucial role in promoting HuR-dependent over-proliferation of HBMEC cells.
An Increase in Mitochondrial MnSOD Expression in Proliferating HBMEC Cells After Tat Treatment
We noted that even after 24 h of GST-Tat treatment, the increase in the oxidative DNA damage marker 8-OHdG was sustained (p < 0.001) in mitochondria (Fig. 2B). To extend these findings, we treated the cells with GST-Tat, and after 24 h, immunohistochemistry (p < 0.001) and western blots (p < 0.01) revealed an increase in mitochondrial MnSOD (Fig. 6A–D). The results suggest the increase in mitochondrial MnSOD in response to the strong oxidative DNA damage in the mitochondria after 24-h Tat treatment, as shown in Fig. 2B, D.

Tat associated with an increase in mitochondrial MnSOD in survival HBMEC cells. Cells were treated with GST or GST-Tat both at 1 µg/ml for 24 h, compared to untreated controls. A, B Immunohistochemistry and C, D western blot analysis of mitochondrial MnSOD. Data are mean ± SE; *, p < .05; **, p < .01; ***, p < .001, ANOVA and Tukey multiple comparison (n = 62–97 cells from 3–4 independent experiments per group) or paired T-test (n = 3 independent experiments per group for western blot). Asterisk(s) over the data bar is/are compared with the control, and asterisks over a line are compared with those 2 data bars between the line
Infection of Lentivirus Carrying Tat Gene Induces Oxidative Stress and HBMEC Cell Loss
To further confirm our data, we endogenously expressed Tat protein in HBMEC cells using lentiviral vectors expressing either fully functional wild-type Tat or partially active mutated Tat (H13L). The according control cells were not infected with the virus, and results are presented as % of control (Fig. 7). Hydroethidine (Fig. 7A) was used to determine changes in O2•− production and cell density. No significant change was observed at 5 h after infection. At 72 h after infection, both WT Tat and mutated H13L Tat viruses increased (p < 0.05) intracellular O2•− level and decreased (p < 0.001) cell density (Fig. 7B, C). Immunohistochemistry (Fig. 7D) showed a temporary increase (p < 0.01) in 8-OHdG in the cytoplasm, indicating the oxidative DNA damage in the mitochondria, at 5 h after both WT and H13L Tat virus infection. Mitochondria can produce O2•− at both sites of the inner mitochondrial membrane into the mitochondrial matrix and release out of the mitochondria [71]. The results suggest that WT Tat and H13L Tat induce excessive intramitochondrial O2•− production that damages mitochondrial DNA without increased cytosolic oxidative stress outside the mitochondria at 5 h after infection. Continuous release of WT Tat or H13L Tat for 72 h results in cytosolic oxidative stress and decreases cell density, although HBMEC cells can prevent the mitochondrial oxidative DNA damage.

Viral expression of Tat induces oxidative stress and HBMEC cell loss. Cultured HBMEC cells were infected with lentivirus carrying either wild-type Tat or mutant (H13L) Tat gene, the uninfected cells were used as control. A Cultured cells were incubated with hydroethidium (2 µg/ml for 10 min) and imaged with a confocal fluorescence microscope to determine B O2•− level and C cell density. D, E Immunohistochemistry of the oxidative DNA damage marker 8-OHdG. Data (mean ± SE) were from 111 HBMEC cells infected with H13L Tat virus and 90 HBMEC cells infected with WT Tat virus as % of control HBMEC cells uninfected (108 cells) from 3 independent experiments per grouAsterisks indicate significant differences as compared with controls. Asterisks over the lines indicate significant differences between groups. *, p < .05; **, p < .01; ***, p < .001; ANOVA and Tukey multiple comparison
Tat Reduces VEGF and Capillaries in Mouse Hippocampus
Mice were injected peritoneally with GST or GST-Tat in normal saline (every day for 12 injections in total) compared to control injected with normal saline without GST and GST-Tat. Vascular endothelial were stained at room temperature for 3 h with the DyLight Fluor 594-conjugated Lycopersicon esculentum (tomato) lectin (Vector Laboratories, Burlingame, CA, 1:50). The area of tomato lectin–stained MV area in the random hippocampal CA1 stratum radiatum areas was not different among GST-Tat, GST, and unstimulated control mice (Fig. 8A, B). Because a capillary is 4 µm in diameter [72], microvessels were divided into 2 groups: small microvessels (< 4 µm diameter, presumably capillaries) and large microvessels (> 4 µm diameter, mostly arterioles and venules). The percentage of small and large microvessels in the total microvessels was calculated (Fig. 8C). GST-Tat reduced the number of small microvessels (p < 0.05) and their VEGF (p < 0.05) but relatively increased the number of large microvessels without any effect on the VEGF (p < 0.05, Fig. 8C–E). GST and GST-Tat had no effect on the number of MV wall cells or the perivascular space (Fig. 8F, G). The results suggest that although Tat does not change the area density of microvessels in the hippocampus, repeated injection of Tat induces the loss of capillaries or small microvessels but increases the number of large microvessels without morphological alteration.

Tat treatment associated with the loss of small diameter microvessels and VEGF in mouse hippocampus. Mice were injected peritoneally with GST or GST-Tat (0.12 µg/kg) in normal saline (5 times a week, 12 injections in total), compared to control injected with normal saline without GST and GST-Tat. A Vascular endothelia were stained at room temperature for 3 h with the DyLight fluor 594-conjugated Lycopersicon esculentum (tomato) lectin (Vector Laboratories, Burlingame, CA, 1:50). B The percentage of microvascular (MV) area per 134.95 × 134.95 µm.2. C % distribution of small or large microvessels in the overall microvessels. D Immunohistochemistry of E cytoplasmic VEGF in the small microvessels (< 4 µm diameter; mainly capillaries) and large microvessels (> 4 µm diameter; arterioles and venules). F The number of MV wall cells per 100 µm perimeter and G perivascular space/MV area ratio. Data (mean ± SE) from 21 control, 25 GST, and 17 Tat microvessels or 30 control, 47 GST, and 27 VEGF-stained cells from 3 mice per grouAsterisks over bars indicate significant differences compared with controls. *, p < .05; paired T-test
Capillary Loss and Change in Large Microvessels in Autopsied Human HIV Hippocampus
Morphological MV changes in the hippocampal CA1 stratum radiatum were studied in the HIV autopsies, compared to age- and sex-matched control (AC) autopsies (Fig. 9A). The microvessels were divided into 4 groups based on their sizes: < 10, 10–20, 20–40, and > 40 µm diameter. The capillary diameter in humans is less than 8–10 µm in diameter; the small microvessels (< 10 µm diameter) are mostly capillary [72]. The AC control hippocampi consisted of a high number of small microvessels rather than large microvessels, while the HIV hippocampi had a low number of small microvessels with a relatively high number of larger microvessels (Fig. 9B). An increase in the number of MV wall cells (p < 0.05) and enlargement of perivascular (Virchow-Robin) space (p < 0.001) were seen in the large, but not small, HIV microvessels (Fig. 9C, D).

A decrease in small microvessels but an increase in large microvessels with increased MV wall cells and perivascular space dilation in HIV hippocampal autopsies. A Confocal microscopy of microvessels was used to determine B % distribution of microvessels at different diameter groups in the total microvessels, C the number of MV wall cells per 100 µm perimeter, and E perivascular space/MV area ratio. Data (mean ± SE) from 192 control microvessels from 4 age/sex-matched control (AC) subjects and 117 HIV microvessels from 2 HIV sub. Asterisks over bars indicate significant differences as compared with controls. *, p < .05; **, p < .01; ***, p < .001; paired T-test
Mild Oxidative Stress and Capillary Loss Are Associated with a Potent Increase in HIF-1α-Dependent Inhibition of TFAM, MnSOD, and VEGF Expression in the HIV Hippocampus
Immunohistochemistry and confocal microscopy were used to study molecular cell signals in the MV wall cells correlated with MV morphology change of different MV diameter groups in the autopsied HIV hippocampi, compared with AC controls. In the capillaries or small microvessels (< 10 µm diameter), an increase in nuclear HIF-1α stability (p < 0.05, Fig. 10A–C) was associated with a reduction (p < 0.05) in cytoplasmic TFAM, a key activator of mitochondrial transcription as well as mitochondrial genome replication (p < 0.001, Fig. 10D, E). Cytoplasmic VEGF (p < 0.05, Fig. 10H–J) and its molecular target PKCε, but not ERK1/2, were decreased (p < 0.001, Fig. 11A–D). Mitochondrial MnSOD was decreased (p < 0.05, Fig. 10F, G) without a rise in the oxidative DNA damage marker 8-OHdG (Fig. 11E–G), suggesting mild oxidative stress. The results suggest that the capillary loss in the human HIV hippocampus (Fig. 9B) is associated with increased HIF-1α that suppresses the mitochondrial transcription TFAM, leading to a decrease in MnSOD, mild oxidative stress, and decreased VEGF and PKCε. This change is similar to the short-term effect of Tat injection in the mice as shown in Fig. 8D, E.

Hypoxia-inducible factor-1α (HIF-1α) is increased in small microvessels but decreased in large microvessels, while a decrease in mitochondrial transcriptional factor A and VEGF in small microvessels is inversely increased in large microvessels in HIV hippocampal autopsies. Immunohistochemistry of A–C cytoplasmic and nuclear HIF-1α, D, E cytoplasmic TFAM, F, G mitochondrial MnSOD, and H–J cytoplasmic and nuclear VEGF. Data (mean ± SE) from 62–205 MV cells from 4 age/sex-matched control (AC) and 196–260 MV cells from 2 HIV patients. Asterisks over bars indicate significant differences compared with controls. *, p < .05; **, p < .01; ***, p < .001; paired T-test

A decrease in PKCε in small microvessels but an increase in PKCε, ERK1/2, oxidative DNA damage marker 8-OHdG, and the oxidative DNA damage repair OGG1 in large microvessels in HIV hippocampal autopsies. Immunohistochemistry of A, B cytoplasmic PKCε, C, D nuclear ERK1/2, E–G the oxidative DNA damage marker 8-OHdG, and H–J the oxidative DNA repair OGG1. Data (mean ± SE) from 62–205 MV cells from 4 age/sex-matched control (AC) and 196–260 MV cells from 2 HIV patients. Asterisks over bars indicate significant differences compared with controls. *, p < .05; **, p < .01; ***, p < .001; paired T-test
Strong Oxidative Stress and Perivascular Degeneration in 10–20 Diameter Microvessels Are Associated with a Weak Increase in HIF-1α-Dependent Inhibition of TFAM in the HIV Hippocampus
An increase in the oxidative DNA damage marker 8-OHdG (p < 0.05, Fig. 11F, G) without a change in MnSOD (Fig. 10F, G) indicated strong oxidative stress in 10–20 µm microvessels. An increase in nuclear HIF-1α (p < 0.01, Fig. 10C) and a decrease in TFAM (p < 0.001, Fig. 10E) in 10–20 µm diameter microvessels were weaker than those in capillaries (< 10 µm diameter microvessels). An increase in nuclear VEGF was seen (p < 0.05, Fig. 10J). The results suggest that strong oxidative stress, a weak HIF-1α increase, and increased VEGF are associated with the beginning of perivascular space dilation in 10–20 diameter microvessels (Fig. 9D).
A Decline in Strong Oxidative Stress and HIF-1α-Dependent Inhibition of TFAM but a Rise of the VEGF Signal via PKCε, MnSOD, and ERK1/2 Are Associated with Increased MV Wall Cells in 20–40 and > 40 µm Diameter Microvessels in the HIV Hippocampus.
The DNA damage induced by ROS involves base modification, base loss, and DNA single-strand breaks, which are usually repaired by the base excision repair (BER) pathway. The human 8-oxoguanine glycosylase 1 (OGG1) is the primary enzyme in the BER pathway, responsible for the excision of 8-oxodG or 8-OHdG to prevent further lesion in the DNA[73, 74]). Change in OGG1 was also investigated. Compared to 10–20 µm diameter microvessels, the increased HIF-1α in the nucleus (p < 0.01, Fig. 10C) and VEGF in the cytoplasm and nucleus (p < 0.01, Fig. 10I, J) declined, while the decrease in cytoplasmic TFAM (p < 0.01, Fig. 10E) was slowly conversed in 20–40 µm and/or > 40 µm diameter microvessels. A gradual increase in cytoplasmic PKCε (p < 0.01, Fig. 11B); nuclear ERK1/2 (p < 0.01, Fig. 11D); mitochondrial MnSOD (p < 0.001, Fig. 10G); and cytoplasmic OGG1 (p < 0.001, Fig. 11I) is inversely correlated with a decline in the cytoplasmic and nuclear oxidative DNA damage marker 8-OHdG in 20–40 µm and/or > 40 µm diameter microvessels (Fig. 11G). The oxidative DNA repair OGG1 is increased (p < 0.05) in the nuclei of all MV groups (Fig. 11J). These data suggest that the transformation of microvessels from 20–40 to > 40 µm diameter is associated with the increase in the downstream VEGF signals involving PKCε, MnSOD, and ERK1/2. That may result in a sustained increase in MV wall cells (Fig. 9C), although oxidative stress is at a low or normal level.
Discussion
The present study supports the hypothesis that upon infection, HIV induces excessive chronic inflammation that can lead to oxidative stress, cell dysfunction, and cell death; however, later compensatory mechanisms become activated and promote recovery and even over-proliferation of endothelial cells [75]. Notably, this compensatory process quantitatively is quite efficient in generating new blood capillaries but qualitatively not that efficient. Therefore, the number of large microvessels increases, but lost smaller microvessels at 10 µm diameter (capillaries) and 10–20 µm diameter (presumably small arterioles) never fully recovered. Consequently, many capillaries among all vasculature in the brain and anatomical sites that are highly dependent on these capillaries for blood and O2 supply, such as the brain, are greatly impacted upon HIV infection. Hence, even after effective HIV control by ART, HIV patients succumb to neurocognitive impairments roughly 10 years earlier than uninfected counterparts [1]. Our results also confirm the dual role of Tat on either vascular endothelial apoptosis or proliferation regarding survival regulation [32, 35,36,37]. We demonstrated that GST-Tat injection induced the loss of VEGF and small microvessels (capillaries) in the mouse hippocampus. We further demonstrated in the human HIV hippocampus that the loss of small microvessels was associated with mild oxidative stress and a decrease in HIF-1α, VEGF, TFAM, and PKCε. In the 10–20 µm diameter microvessels with dilated perivascular space, a sustained increase in strong oxidative stress and HIF-1α is associated with an increase in VEGF. In the 20–40 and > 40 µm diameter microvessels, the oxidative stress, HIF-1α, and VEGF declined, whereas a gradual surge in PKCε, ERK1/2, and MnSOD was associated with an increase in the number of large microvessels with over-proliferation of MV wall cells.
The experiment in cultured cells, Tat protein–induced mitochondrial oxidative damage apoptosis, resulted in a decrease in cell density at 5 h after treatment. During the decay of Tat protein in the cultured medium, the compensatory system was active and even overshoots resulting in over-proliferation of HBMEC cells at 72 h. Continuous releasing of Tat protein in HBMEC infected with lentivirus carrying Tat gene induced mitochondrial oxidative damage at 5 h but mild oxidative stress and decreased HBMEC cell density at 72 h. In mice, repeated injection of Tat 12 times induced capillary loss in the CA1 hippocampus stratum radiatum without any change in large microvessel morphology. The change in capillaries and large microvessels in the HIV autopsy may reflect the effect of VEGF against the long-term HIV infection and/or long-term therapeutic effect in PLWHAs.
Our data may reflect pathogenesis of MV enlargement from capillaries to large microvessels. These microvessels have a morphology similar to microvascular dysplasia, which is atypical hyperplasia with an increase in the numbers of immature cells (with different size and morphology). An increase in local angiogenic factors (e.g., growth factors such as VEGF) and inflammation (cytokines and chemokines) can turn capillaries and/or small arterioles into microvascular dysplasia [76, 77].
The possibility of distinct pathogenesis in small microvessels (capillaries) and large microvessels (arterioles and/or venules) could not be ruled out. Hyperplastic arteriolosclerosis is mostly correlated to severe (malignant) hypertension with markedly thickened walls due to cell proliferation and infiltration of lymphocytes in the tunica intima and concentric ring of smooth muscles in the thickened tunica media. Unlike an increase in cells with the same size and shape in hyperplastic arteriolosclerosis, microvascular dysplasia is comprised of hyperplastic cells with different sizes and shapes. The present results may suggest that HIV induces capillary loss but enlarges normal arterioles to hyperplastic arteriolosclerosis.
The present study demonstrates that HIV may activate early aging in capillaries (< 10 µm diameter in humans). Vascular endothelial dysfunction and capillary loss are currently considered to be a primary phenotype of normal human aging and Alzheimer’s disease (AD) [13, 16, 78,79,80,81,82]. During the aging process, the activity of sirtuin1 (SIRT1) in the nucleus is reduced, which decreases pVHL levels and subsequently stabilizes HIF-1α. Activated HIF-1α reduces c-Myc that targets genes regulating processes required for cell growth including rRNA transcription and processing, ribosomal protein transcription and translation, and translation initiation [83]. The reduction of c-Myc activity may elicit the loss of VEGF and small microvessels [84, 85].
HuR protein prevents HIF-1α, MnSOD, and VEGF mRNA degradation and enhances HIF-1α, MnSOD, and VEGF protein synthesis in vascular endothelial cells [50,51,52,53,54,55]. The downstream VEGF molecular target PKCε was also decreased in < 10 and 10–20 µm diameter microvessels. It is likely that the HIV-induced inflammation activates HIF-1α [43]. HIF-1α subsequently reduces TFAM, which is required for replication, transcription, and maintenance of mitochondrial biogenesis. HIF-1α also inhibits PGC-1β activity, resulting in the downregulation of mitochondrial genes and MnSOD [42, 83]. Decreased PKCε expression during aging [86] may result in the reduction of the mRNA stabilizing protein Hu on SIRT1 and MnSOD expression [87, 88]. The decrease in MnSOD results in mitochondria-induced oxidative damage and mitochondrial dysfunction, leading to ROS production and inflicting further oxidative injury to the mitochondria and/or nucleus [89, 90].
The present study verifies that Tat activates apoptosis in HBMEC cells. This is in agreement with previous studies’ findings that Tat-induced apoptosis in HBMEC cells is involved in endoplasmic reticulum stress and mitochondrial dysfunction [40, 91]. Mitochondrial oxidative damage activates cytochrome C oxidase/caspase/apoptosis and a decrease in cell density [92, 93]. Under healthy condition, the direct cell-to-cell contact activates notch receptors 1 that suppresses VEGFR-2 activity [40]. Apoptotic cells reduce the cell-to-cell contact and notch receptors, resulting in an increase in VEGFR-2 sensitivity in surviving cells, over-proliferation of endothelial cells, exaggerated wound repair, vascular wall thickening (arteriolosclerosis), and endothelial dysregulation [92, 93]. Apoptosis can upregulate VEGF autocrine via Akt-dependent cell signal [44].
Increased HIF-1α stability in the nuclei of 10–20 µm diameter microvessels was associated with a rise of VEGF expression in 10–20 and 20–40 µm diameter microvessels (Figs. 10F and 11A, B). The HIF-1α forms a heterodimer with HIF-1β to function as a transcription factor resulting in an overshoot of VEGF [94] and subsequent increase in the number of 20–40 µm diameter microvessels with the over-proliferation of MV wall cells (Fig. 9A, C). The present study showed that an increase in the diameter of microvessels is correlated with the decline of HIF-1α-dependent inhibition of TFAM and a temporal increase in VEGF. During the proliferation of surviving HBMEC cells after 24-h treatment of Tat, the study showed that the HIF-1α activator DMOG decreased TFAM and VEGF (Fig. 3), indicating the gradual decline of HIF-1α and the recovery of HIF-1α inhibition of TFAM expression that regulated VEGF translation. Our results support the dual function of HIF-1α on VEGF expression: (1) high HIF-1α stabilization may inhibit the c-Myc-dependent VEGF expression rather than activate HIF-1α/HIF-1β-dependent VEGF expression, resulting in VEGF reduction, (2) weak HIF-1α stabilization may elicit HIF-1β-α/HIF-1-dependent VEGF expression rather than the c-Myc-dependent VEGF expression, leading to an increase in VEGF expression.
The present study demonstrates that the downstream molecular targets of VEGFR-2 receptors on PKCε, MnSOD, and ERK1/2 were active in the large > 20 µm diameter microvessels without a sustained increase in VEGF. The data suggest that VEGFR-2 receptors are hyperactive. Although TFAM expression is inhibited by HIF-1α in the small microvessels [42, 83], the TFAM is not different between HIV and control microvessels larger than 40 µm diameter, possibly due to the recovery of strong oxidative DNA damage and associated with an increase in MnSOD. The data are in agreement with cultured HBMEC cells that after 24 h of treatment, TFAM in GST-Tat-treated cells was not different from control cells. As shown in the HBMEC cells, it is likely that PKCε/HuR/MnSOD and PKCε/VEGF cascades enhance MV cell over-proliferation and enlargement, resulting in the decline of perivascular space/MV area ratio versus an increase in MV diameter (Fig. 9D).
Proper functioning of the brain is dependent on the integrity of the cerebral vasculature. Cardiovascular and cerebrovascular diseases increasingly afflict PLWHAs in the contemporary era of cART [3, 95, 96]. The degree of involvement of the heart and extracranial vessels and the consequent location of tissue pathology may vary considerably. In the present study, we demonstrated that HIV promotes the thickening of the arteriolar wall. Arteriolar wall thickening (arteriolosclerosis), characterized as a prominent feature of small vessel disease, is one of the first changes to occur during the natural history of cerebrovascular pathology [97].
Circulating gp120 and Tat alter BBB permeability in the capillaries [6, 33, 34, 98]. In the present study, we demonstrate that HIV increases hypoxia and HIF-α that inhibits TFAM expression in the capillaries (< 10 µm diameter) and the adjacent arterioles (10–20 µm diameter). This suggests that HIV inhibits brain functioning by restricting proper oxygen supply. Moreover, TFAM inhibition by HIV results in restricted replication and transcription of mitochondria DNA, besides distortion of the homeostatic balance of mitochondrial metabolism and biogenesis of MnSOD [99]. A decrease in MNSOD results in oxidative stress that degrades and activates autophagy of the tight junction protein zonula occludens-1 (ZO-1) and basal membrane in the BBB of microvessels [100, 101]. HIV also directly damages BBB pericytes [33]. These may result in the reduction of astrocyte-vascular coupling, retraction of astrocytic endfeet, and disruption of the neurovascular unit.
Previous studies showed that HIV-1 Tat exposure is sufficient to destabilize BBB integrity and to increase the presence of activated, phagocytic, perivascular macrophages, and microglia [34, 102]. This may result in perivascular space dilation around arterioles, as shown in the present study. The consequences of these microvascular changes and perivascular degeneration are lacunar infarcts, cortical and subcortical microinfarcts, microbleeds, and diffuse white matter disease, which involves oligodendrocytic malfunction, myelin loss, and axonal abnormalities [97].
18F-fluorodeoxyglucose positron emission tomography (18F -FDG-PET) and/or arterial spin-labeling MRI (ASL-MRI) are used to detect cerebral hypoperfusion and hypometabolism for several neurological diseases, including mild cognitive impairment, early-onset Alzheimer’s disease, and HAND [103,104,105]. Arterial spin-labeling MRI, with a cheaper cost than PET and does not involve radioactive, showed reasonable test characteristics in HIV-associated neurocognitive disorder (hypoperfusion of bilateral temporal, parietal, and occipital brain regions) from healthy controls and participants with mild cognitive impairment [105]. In general, thickening of the arteriolar wall (arteriolosclerosis) can reduce blood flow in the capillaries, resulting in hypoperfusion and then cortical and subcortical microinfarcts, which appear to be the most robust substrates of cognitive impairment [106,107,108]. Future research is required to prove whether the arteriolosclerosis, perivascular space dilation, and/or microinfarcts, determined by, e.g., high-resolution MRI of angiography, can be a marker for HAND.
The present study demonstrates that HIV can activate the pathogenesis of microvessels, including capillary loss, large MV wall change, and perivascular space dilation. Perivascular space enlargement is an emerging marker of cerebral small vessel disease that is associated with cognitive decline, depression, increased mortality, and disability in stroke patients [55, 109]. An increase in cells in the MV walls (microvascular dysplasia) and perivascular space dilation may induce BBB dysfunction. Aging and heart and blood vessel disease are the risks for thickening of cerebral atherosclerosis and arteriosclerosis, which cause focal ischemia and infarcts in the brain, which are related to dementia[77, 79, 110,111,112,113]. During the disease process in cases such as brain trauma, ischemia, or inflammation, microvascular dysplasia is greatly increased [76]. Addition of HIV and other risks for cerebrovascular damage may reduce cerebral blood flow, which can induce perivascular change and cerebrovascular disease (microinfarcts and infarcts) that correlates with global decline in cognitive performance and dementia [16, 112, 113], leading to ischemic stroke in HIV.
Data Availability
The datasets generated from this study is included in this manuscript and are also available from the corresponding author on reasonable request.
References
Clifford DBBM (2013) Ances, HIV-associated neurocognitive disorder. Lancet Infect Dis 13(11):976–986
Anand AR, Rachel GD (2018) Parthasarathy HIV proteins and endothelial dysfunction implications in cardiovascular disease. Front Cardiovasc Med 5:185
Barnes RP, Lacson JC, Bahrami H (2017) HIV infection and risk of cardiovascular diseases beyond coronary artery disease. Curr Atheroscler Rep 19(5):20
Cysique LABJ (2019) Brew, Vascular cognitive impairment and HIV-associated neurocognitive disorder: a new paradigm. J Neurovirol 25(5):710–721
Ovbiagele BA (2011) Nath, Increasing incidence of ischemic stroke in patients with HIV infection. Neurology 76(5):444–450
Di Napoli MIM (2011) Shah, Neuroinflammation and cerebrovascular disease in old age: a translational medicine perspective. J Aging Res 2011:857484
Masia M et al (2019) Evolving understanding of cardiovascular, cerebrovascular and peripheral arterial disease in people living with HIV and role of novel biomarkers A study of the Spanish CoRIS cohort, 2004–2015. PLoS One 14(4):e0215507
Dobbs MRJR (2009) Berger, Stroke in HIV infection and AIDS. Expert Rev Cardiovasc Ther 7(10):1263–1271
Sen S et al (2012) Recent developments regarding human immunodeficiency virus infection and stroke. Cerebrovasc Dis 33(3):209–218
Silva JN et al (2014) Chronic central nervous system expression of HIV-1 Tat leads to accelerated rarefaction of neocortical capillaries and loss of red blood cell velocity heterogeneity. Microcirculation 21(7):664–676
Antonios TF (2006) Microvascular rarefaction in hypertension–reversal or over-correction by treatment? Am J Hypertens 19(5):484–485
Boudier HA (1999) Arteriolar and capillary remodelling in hypertension. Drugs 58(Spec No 1):37–40
Brown WRCR (2011) Thore, Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol 37(1):56–74
Troen AM et al (2008) B-vitamin deficiency causes hyperhomocysteinemia and vascular cognitive impairment in mice. Proc Natl Acad Sci U S A 105(34):12474–12479
Attems JKA (2014) Jellinger, The overlap between vascular disease and Alzheimer’s disease–lessons from pathology. BMC Med 12:206
Arvanitakis Z et al (2011) Microinfarct pathology, dementia, and cognitive systems. Stroke 42(3):722–727
Hill LK et al (2020) Detection of cerebrovascular loss in the normal aging C57BL/6 mouse brain using in vivo contrast-enhanced magnetic resonance angiography. Front Aging Neurosci 12:585218
Paradise M et al (2021) Association of dilated perivascular spaces with cognitive decline and incident dementia. Neurology 96(11):e1501–e1511
Smeijer D, Ikram MK, Hilal S (2019) Enlarged perivascular spaces and dementia: a systematic review. J Alzheimers Dis 72(1):247–256
Castelo JM et al (2006) Altered hippocampal-prefrontal activation in HIV patients during episodic memory encoding. Neurology 66(11):1688–1695
Kelschenbach J et al (2019) Efficient expression of HIV in immunocompetent mouse brain reveals a novel nonneurotoxic viral function in hippocampal synaptodendritic injury and memory impairment. mBio 10:4
Johnson AC, Miller JE, Cipolla MJ (2020) Memory impairment in spontaneously hypertensive rats is associated with hippocampal hypoperfusion and hippocampal vascular dysfunction. J Cereb Blood Flow Metab 40(4):845–859
Hokello J et al (2021) Human immunodeficiency virus type-1 (HIV-1) transcriptional regulation, latency and therapy in the central nervous system. Vaccines (Basel) 9:11
Karn J (1999) Tackling Tat. J Mol Biol 293(2):235–254
Marzio G et al (1998) HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc Natl Acad Sci U S A 95(23):13519–13524
Peterlin BMDH (2006) Price, Controlling the elongation phase of transcription with P-TEFb. Mol Cell 23(3):297–305
Rice ACH (2003) Herrmann, Regulation of TAK/P-TEFb in CD4+ T lymphocytes and macrophages. Curr HIV Res 1(4):395–404
Mediouni S et al (2012) Antiretroviral therapy does not block the secretion of the human immunodeficiency virus tat protein. Infect Disord Drug Targets 12(1):81–86
Poggi A et al (2004) Migration of V delta 1 and V delta 2 T cells in response to CXCR3 and CXCR4 ligands in healthy donors and HIV-1-infected patients: competition by HIV-1 Tat. Blood 103(6):2205–2213
Silhol M et al (2002) Different mechanisms for cellular internalization of the HIV-1 Tat-derived cell penetrating peptide and recombinant proteins fused to Tat. Eur J Biochem 269(2):494–501
Tyagi M et al (2001) Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem 276(5):3254–3261
Barillari G et al (1993) The Tat protein of human immunodeficiency virus type 1, a growth factor for AIDS Kaposi sarcoma and cytokine-activated vascular cells, induces adhesion of the same cell types by using integrin receptors recognizing the RGD amino acid sequence. Proc Natl Acad Sci U S A 90(17):7941–7945
Bertrand L, Cho HJ, Toborek M (2019) Blood-brain barrier pericytes as a target for HIV-1 infection. Brain 142(3):502–511
Leibrand CR et al (2017) HIV-1 Tat disrupts blood-brain barrier integrity and increases phagocytic perivascular macrophages and microglia in the dorsal striatum of transgenic mice. Neurosci Lett 640:136–143
Albini A et al (1996) The angiogenesis induced by HIV-1 tat protein is mediated by the Flk-1/KDR receptor on vascular endothelial cells. Nat Med 2(12):1371–1375
Nyagol J et al (2008) HIV-1 Tat mimetic of VEGF correlates with increased microvessels density in AIDS-related diffuse large B-cell and Burkitt lymphomas. J Hematop 1(1):3–10
Rusnati MM (2002) Presta, HIV-1 Tat protein and endothelium: from protein/cell interaction to AIDS-associated pathologies. Angiogenesis 5(3):141–151
Sanchez-Alavez M et al (2000) HIV- and FIV-derived gp120 alter spatial memory, LTP, and sleep in rats. Neurobiol Dis 7(4):384–394
Tang H et al (2009) Curcumin improves spatial memory impairment induced by human immunodeficiency virus type 1 glycoprotein 120 V3 loop peptide in rats. Life Sci 85(1–2):1–10
Kim S, Lee M, Choi YK (2020) The role of a neurovascular signaling pathway involving hypoxia-inducible factor and notch in the function of the central nervous system. Biomol Ther (Seoul) 28(1):45–57
Wu LY, He YL, Zhu LL (2018) Possible role of PHD inhibitors as hypoxia-mimicking agents in the maintenance of neural stem cells’ self-renewal properties. Front Cell Dev Biol 6:169
Yeo EJ (2019) Hypoxia and aging. Exp Mol Med 51(6):1–15
Piret JP et al (2002) Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem Pharmacol 64(5–6):889–892
Ji Y et al (2014) Upregulated autocrine vascular endothelial growth factor (VEGF)/VEGF receptor-2 loop prevents apoptosis in haemangioma-derived endothelial cells. Br J Dermatol 170(1):78–86
Masoumi Moghaddam S et al (2012) Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev 31(1–2):143–162
Nicolau Y et al (2018) SiRNA silencing of VEGF, IGFs, and their receptors in human retinal microvascular endothelial cells. Am J Transl Res 10(7):1990–2003
Antic DJD (1998) Keene, Messenger ribonucleoprotein complexes containing human ELAV proteins: interactions with cytoskeleton and translational apparatus. J Cell Sci 111(Pt 2):183–197
Pascale A et al (2005) Neuronal ELAV proteins enhance mRNA stability by a PKCalpha-dependent pathway. Proc Natl Acad Sci U S A 102(34):12065–12070
Quattrone A et al (2001) Posttranscriptional regulation of gene expression in learning by the neuronal ELAV-like mRNA-stabilizing proteins. Proc Natl Acad Sci U S A 98(20):11668–11673
Arcondeguy T et al (2013) VEGF-A mRNA processing, stability and translation: a paradigm for intricate regulation of gene expression at the post-transcriptional level. Nucleic Acids Res 41(17):7997–8010
Chamboredon S et al (2011) Hypoxia-inducible factor-1alpha mRNA: a new target for destabilization by tristetraprolin in endothelial cells. Mol Biol Cell 22(18):3366–3378
Chaudhuri L et al (2012) Preferential selection of MnSOD transcripts in proliferating normal and cancer cells. Oncogene 31(10):1207–1216
King PH (2000) RNA-binding analyses of HuC and HuD with the VEGF and c-myc 3′-untranslated regions using a novel ELISA-based assay. Nucleic Acids Res 28(7):E20
Rahat MA, Bitterman H, Lahat N (2011) Molecular mechanisms regulating macrophage response to hypoxia. Front Immunol 2:45
Wang W et al (2001) Loss of HuR is linked to reduced expression of proliferative genes during replicative senescence. Mol Cell Biol 21(17):5889–5898
O’Shea A et al (2016) Cognitive aging and the hippocampus in older adults. Front Aging Neurosci 8:298
Cotto B, Natarajanseenivasan K, Langford D (2019) HIV-1 infection alters energy metabolism in the brain: contributions to HIV-associated neurocognitive disorders. Prog Neurobiol 181:101616
Wilhelm I et al (2016) Heterogeneity of the blood-brain barrier. Tissue Barriers 4(1):e1143544
Sen A et al (2016) Protein kinase C (PKC) promotes synaptogenesis through membrane accumulation of the postsynaptic density protein PSD-95. J Biol Chem 291(32):16462–16476
Zielonka JB (2010) Kalyanaraman, Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic Biol Med 48(8):983–1001
Sen A, Nelson TJ, Alkon DL (2015) ApoE4 and Aβ oligomers reduce BDNF expression via HDAC nuclear translocation. J Neurosci 35(19):7538–51
World Medical, A. (2013) World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 310(20):2191–2194
Kasai H (1997) Analysis of a form of oxidative DNA damage, 8-hydroxy-2′-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat Res 387(3):147–163
Zille M et al (2019) The impact of endothelial cell death in the brain and its role after stroke: A systematic review. Cell Stress 3(11):330–347
Heikal L et al (2018) Assessment of HIF-1alpha expression and release following endothelial injury in-vitro and in-vivo. Mol Med 24(1):22
Holmes K et al (2007) Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 19(10):2003–2012
Zhou Y et al (2013) Reactive oxygen species in vascular formation and development. Oxid Med Cell Longev 2013:374963
Mebratu YY (2009) Tesfaigzi, How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle 8(8):1168–1175
Amadio M et al (2012) Protein kinase C activation affects, via the mRNA-binding Hu-antigen R/ELAV protein, vascular endothelial growth factor expression in a pericytic/endothelial coculture model. Mol Vis 18:2153–2164
Rask-Madsen C, King GL (2008) Differential regulation of VEGF signaling by PKC-alpha and PKC-epsilon in endothelial cells. Arterioscler Thromb Vasc Biol 28(5):919–924
Muller FL, Liu Y, VanRemmen H (2004) Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279(47):49064–73
Liu G et al (2012) High-resolution imaging of microvasculature in human skin in-vivo with optical coherence tomography. Opt Express 20(7):7694–7705
Lillenes MS et al (2016) Altered DNA base excision repair profile in brain tissue and blood in Alzheimer’s disease. Mol Brain 9(1):61
Santos RX et al (2013) Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid Redox Signal 18(18):2444–2457
Dinh QN et al (2014) Roles of inflammation, oxidative stress, and vascular dysfunction in hypertension. Biomed Res Int 2014:406960
Fan YGY (2007) Yang, Therapeutic angiogenesis for brain ischemia: a brief review. J Neuroimmune Pharmacol 2(3):284–289
Su H et al (2008) Development of a cerebral microvascular dysplasia model in rodents. Acta Neurochir Suppl 105:185–189
Desai BS et al (2009) Evidence of angiogenic vessels in Alzheimer’s disease. J Neural Transm (Vienna) 116(5):587–597
Kemper T et al (1999) Microinfarction as a result of hypertension in a primate model of cerebrovascular disease. Acta Neuropathol 98(3):295–303
Riddle DR, Sonntag WE, Lichtenwalner RJ (2003) Microvascular plasticity in aging. Ageing Res Rev 2(2):149–68
Toda N (2012) Age-related changes in endothelial function and blood flow regulation. Pharmacol Ther 133(2):159–176
Ungvari Z et al (2018) Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol 15(9):555–565
Lu Z et al (2010) PGC-1 alpha regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal 13(7):1011–1022
Baudino TA et al (2002) c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 16(19):2530–2543
Florea V et al (2013) c-Myc is essential to prevent endothelial pro-inflammatory senescent phenotype. PLoS ONE 8(9):e73146
Hongpaisan J et al (2013) PKC activation during training restores mushroom spine synapses and memory in the aged rat. Neurobiol Dis 55:44–62
Otsuka H et al (2019) Emerging evidence of translational control by AU-rich element-binding proteins. Front Genet 10:332
Sen A et al (2018) Loss in PKC epsilon causes downregulation of MnSOD and BDNF expression in neurons of Alzheimer’s disease hippocampus. J Alzheimers Dis 63(3):1173–1189
Bereiter-Hahn J (2014) Do we age because we have mitochondria? Protoplasma 251(1):3–23
Lee HCYH (2012) Wei, Mitochondria and aging. Adv Exp Med Biol 942:311–327
Ma R et al (2016) HIV Tat-mediated induction of human brain microvascular endothelial cell apoptosis involves endoplasmic reticulum stress and mitochondrial dysfunction. Mol Neurobiol 53(1):132–142
Tian DY et al (2017) Notch signaling in endothelial cells: is it the therapeutic target for vascular neointimal hyperplasia? Int J Mol Sci 18:8
Zhu M et al (2014) L-cystathionine inhibits the mitochondria-mediated macrophage apoptosis induced by oxidized low density lipoprotein. Int J Mol Sci 15(12):23059–23073
Forsythe JA et al (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16(9):4604–4613
Moulignier A et al (2018) Silent cerebral small-vessel disease is twice as prevalent in middle-aged individuals with well-controlled, combination antiretroviral therapy-treated human immunodeficiency virus (HIV) than in HIV-uninfected individuals. Clin Infect Dis 66(11):1762–1769
Su T et al (2017) Cerebral blood flow and cognitive function in HIV-infected men with sustained suppressed viremia on combination antiretroviral therapy. AIDS 31(6):847–856
Kalaria RNY (2019) Hase, Neurovascular ageing and age-related diseases. Subcell Biochem 91:477–499
Cioni C (2002) Annunziata, Circulating gp120 alters the blood-brain barrier permeability in HIV-1 gp120 transgenic mice. Neurosci Lett 330(3):299–301
Picca AAM (2015) Lezza, Regulation of mitochondrial biogenesis through TFAM-mitochondrial DNA interactions: useful insights from aging and calorie restriction studies. Mitochondrion 25:67–75
Liao K et al (2020) HIV Tat-mediated induction of autophagy regulates the disruption of ZO-1 in brain endothelial cells. Tissue Barriers 8(2):1748983
Louboutin J, Strayer DS (2012) Blood-brain barrier abnormalities caused by HIV-1 gp120: mechanistic and therapeutic implications. ScientificWorldJournal 2012:482575
Pu H et al (2003) HIV-1 Tat protein upregulates inflammatory mediators and induces monocyte invasion into the brain. Mol Cell Neurosci 24(1):224–237
Dolui S et al (2020) Arterial spin labeling versus (18)F-FDG-PET to identify mild cognitive impairment. Neuroimage Clin 25:102146
Verclytte S et al (2016) Cerebral hypoperfusion and hypometabolism detected by arterial spin labeling MRI and FDG-PET in early-onset Alzheimer’s disease. J Neuroimaging 26(2):207–212
Narvid J et al (2018) Abnormal cerebral perfusion profile in older adults with HIV-associated neurocognitive disorder: discriminative power of arterial spin-labeling. AJNR Am J Neuroradiol 39(12):2211–2217
Arvanitakis Z et al (2017) The relationship of cerebral vessel pathology to brain microinfarcts. Brain Pathol 27(1):77–85
Kalaria RN (2016) Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol 131(5):659–685
Kalaria RN (2012) Cerebrovascular disease and mechanisms of cognitive impairment: evidence from clinicopathological studies in humans. Stroke 43(9):2526–2534
Wang X et al (2019) Brain grey-matter volume alteration in adult patients with bipolar disorder under different conditions: a voxel-based meta-analysis. J Psychiatry Neurosci 44(2):89–101
Banerjee G et al (2016) Novel imaging techniques in cerebral small vessel diseases and vascular cognitive impairment. Biochim Biophys Acta 1862(5):926–938
Kalaria RNC (1999) Ballard, Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord 13(Suppl 3):S115–S123
Kovari E et al (2013) The relationship between cerebral amyloid angiopathy and cortical microinfarcts in brain ageing and Alzheimer’s disease. Neuropathol Appl Neurobiol 39(5):498–509
Schneider JA (2009) High blood pressure and microinfarcts: a link between vascular risk factors, dementia, and clinical Alzheimer’s disease. J Am Geriatr Soc 57(11):2146–2147
Acknowledgements
Human tissue specimens were obtained from the NIH NeuroBioBank: University of Maryland Brain and Tissue Bank (A Brain and Tissue Bank Reposition of the NIH NeuroBioBank) and NIH Brain & Tissue Repository-California, Human Brain & Spinal Fluid Resource Center, VA West Los Angeles Medical Center, Los Angeles, California, which is supported in part by National Institutes of Health and the US Department of Veterans Affairs.
Funding
Research reported in this publication was funded by the National Institutes of Health, an R01, R01AG058884, from National Institute on Aging to JH and an R01, R01DA041746, National Institute on Drug Abuse to MT.
Author information
Authors and Affiliations
Contributions
A.L.S., H.W, Z.Z., G.M., and J.H. performed the experiments; J.H. and M.T. analyzed the data and wrote the first draft of the manuscript; J.H., A.L.S., and M.T. reviewed and edited the manuscript; J.H. and M.T. supervised the project; J.H. and M.T. acquired the funding. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethical Statement/Informed Consent
The present work was carried out in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. All patients (or relatives/representatives who had the power of attorney) signed informed consent forms.
Consent of Publication
Not applicable.
Consent to Participate
Not applicable.
Conflict of Interest.
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sharma, A.L., Wang, H., Zhang, Z. et al. HIV Promotes Neurocognitive Impairment by Damaging the Hippocampal Microvessels. Mol Neurobiol 59, 4966–4986 (2022). https://doi.org/10.1007/s12035-022-02890-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02890-8