
Abstract
Alzheimer’s disease (AD) is the most common late-onset dementia characterized by the deposition of extracellular amyloid plaques and formation of intracellular neurofibrillary tangles, which eventually lead to neuronal loss and cognitive deficits. Multiple lines of evidence indicate that mitochondrial dysfunction is involved in the initiation and progression of AD. As essential machinery for mitochondrial quality control, mitophagy plays a housekeeping role in neuronal cells by eliminating dysfunctional or excessive mitochondria. At present, mounting evidence support that the activity of mitophagy markedly declines in human brains during aging. Impaired mitophagy and mitochondrial dysfunction were causally linked to bioenergetic deficiency, oxidative stress, microglial activation, and chronic inflammation, thereby aggravating the Aβ and tau pathologies and leading to neuron loss in AD. This review summarizes recent evidence for age-associated mitophagy decline during human aging and provides an overview of mitochondrial dysfunction involved in the process of AD. It also discusses the underlying mechanisms through which defective mitophagy leads to neuronal cell death in AD. Therapeutic interventions aiming to restore mitophagy functions can be used as a strategy for ameliorating AD pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rapid aging of the population is accompanied by a rising prevalence of aging-related neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and frontotemporal dementia (FTD). Neurodegenerative diseases are generally characterized by synaptic loss and neuronal death, resulting in cognitive decline, dementia, and loss of motor function. Alzheimer’s disease (AD) is the most common cause of dementia. Hallmark pathological changes in AD include extensive neuronal loss, synaptic dysfunction, and accumulation of β-amyloid (Aβ) plaques and neurofibrillary tangles (NFTs) in the neocortex and hippocampus [1,2,3].
Knowledge of genetic susceptibilities and risk factors for AD has provided insight into its pathogenic pathways, which include mitochondrial dysfunction, disturbed intracellular protein quality control, and chronic inflammation. Mitochondrial dysfunction is a common pathological feature and contributes to the neurodegeneration in both transgenic animal models and human AD patients [4,5,6]. Mitophagy is a selective form of autophagy responsible for the removal of damaged mitochondria. In the past decade, the impairment of mitophagy has been proposed as a critical component of susceptibility to AD and received increasing attention [7,8,9]. In the human brain, mitophagy tends to decline with age, which inevitably disrupts the maintenance of mitochondrial homeostasis. Mitochondrial dysfunction not only exacerbates β-amyloid-associated AD pathologies, but also triggers a cascade of pathophysiological events, including oxidative stress, inflammatory response, and programmed cell death [4, 10]. In particular, microglia-mediated chronic inflammation can further aggravate neuronal damage and promote the development of AD.
In this review, we provide a concise overview of studies that support the causal linkage between aging-dependent mitophagy impairment and AD and offer insights into the underlying mechanisms for impaired mitophagy in the pathophysiology of AD. Given that dysregulation of mitophagy is causally involved in AD pathologies, it is likely that correcting neuronal mitochondrial dysfunction through regulating mitophagy could help to develop a promising therapeutic and preventive strategy for AD patients.
The Epidemiology of Alzheimer’s Disease and the Amyloid Cascade Hypothesis
Dementia can be defined as a clinical syndrome characterized by a cluster of symptoms and signs manifested by difficulties in memory and cognitive functions, changes in behaviors, and impairments in activities of daily living. According to the World Alzheimer Report 2015, 46.8 million people worldwide are living with dementia in 2015. This number will double every 20 years due to increases in population aging and growth, reaching 131.5 million in 2050 [11]. In 2016, dementia was the fifth leading cause of death globally, accounting for 2.4 million deaths [12]. It is estimated that among individual over 60 years of age dementia contribute 11.2% of years with disability, surpassing stroke, musculoskeletal disease, and cardiovascular disease [13].
AD is the most common cause of dementia in the elderly, accounting for up to 75% of all dementia cases. At present, the etiology of AD is not completely clarified, it is generally considered to be the combined effects of genetic and environmental factors. Mutations in key genes such as amyloid precursor protein (APP) or presenillin1/2 (PS1/2) have been shown to significantly increase Aβ production and impair its clearance in the neuronal cells, leading to early-onset AD [14]. Furthermore, the polymorphism in the apolipoprotein E (APOE) gene is a major risk factor for sporadic AD [15]. In addition, infections, drugs, and exposure to environmental toxicants can increase the risk of AD occurrence in the population. Pathologically, the most significant pathological changes in AD are the accumulation of β-amyloid (Aβ) plaques and neurofibrillary tangles (NFTs). A growing body of evidence supports that these pathogenic protein aggregates compromise the function and viability of neurons [16]. Aβ peptide is generated from the cleavage of amyloid precursor protein (APP) by secretases while NFTs comprised primarily of tau protein. The mutation in AD-related genes such as APP, presenilins, and APOE4 variant has effects on APP processing and Αβ aggregation [17]. In contrast, hyperphosphorylated forms of tau are responsible for their aggregation into paired helical filaments and subsequent accumulation into neurofibrillary tangles.
Since the discovery of amyloid plaques by Alois Alzheimer in the 1920s, the role of Aβ in the etiology of AD has always been the focus of AD research. The amyloid cascade hypothesis was first proposed by Hardy and Higgins [18]. According to this hypothesis, Aβ peptide deposition is the underlying mechanism leading to the pathogenesis of AD, and the occurrence of NFTs, neuronal cell loss, vascular damage, and dementia is a direct result of the deposits. Further research has found that the soluble oligomers have a higher neurotoxicity than monomers; injection of Aβ oligomers into rat brains can induce memory deficits [19]. Nonetheless, later studies using Aβ 42 overexpressing BRI2-Aβ mice showed that despite the presence of Aβ oligomers and Aβ amyloid fibrils, there was no impairment in cognitive function or degeneration of neurons [20, 21]. Meanwhile, vast deposits of Aβ in the brains of elderly non-AD patients were also observed, suggesting that Aβ deposition might not be specific to AD [22]. Furthermore, extensive clinical trials of drugs targeting Aβ have been carried out, including vaccines, drugs to promote Aβ clearance, and inhibitors of β-site amyloid precursor protein-cleaving enzyme 1 (BACE-1), γ-secretase and β-secretase. However, they did not prove clinically effective [23, 24]. In addition, clinical trials with Aβ monoclonal antibodies bapineuzumab, gantenerumab, solanezumab and crenezumab could not significantly improve the cognitive function of AD patients, although they might lower the level of oligomeric Aβ in the large majority of treated patients [25,26,27,28]. Therefore, although senile plaques and neurofibrillary tangles are the dominant neuropathological findings in AD, their relative importance in the pathogenesis of AD has been the subject of debate.
The Role of Mitochondrial Damage in Pathogenesis of AD
Mitochondrial Damage in AD
As mentioned above, although the theory of toxic protein deposits can well explain the characteristic pathological manifestations of AD, it is still very limited in understanding the molecular mechanisms of AD. At present, the research focus is slowly shifting to the mitochondrial dysfunction. Mitochondria are the cellular energy powerhouses, supplying most of ATP through oxidative phosphorylation (OXPHOS). Moreover, mitochondria also perform the pivotal role in a variety of cellular functions including biosynthesis of steroids, maintenance of calcium homeostasis, and regulation of cellular life/death decisions.
Aging is a multifaceted process that leads to time-dependent alterations in tissue structure and the progressive decline of many functions in living organisms. As organisms age, the efficacy of the mitochondrial respiratory chain tends to diminish, thus reducing ATP generation and increasing electron leakage and mitochondria-derived reactive oxygen species (ROS) production. Progressive mitochondrial dysfunction will result in an increased level of ROS, which in turn causes further mitochondrial deterioration and global cellular damage [29]. Neurons are heavily dependent on mitochondria owing to their high bioenergetic demand. Aged brains show a significant decline in mitochondrial function, characterized as a loss of oxidative phosphorylation capacity, decreased mitochondrial membrane potential, and activation of the mitochondrial permeability transition pore [30]. Furthermore, markers of oxidative stress such as protein carbonylation, lipid oxidation, and the oxidation of the mitochondrial genome are also increased with age, which appears to be particularly noticeable in AD [31].
Multiple studies have found that neurons in AD patients demonstrated dysfunctional mitochondria, which are characterized by structural abnormalities, functional defects, and changes in mitochondrial dynamics [32, 33]. Impairment of mitochondrial function causes bioenergetic deficiency, intracellular calcium imbalance, and oxidative stress, thereby aggravating the effect of Aβ and tau pathologies, leading to synaptic dysfunction, cognitive impairment, and memory loss [34,35,36,37]. Importantly, mitochondrial dysfunction was observed prior to Aβ deposition and tau aggregation, suggesting that mitochondrial damage is a pathological event upstream of the formation of toxic protein aggregates in AD [6, 38, 39]. Hence, the mitochondrial cascade hypothesis was proposed by Swerdlow and Khan in 2004 [40, 41]. They have hypothesized that the age-related decline in mitochondrial function is an early initiating event in AD, which leads to various pathophysiological changes in the neurons and contributes to the progression of the disease.
Causal Linkage Between Mitochondrial Dysfunction and Aβ Deposition in AD
At present, a large number of studies using cell lines and mouse models have provided the compelling evidence that mitochondrial dysfunction is the etiology of Aβ-related AD pathology [4, 42, 43]. For example, in cybrid (cytoplasmic hybrid) cell lines containing mitochondria from AD patients, a markedly increase in Aβ protein level was observed, which was a characteristic pathological hallmark of AD [42]. Furthermore, multiple mouse models of AD have exhibited that mitochondrial dysfunction not only induces AD-like symptoms but also causes AD pathology characterized by tau hyperphosphorylation and neurofibrillary tangles [44,45,46]. These studies have substantiated the mitochondrial cascade hypothesis in the pathogenesis of AD.
However, the causal relationship between mitochondrial damage and toxic protein aggregation is still controversial, because some researchers believe that Aβ and p-tau protein accumulation is the cause of mitochondrial dysfunction, not the result of mitochondrial dysfunction [9, 47, 48]. Contrary to the evidence highlighting that mitochondrial dysfunction drives Aβ-related AD pathology, a large number of studies demonstrated the role of toxic protein aggregates in abrogating mitochondrial function. In this respect, studies with both cultured cells and in vivo models have shown the detrimental effects of Aβ peptides on mitochondria in neurons [49,50,51,52]. Moreover, post-mortem brain autopsies of AD patients showed the presence of Aβ in mitochondria of neurons of cortical and hippocampal regions [49]. Mechanistically, the full-length and C-terminus truncated forms of APP have been demonstrated to cause mitochondrial dysfunction through APP-mitochondrial interactions, insertion into mitochondrial membrane pore, or inhibiting the mitochondrial fission-fusion machinery [50, 53,54,55,56]. Furthermore, AD-related toxic proteins Aβ and tau can lead to defects in axonal transport in neurons, resulting in ATP depletion and synaptic dysfunction [57, 58].
Taken together, whether mitochondrial dysfunction is the cause of AD pathologies or the pathophysiological manifestations in AD progression is still a debated topic. However, they draw attention to the fact that a healthy mitochondrial pool is essential for maintaining the integrity of neurons and that severe insults to mitochondria will inevitably lead to synaptic damage, neuronal death, and neurodegeneration [4, 59,60,61]. Here, we speculate that there exists a feedback loop between mitochondrial damage and the accumulation of toxic protein aggregates in the pathogenesis of AD. On the one hand, mitochondrial dysfunction in the early stage of AD will promote the pathological changes characterized by the accumulation of Aβ and tau; on the other hand, the formation of protein aggregates will further aggravate mitochondrial damage, forming a vicious circle, which eventually leads to neuronal death.
Decline in Neuronal Mitophagy During the Development of AD
Neuronal Autophagy Declines During Aging
As a highly conserved cellular degradative pathway, autophagy is responsible for the removal of most long-lived proteins and damaged organelles in the cell. In the past decades, the autophagy machinery has been investigated in depth. Autophagy initiates as an isolation membrane, then gradually grows into a double-membrane autophagosome, and subsequently matures into an autolysosome after fusion with lysosomes. Finally, the autophagosome-containing cytoplasmic materials are degraded by lysosomal enzymes [62]. In neurons, the vast majority of autophagosomes formed in the distal axons [63,64,65]. The autophagosomes in the distal axons need to be delivered to the soma where lysosomes accumulate for degradation [66]. At present, multiple studies have found that the expression of autophagy-related genes LC3, p62, Atg1, Atg5, Atg6, Atg7, and Atg12 are significantly downregulated in aging animals and human tissues, especially in the central nervous system (CNS) [67, 68]. Furthermore, protein levels of ATG5, VPS34, and Beclin1 in mouse hippocampus gradually declined from 3 months to 16 months of age, and the ratio of LC3-II/LC3-I also decreased [69]. Consistent with the change in an animal model, the mRNA levels of ATG5, ATG7, and Beclin1 in human brains also declined with age [70]. Similarly, Caenorhabditis elegans experiment also proved that the autophagy activity gradually decreases with age, which is primarily due to the post-translational modification of autophagy-related proteins. Compared with other types of cells, neuronal autophagy activity is more susceptible [71].
Basal autophagy is essential for the maintenance of survival and normal function of neurons. Autophagy goes awry at various points along the pathway, giving rise to distinct pathologic patterns. A defect in one or more stages of autophagy contributes to pathogenesis in different neurodegenerative disorders [64, 72]. In 2002, Ravikumar first proposed that the pathogenesis of neurodegenerative diseases was related to the dysregulated autophagy in neurons [73]. Marked change in neuronal autophagy activity was observed in many neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease [74]. Moreover, tissue-specific autophagy-related gene deletion (Atg5 or Atg7) in mice resulted in an aberrant polyubiquitinated proteins accumulation in the axons of autophagy-deficient neurons, axon degeneration, and eventually loss of neurons [75, 76].
Impairment of Mitophagy in Alzheimer’s Disease
Neurons are highly specialized post-mitotic cells that have large expanses of dendritic and axonal cytoplasm. They are heavily dependent on mitochondrial distribution and function. Meanwhile, mitochondria are highly susceptible to the genetic lesions and environmental factors due to their highly active metabolism and finite repair mechanisms. Therefore, neurons host a set of quality control machinery to maintain the pool of functional mitochondria, including mitochondrial biogenesis, mitochondrial dynamics, mitochondrial unfolded protein response (UPRmt), and mitophagy [77, 78]. Mitophagy is one type of selective autophagy responsible for the selective removal of damaged mitochondria. At present, it is acknowledged that mitophagy can be completed via ubiquitin-dependent pathways such as PINK1-PARKIN-mediated mitophagy, or ubiquitin-independent pathways such as BNIP-NIX, FUNDC1, and Bcl2L13-mediated mitophagy [79]. Among them, PINK1-PARKIN-dependent mitophagy is the most studied and best-characterized mitophagy. In this pathway, dysfunctional mitochondria stabilize PTEN-induced putative kinase (PINK1) on the outer mitochondrial membrane (OMM) [80]. Accumulated PINK1 can phosphorylate ubiquitin and Parkin. As an E3 ubiquitin ligase, activated Parkin mediates ubiquitination of the outer mitochondrial membrane proteins, which serve as a signal to recruit the autophagy adaptors such as OPTN, NDP52, and p62. Consequently, the autophagy machinery is recruited to damaged mitochondria for degradation [81, 82].
As an essential mitochondrial quality control mechanism, mitophagy plays a critical role in maintaining neuronal health and function. There is increasing evidence that the level of mitophagy markedly declines in mammalian tissues during aging [83, 84]. For example, Sun et al. used an mt-Keima transgenic mouse model to characterize the influence of physiological aging on mitophagy of hippocampal neurons. In 3-month-old mice, the neurons in DG region of the hippocampus demonstrated a high level of mitophagy. By contrast, hippocampal neurons in older mice (age 21 months) presented approximately 70% reduction of mitophagic activity [84]. Furthermore, the decline in the expression of autophagy genes with age was further exacerbated in AD patients [70]. In the early studies, post-mortem examinations of hippocampal CA1 neurons in AD patients found that the expression of autophagy-related genes and lysosomal genes might be upregulated in the early stage of the disease, which might be a compensatory response of neurons to toxic protein aggregates stimulation. Furthermore, increased recruitment of Parkin, LC3, and p62 to damaged mitochondria was observed in mutant hAPPTg neurons and AD patient brains [85].
However, the elimination of substrates from these autolysosomes was always defective throughout the process of AD, manifested by the aberrant accumulation of mitophagosomes and increased retention of damaged mitochondria in LAMP1-positive vesicles [9, 86]. Therefore, abnormal accumulation of autophagosomes observed in AD did not indicate an enhanced autophagic activity of neurons, but the result from blocked autophagic flux [9, 72, 87]. Importantly, with disease progression, there is a significant reduction in cytosolic Parkin levels in AD patient brain [85, 88]. Recent studies provide compelling evidence that mitophagy is impaired in the hippocampus of AD patients, in induced pluripotent stem cell-derived human AD neurons, and in APP/PS1 mice models. The levels of mitophagy-related proteins PINK1, Bcl2L13, and BNIP3L/NIX were reduced and mitophagy initiation proteins such as phospho-ULK1 and phospho-TBK1 were inactivated in AD patient samples [8]. Furthermore, Martin-Maestro et al. [48] also reported a reduction in PINK1 and Parkin translocation to damaged mitochondria in APP and tau overexpression models, suggesting the role of compromised mitophagy in the accumulation of damaged mitochondria in AD models. These studies support that mitophagy impairment could be the result of the combination of compromised lysosomal function and decreased autophagy-related proteins.
Indeed, it has been reported that several genetic risk factors and AD-associated proteins directly impair lysosome function [89]. Presenilin 1, the most common cause of early-onset familial Alzheimer’s disease, is required for lysosome acidification and protease activation. The mutations in presenilin 1 can disrupt lysosomal functions and markedly accelerate disease onset and neuropathological severity [90, 91]. Moreover, ApoE4, a genetic risk factor of late-onset AD APOE, can disrupt the integrity of lysosomal membranes in an allele-specific manner [92]. Likewise, other factors contributing to Alzheimer’s disease pathogenesis such as reactive oxygen species and accumulated Aβ peptide similarly impede lysosomal proteolysis, damage lysosomal membranes, and disrupt lysosomal integrity.
Studies in AD animal models, cell models, and AD patients all support that impaired mitophagy can promote the accumulation of Aβ and tau proteins, aggravate synaptic deficiency, and cognitive disorders [9, 93]. For example, PINK1-deficient mAPP mice appear to develop Aβ plaques, mitochondrial abnormalities, and memory impairment earlier. In contrast, PINK1 overexpression promotes the clearance of damaged mitochondria, thereby alleviating Aβ-induced loss of synapses and cognitive decline in AD mice [93]. Furthermore, the restoration or enhancement of mitophagy with pharmacological methods can ameliorate pathological damage and memory loss in AD animals through the inhibition of Aβ plaques [8, 94]. For example, autophagy-inducing agent rapamycin ameliorated cognitive deficits and reduced Aβpathology in an APP-mutant mouse AD model [94]. Moreover, a recent study has reported that induction of mitophagy with NAD+ supplementation, urolithin A, and actinonin can significantly inhibit Aβ pathology and reverse cognitive deficits in both Caenorhabditis elegans and mouse models of AD [8]. These findings suggest that impaired removal of defective mitochondria is a pivotal event in AD pathogenesis and that mitophagy represents a potential therapeutic intervention.
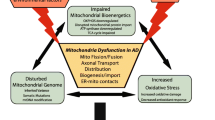
More importantly, it is recognized that the deficiency in mitophagy alone is not sufficient to lead to neurodegenerative disorders. For example, genetic mouse models of human PD, harboring mutations in PARK2 or PARK6 exhibited defects in mitophagy but failed to recapitulate the neurological defects typical of PD patients. In contrast, PD symptoms gradually appeared only under mitochondrial stress [95, 96]. These studies highlight the importance of mitochondrial damage caused by Aβ deposit and environmental factors in the pathogenesis of AD. In summary, the initiation and progression of AD may be the result of the combined action of mitophagy impairment and mitochondrial dysfunction during inflammatory aging. Among them, the progressive decline of mitophagy interferes with the mechanisms for neurons to maintain homeostasis, while the accumulation of pathogenic protein aggregates and exposure to environmental toxicants further aggravates the mitochondrial damage, which ultimately leads to neuronal death in AD (Fig. 1).

Possible mechanisms underlying the occurrence of Alzheimer’s disease. With the aging of human beings, mitophagy in neurons progressively declines, which compromises the capability of neurons to maintain the mitochondrial homeostasis. In the meantime, the accumulation of pathogenic protein aggregates in neurons and exposure to environmental toxicants further exacerbate mitochondrial damage. Both of them work together in a synergistic manner, ultimately leading to neuronal death in brains and contributing to the development of Alzheimer’s disease.
The Molecular Mechanisms Underlying Mitophagy Defects in AD
Normally, autophagy constitutively occurs at the basal level, however. However, upon a wide range of stimuli including nutrient deprivation, oxidative stress, and damage to organelles, autophagy activity will change dramatically. In the past two decades, the regulatory mechanisms of autophagy have been thoroughly elucidated. It is generally believed that transcriptional activation and post-translational modification of autophagy-related proteins are the primary mechanisms responsible for the regulation of autophagy [97, 98].
Recent studies suggested that reduced autophagy in AD patients and animal models is likely caused by the hyperactivation of the PI3K/Akt/mTOR axis [99, 100]. The mammalian target of rapamycin (mTOR) is a multidomain protein kinase that integrates cellular metabolism with cell growth and proliferation. In higher eukaryotes, mTOR, together with other molecules such as RAPTOR and DEPTOR, forms the complex of mTORC1. mTORC1 is a pivotal mechanism that negatively regulates the autophagic activity. It can suppress the ULK1 complex by phosphorylation, thus inhibiting the formation of autophagosomes [98].
The mTOR pathway was shown to be hyperactivated in AD in both mouse models and humans [100]. The cognitive performance of APP/PS1 mice had a strong inverse correlation with Aβ plaque load and mTOR activation. Mechanistically, mTOR signaling may account for the reduction in autophagy activation and cognitive performance in APP/PS1 mice [101]. Furthermore, when administered in the early stages of the disease, rapamycin and its analogs were shown to prevent cognitive decline in AD mouse models, which correlated with a decrease in aggregated beta-amyloid plaques, tau tangles, and microglial activation in AD [94, 102]. Additionally, several studies have shown that monomeric and oligomeric Aβ peptides can over-activate the PI3K/Akt/mTOR axis [100, 103]. Levels of Aβ oligomers in AD brain have been correlated with the increased activation of kinases including Akt and mTOR [99, 104].
Transcription factor EB (TFEB) is the major regulator responsible for the transcription of autophagy-lysosomal genes [105]. At present, there has been increasing evidence that supports that TFEB is associated with PINK1/Parkin-mediated mitophagy. For example, TFEB overexpression can significantly enhance the clearance of damaged mitochondria [106]. Moreover, pharmacological induction of TFEB not only facilitates the sequestration of the damaged mitochondria by autophagosomes but also promotes the recruitment of PINK1 and Parkin to mitochondria to enhance mitophagy[107]. More importantly, the beneficial effects of TFEB have also been observed in multiple AD mouse models [108,109,110,111]. TFEB overexpression in the mice brain dramatically ameliorates phosphorylated tau and neurofibrillary tangle–associated neuropathology and rescues behavioral and synaptic deficits and neurodegeneration in the rTg4510 AD mouse model [109]. The specificity and efficacy of TFEB in mediating the clearance of toxic Aβ and tau peptide makes it a candidate therapeutic target for AD. Indeed, curcumin analog C1 could significantly enhance the efficiency of autophagic-degradative pathway in AD models through TFEB-mediated transcriptional machinery, which significantly alleviates the neuropathological changes of animals and improved their cognition functions [112].
Interestingly, there exists a mechanistic linkage between TFEB and mTOR signaling pathway. Specifically, activated mTORC1 phosphorylates TFEB and inhibits its transport into the nucleus. Conversely, inhibition of mTORC1 reduces TFEB phosphorylation and promotes the translocation of TFEB into the nucleus [109, 113]. Therefore, in the setting of AD, it appears that activated mTORC1 can regulate autophagy activity at two levels: post-translational modification of autophagy-related proteins and the transcription of autophagy-related genes. In addition, intracellular calcium signaling pathways including calcium/calmodulin-dependent protein kinase 훽 (CaMKK훽) and calcium-dependent protein phosphatase calcineurin are thought to be involved in TFEB-mediated transcription regulation [114,115,116]. For example, CaMKK훽 can phosphorylate and activate AMPK to inactivate mTORC1, releasing its brake on TFEB-mediated transcription of autophagy [117], whereas calcineurin directly dephosphorylates TFEB and promotes its nuclear translocation [115]. Hence, an in-depth study of calcium-mediated autophagy-lysosomal gene transcription will provide a better understanding of the mechanism of AD.
The Underlying Mechanisms for Impaired Mitophagy in AD Disease: Mitochondrial Dysfunction, Neuroinflammation, and Neuronal Cell Death
As an essential stress-response mechanism in cells, mitophagy could effectively remove damaged mitochondria, which can not only reduce the generation of reactive oxygen species (ROS) and control the level of intracellular oxidative stress but also inhibit the activation of NLRP3 inflammasome and suppress the inflammatory response [118,119,120,121]. Therefore, it is reasonable to assume that age-dependent decline of mitophagy will inevitably cause the accumulation of damaged mitochondria, leading to a series of AD-related pathophysiological events. Indeed, commensurate with an age-dependent increase in the incidence of AD, there is also an age-dependent accumulation of dysfunctional mitochondria, chronic neuroinflammation, and neuronal cell death [5, 9]. Here, we propose a mechanistic diagram for the role of impaired mitophagy in the “mitochondrial dysfunction-neuroinflammation-neuron death axis” during the initiation and progression of AD (Fig. 2).

Proposed mechanistic pathways by which mitophagy are implicated in Alzheimer’s disease. During the process of Alzheimer’s disease, the accumulation of pathogenic proteins and the exposure of environmental toxicants can cause a cascade of pathophysiological events, including mitochondrial damage, neuroinflammation, and neuron death. As an important quality control mechanism, mitophagy can not only maintain the healthy mitochondrial pool in neurons but also alleviate oxidative stress, neuroinflammation, and neuronal cell death. Conversely, the decline in mitophagy activity with age can aggravate mitochondrial dysfunction and neuroinflammation, and promote neuronal death through apoptosis or other programmed cell death.
Changes in Mitochondrial Function and Mitochondrial Dynamics During AD
Current research on post-mortem tissues from AD patients and mouse models of AD has led to an important insight into AD pathophysiology. Abnormal mitochondrial dynamics and bioenergetics are the early pathophysiological events of neuronal damage in AD patients [88]. As a highly energy-consuming organ, the brain is vulnerable to impaired energy metabolism. Before AD patients demonstrated any pathological changes or clinical symptoms, neurons showed obvious abnormality in mitochondrial energy metabolism [88, 122]. The bioinformatics analysis of AD patient hippocampus identified OXPHOS pathway as one of most significant pathways involved in AD [123]. Moreover, as mentioned above, increasing evidence supports that mitophagy activity is declined with age in mice and human cells [83, 84]. Considered the pivotal role of mitophagy for the maintenance of mitochondrial homeostasis, it is not surprising that alterations in mitophagy will significantly impair mitochondrial bioenergetics in neurons.
Besides defects in mitochondrial function, mitochondrial structural abnormality was also observed in injured brain regions of familial and sporadic AD patients [41, 124]. Mitochondria are highly dynamic organelles. They continuously change their size, shape, and cristae architecture via undergoing fusion and fission events. In mammals, mitochondrial fusion is mediated by mitofusion 1/2 (MFN1/2) and opal, while mitochondrial division is mediated by Drp1, Fisl, and Mff [125]. In general, mitochondrial fusion contributes to the integrity of the mitochondrial network, whereas mitochondrial fission promotes the removal of the dysfunctional organelles by mitophagy [126, 127]. As a pro-fission signal, PINK1 promoted Drp1-associated fission machinery to selectively segregate damaged mitochondria and ensured the requisite division of damaged mitochondria for organelle degradation [128]. Furthermore, Mfn1 and Mfn2 were found to be a target for Parkin-mediated ubiquitination. The recruitment of Parkin to depolarized mitochondria modulated the UPS-dependent degradation of Mfn1/2, to further promote mitophagy [129, 130]. In addition, PINK1 also regulated mitochondrial fission via directly phosphorylating Drp1 on Ser616, which was independent of Parkin and autophagy [131]. Taken together, the interplay between mitophagy and mitochondrial dynamics is important for the functional homeostasis of mitochondria.
Manczak et al. evaluated the mitochondrial proteins in post-mortem AD brain tissues at different stages of the pathology and found a reduction in the expression levels of mitochondrial fusion genes (Mfn1/2 and OPA1) and an increase of fission genes (Drp1) in AD [49]. Likewise, Wang et al. also proved that the level of mitochondrial fusion proteins is significantly reduced in the AD brain, especially in the hippocampal region [132]. Moreover, Drp1 translocation to mitochondria was increased and Mfn2 expression was decreased in AD cybrid cells, suggesting an imbalance of mitochondrial dynamics [133]. Additionally, Tyumentsev et al. used accelerated senescence OXYS rats to investigate the mitochondrial change of pyramidal neurons in the hippocampus CA1 region and demonstrated a shift from mitochondrial fusion toward fission from 4 to 24 months of age [134]. Taken together, aberrant mitochondrial dynamics, especially increased mitochondrial division may be considered a critical molecular event in AD progression. Surprisingly, with imaging techniques to visualize mitochondrial structure in the brain tissue from patients and mouse models of AD, Zhang et al. found that fission arrest and mitochondrial elongation may occur at different disease stages [135]. Moreover, the cybrid model with mitochondria derived from mild cognitive impairment (MCI) patients, a prodromal stage of AD, has revealed a significant rise in Mfn2 expression, but not in Drp1 [136]. Similarly, a significant increase in Mfn2 level was also observed in 3-month-old APP/PS1 mice [137]. These data support that the enhanced mitochondrial fusion could be considered an early event in AD.
At present, the underlying mechanism for changes in mitochondrial dynamics in AD has not been fully elucidated. Recent studies suggested that Aβ may directly disrupt the mitochondrial fission-fusion machinery. In fact, Aβ deposition was evident in synaptic mitochondria isolated from APP transgenic mice early in life and prior to extracellular Aβ accumulation [36]. Furthermore, in post-mortem AD patient brains and brain tissues from APP mice, the colocalization of Drp1 and Aβ was observed. Further investigation revealed that Drp1 interacts with Aβ, and these abnormal interactions are increased with disease progression [49, 138, 139]. Moreover, overexpression of APP mutants or administration of Aβ can induce mitochondrial fragmentation and disrupted mitochondrial distribution in in vivo and in vitro experiments [36, 140,141,142]. In contrast, inhibition of mitochondrial division with Drp1 inhibitor Mdivi-1 can alleviate Aβ-induced mitochondrial dysfunction and memory deficits in AD mice [143]. These findings suggest that Drp1 interactions with AD-associated pathogenic proteins likely lead to excessive mitochondrial fragmentation and synaptic deficiencies, ultimately possibly leading to neuronal damage and cognitive decline.
Moreover, increased intracellular calcium might be involved in the imbalance of mitochondrial fusion-fission machinery in AD. Several lines of evidence from experimental and human systems support that neuronal Ca2+ homeostasis is disrupted in AD [144, 145]. For instance, Aβ oligomers can induce a sustained rise of intracellular Ca2+ in neurons [146]. Likewise, mutations in the presenilin gene also affect the intracellular calcium homeostasis and interfere with calcium-mediated cascade signaling [144]. Importantly, the significant changes in intracellular Ca2+ signaling precede neuronal death and cognitive deterioration in AD [145]. Calpains are calcium-dependent neutral cysteine proteases. Ubiquitous calpain can be divided into two major forms: μ-calpain and m-calpain, depending on the level of calcium required for their activation [147, 148]. There is increasing evidence supporting the pathological role of calpain in the early stage of Alzheimer’s disease because μ-calpain was suggested to be involved in the processing of AD-associated pathogenic proteins [149, 150]. In this regard, post-mortem examination of AD patients found that widespread activation of μ-calpain occurred in the brains, showing a 3-7-fold increase. Meanwhile, the cleavage of calpain substrates was also increased [151,152,153,154]. Importantly, activated calpain can degrade Drp1, MFN1/2, and other mitochondrial regulatory proteins [155, 156]. For instance, Drp1 is a substrate of calpain, which can be cleaved to produce specific N-terminal Drp1 cleavage fragments. Various AD-related insults such as exposure to glutamate, soluble Aβ oligomers, or reagents inducing tau hyperphosphorylation led to calpain-dependent cleavage of Drp1 in primary cortical neurons. Likewise, N-terminal Drp1 cleavage fragments were also present in cortical neurons of CRND8 APP transgenic mice and human AD brains [156].
Microglia-Mediated Oxidative Stress and Neuroinflammation in AD
The brain has been considered an immune-privileged organ owing to the existence of the blood-brain barrier. Immune responses in CNS primarily depend on the innate immune system. Microglia are the main innate immune cells present in the brain and protect the CNS through maintaining neuronal homeostasis. Under normal conditions, microglia retain a relative quiescent phenotype with their processes extending into the surrounding microenvironment. When microglia sense a change in the microenvironment, they can rapidly become activated. Activated microglia can not only clear dead cells and tissue debris but also secrete pro-inflammatory factors such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and ROS to induce neuroinflammatory response [157, 158]. Nowadays, it is widely acknowledged that the neuroinflammatory response is a double-edged sword, which can exert both beneficial and potentially deleterious effects on the progression of neurodegenerative diseases [159]. Short-term and acute inflammation usually promotes the elimination of the pathogenic stimuli and tissue repair, whereas a prolonged and non-resolving inflammation can lead to the amplification of pathogenic events [160, 161].
There is a growing body of evidence supporting microglia-mediated inflammatory responses plays a key role in age-related neurodegenerative diseases. During aging, microglia may become hyperresponsive with increased proinflammatory cytokine output and/or assume a dysfunctional state with loss of phagocytic functionality [162]. Transcriptomics analysis of the elderly rodents demonstrated that 50% of upregulated genes in brain tissue were associated with inflammation and oxidative stress. This profile was similar to the neuroinflammatory phenotype in AD patients and animal models [163, 164]. The sustained inflammatory response is likely to be mediated by endogenous danger-associated molecular patterns (DAMPs). Aging can be associated with the accumulation of endogenous DAMPs such as ATP, uric acid, oxidatively modified DNA, or aggregated proteins [165,166,167,168]. Microglia can rapidly detect the DAMPs with subsequent initiation of an inflammatory response [157, 158]. More importantly, the elevated proinflammatory cytokines can interact with the processing and production of Aβ peptide in AD [160, 161].
Pathogenic Protein Aggregates and Mitochondrial Damage Induce Microglial Activation
In AD, the formation of Aβ plaque and neurofibrillary tangles can reduce the level of Aβ and tau oligomers in the brain; it is considered to be a protective mechanism for relieving the protein toxicity. However, the aggregates deposited in the brain are an important factor inducing an inflammatory response [169]. Moreover, mtDNA and mtROS released from damaged mitochondria are known to stimulate the innate immune system [170, 171]. Therefore, toxic protein aggregates and mitochondrial damage in AD can not only exert direct insults to neurons by themselves but also indirectly cause neuronal damage through the activation of microglia (Fig. 3). According to current knowledge, activated microglia can cause neuronal damage through two interconnecting processes: NADPH oxidase (PHOX)–mediated oxidative stress and NLR family pyrin domain containing 3 (NLRP3) inflammasome-mediated neuroinflammation.

Mitochondrial dysfunction mediates the cross-talk between microglia and neurons in Alzheimer’s disease. The decline of mitophagy and the aggregation of toxic proteins during aging cause the dysregulation of mitochondrial quality control and eventually lead to the accumulation of damaged mitochondria in neuronal cells. Mitochondrial dysfunction can not only directly influence the function of neurons but also induce oxidative stress and neuroinflammation by inducing microglial activation, thereby further aggravating neuronal damage. Mechanistically, following microglial activation, PHOX in microglia was triggered, resulting in robust induction of reactive oxygen species. In the meantime, NLRP3 inflammasome was also activated, leading to the production of the proinflammatory cytokines including IL-1β. Abbreviations: IL-1β, interleukin-1β; NLRP3, NLR family pyrin domain containing 3; PHOX, NADPH oxidase
PHOX is highly expressed in microglia and responsible for the production of ROS following the activation of microglia. ROS and nitric oxide derived from microglia are directly cytotoxic to neurons [172]. In turn, the excessive accumulation of ROS in the AD brain can further induce mitochondrial dysfunction and continued release of ROS [173]. Furthermore, Aβ accumulation also increases oxidative stress even at the early stages of AD [174]. Consistent with the findings in the human brain, increased ROS levels are found in APP/PS1 mice [174, 175]. Neurons are highly susceptible to the damaging effects of these radicals for several reasons including their high oxidative metabolic activity and relatively low antioxidant capacity. Therefore, it is not surprising that oxidative stress in the brain can result in neuronal damage [176]. Removal of damaged mitochondria by mitophagy is important not only for maintaining a functional mitochondrial pool but also for limiting oxidative cell damage. Indeed, the accumulation of dysfunctional mitochondria and a marked rise in ROS levels were observed in Atg7-knockout animal models [83]. These results support that a decline in mitophagy might fuel the vicious circle of oxidative stress-induced, age-related tissue damage.
NLRP3 inflammasome is one of the most well-characterized inflammasomes, which can be activated by a wide spectrum of PAMPs and sterile DAMPs. As mentioned previously, high levels of proinflammatory cytokines including TNF and IL-1β are detected in the brains, cerebrospinal fluid, and serum of patients with AD [169]. There is extensive evidence that fibrillar Aβ can act as DAMPs within the CNS, but dying neurons can give rise to a multitude of other DAMPs, including ATP and lysophosphatidylcholine [177]. Furthermore, damaged mitochondria can release non-methylated mtDNA and formyl peptides. They can be viewed as mitochondrial-derived DAMPs that are known to activate NLRP3 inflammasome [165, 170, 171, 178]. Microglia express several classes of pattern recognition receptors (PRRs). Among them, Toll-like receptors (TLRs) are the best-characterized PRRs. Following the activation of TLRs after binding ligands, the downstream signaling cascades result in activation of nuclear factor κB (NF-κB) and subsequent induction of proinflammatory cytokines. NLRP3 inflammasomes are cytosolic multimeric signaling platforms that activate caspase-1 and facilitate the maturation and secretion of the inflammatory mediators. In this aspect, high levels of full-length caspase-1 and cleaved caspase-1 were detected in the brains from patients with AD. More importantly, this activation phenotype is fully replicated in the brains of APP/PS1 mice model [167, 179, 180].
Recent studies highlighted an important role of NLRP3 inflammasome activation in the pathogenesis of Alzheimer’s disease. Intrahippocampal injection of ASC specks resulted in the spreading of Aβ pathology in APP/PS1 transgenic mice while the deficiency in NLRP3 inflammasome protected APP/PS1 mice from Aβ-related pathology and development of cognitive decline [167, 181]. Moreover, intracerebral injection of fibrillar Aβ induced tau pathology in an NLRP3-dependent manner. By contrast, loss of NLRP3 inflammasome function reduced tau hyperphosphorylation and aggregation [182]. As an important mechanism for mitochondrial quality control, mitophagy can effectively eliminate damaged mitochondria and limits activation of the NLRP3 inflammasome in a feedback loop [183, 184]. The induction of mitophagy by pharmacological treatment can suppress the neuroinflammation and improve memory performance in AD mice model [8, 185]. For example, in APP/PS1 AD mice, elevated expression and activity of NLRP3 inflammasome were observed in the brain tissues. By contrast, restoration of neuronal mitophagy with mitophagy-inducing compounds urolithin A and actinonin mitigated the neuroinflammation, as indicated by lower levels of cleaved caspase 1, proinflammatory IL-1β, and active IL-1β [8].
Mitophagy Impairment in Microglia Contributes to the Neuroinflammation in AD
In a recent study, it was reported that mitophagic activity in microglia was decreased by about 60% in the hippocampal region of AD mouse [8]. The induction of mitophagy with urolithin A and actinonin could enhance the phagocytic function of microglia and promote the clearance of Aβ deposits in APP/PS1 mice [8]. Moreover, mitophagy induction could suppress the activation of NLRP3 inflammasomes and reduce the level of proinflammatory cytokines in AD models [8]. By contrast, loss of mitophagy-related gene Parkin exacerbates the chronic neuroinflammation in neurodegenerative diseases [186]. Taken together, these results support that restoration of mitophagy plays a neuroprotective role in AD through mitigating NLRP3-dependent neuroinflammation and ameliorating AD pathology.
Mitochondrial Dysfunction and Neuron Death in AD
Imaging studies and post-mortem examinations of AD patients showed a significant decrease in brain volume and the number of neurons, suggesting the presence of severe neuronal loss [187, 188]. Despite this indisputable evidence, the exact mechanism by which neurons die remains largely unknown. In this respect, early studies mainly focused on the role of apoptotic machinery in neuronal death and found that dying neurons in the brain of AD patients showed morphological characteristics of apoptosis [189, 190]. Furthermore, DNA fragmentation and caspases activation have been detected in post-mortem human brains [189, 191].
Mitochondria play an important role in integrating different apoptotic signals by the release of proapoptotic factors. Although the intrinsic and extrinsic apoptotic pathways may be activated by different signals, they converge on caspase-3 activation. As opposed to the death receptor-mediated apoptosis, the intrinsic programmed cell death is initiated by the release of apoptotic factors such as cytochrome c from the mitochondria, with subsequent activation of caspase-9 and caspase-3. The release of these apoptotic factors requires mitochondrial outer membrane permeabilization (MOMP) modulated by various pro- and anti-apoptotic proteins. Due to the long-term and chronic properties of AD, only a limited number of apoptotic neurons can be detected at different time-points during AD progression, so the exact contribution of apoptosis to the loss of neurons in Alzheimer’s disease remains to be determined [192]. Moreover, many degenerated neurons in AD did not show apoptosis features, suggesting that apoptosis is not the only mechanism responsible for neuronal death in Alzheimer’s disease [193, 194].
Necroptosis is a newly identified form of regulated necrotic cell death under apoptotic deficient conditions [195]. Necroptosis can be triggered by the activation of death receptors such as TNF receptor 1 under certain conditions [196]. The activation of receptor-interacting protein kinase 1 (RIPK1) is required for the induction of necroptosis, which subsequently leads to the formation of RIPK1-RIPK3-MLKL complex. The oligomerization of MLKL molecules ultimately initiates the necroptosis through disruption of the integrity of the plasma membrane [197]. Necroptosis and apoptosis can both be triggered by the same stimuli, including death receptor ligation, DNA damage, and mitochondrial dysfunction. In general, the severity of the insult determines the mode of cell death. Specifically, an extensive and severe insult usually causes necrotic cell death rather than apoptosis because it incapacitates the cell to prevent it from a more deliberate form of programmed cell death. This may result from the increasing number of mitochondria undergoing MPT and depletion of ATP. Based on the mechanistic understanding of necroptosis, necroptotic cell death was also dependent on altered mitochondrial function. For example, mitochondrial ROS can facilitate the initiation of necroptosis by promoting RIPK1 autophosphorylation, leading to necrosome formation [198, 199]. Moreover, in the absence of Drp1, the disruption of mitochondrial dynamics resulted in the degeneration of cerebellar Purkinje neurons in mice via necroptotic cell death [200].
Abnormal activation of necroptosis has been extensively reported to be involved in the etiology of multiple neurodegenerative diseases including AD, ALS, and PD [196]. In AD, the activation of the RIPK1-RIPK3-MLKL signaling pathway was detected in human AD brains, especially in neurons and microglia [201, 202]. In ALS, RIPK1-regulated necroptosis not only resulted in the death of neuronal cell bodies but also led to progressive demyelination and axonal degeneration through engagement of necroptotic machinery in the CNS [203, 204]. In contrast, anti-necroptotic molecule necrostatin-1 (Nec-1) attenuated p-MLKL recruitment and necroptosis-related neurodegeneration in rodent models of chronic neurological conditions [201, 205,206,207]. Furthermore, inhibition of RIPK1 activity by either Nec-1s or RIPK1 D138N mutation in APP/PS1 mice obviously reduced the levels of Aβ oligomers, plaques, and hyperphosphorylated tau, alleviated the levels of inflammatory cytokines, and ameliorated cognitive deficits [202, 207].
At present, it is generally accepted that autophagy is the key factor that determines the cell fate switch between cell survival and death. The mechanistic linkage between autophagy and necroptosis is gradually being elucidated. On the one hand, autophagy can inhibit necroptotic cell death by maintaining intracellular mitochondrial homeostasis [208]. On the other hand, autophagic machinery can provide a scaffold for necrosome formation under specific setting. For example, Mizumura et al. found that PINK1 was required for RIPK3 expression in cigarette smoke–exposed cultured pulmonary epithelial cells and mice, suggesting mitophagy may regulate stress-induced necroptosis [209]. Furthermore, p62/SQSTM1 can recruit RIPK1 and mediate the assembly of necrosome and its combination with autophagosomes, thus inducing the occurrence of programmed necrosis [210]. However, so far there is no report on the relationship between mitophagy and necroptotic neuron death in AD. Therefore, in-depth research on the causal relationship between mitophagy and necroptosis will not only help decipher the role and its possible mechanism for mitophagy in AD but also promote the development of the promising AD intervention strategy.
Conclusion
Neuronal mitophagy in the CNS progressively declines as human beings age. Impairment of mitophagy will disrupt the maintenance of mitochondrial homeostasis and lead to a cascade of AD-associated pathogenic events, including mitochondrial dysfunction, chronic neuroinflammation, and eventually neuronal death. Therefore, an in-depth investigation of mitochondrial dysfunction-inflammation response-programmed cell death axis in AD, especially the interaction between neuron and microglia, is helpful to elucidate the underlying mechanism for the progressive neuron loss in AD. Moreover, given that multiple beneficial modes of mitophagy are involved in AD pathologies, it is likely that correcting the mitochondrial dysregulation with chemical agents specific to mitophagy could help to develop a promising therapeutic and preventive strategy for AD patients.
Abbreviations
- Aβ:
-
Amyloid beta
- AD:
-
Alzheimer disease
- APP:
-
Amyloid precursor protein
- IL-1β:
-
Interleukin-1β
- ROS:
-
Reactive oxygen species
- NFT:
-
Neurofibrillary tangles
- TNF:
-
Tumor necrosis factor
References
Hampel H, Prvulovic D, Teipel S, Jessen F, Luckhaus C, Frolich L, Riepe MW, Dodel R et al (2011) The future of Alzheimer’s disease: the next 10 years. Prog Neurobiol 95:718–728
Price JL, Davis PB, Morris JC, White DL (1991) The distribution of tangles, plaques and related immunohistochemical markers in healthy aging and Alzheimer’s disease. Neurobiol Aging 12:295–312
Citron M (2010) Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov 9:387–398
Chakravorty A, Jetto CT, Manjithaya R (2019) Dysfunctional mitochondria and mitophagy as drivers of Alzheimer’s disease pathogenesis. Front Aging Neurosci 11:311
Wang W, Zhao F, Ma X, Perry G, Zhu X (2020) Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol Neurodegener 15:30
Swerdlow RH (2018) Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis 62:1403–1416
Cai Q, Jeong YY (2020) Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells 9:150
Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM et al (2019) Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22:401–412
Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF (2017) Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci 40:151–166
Rai SN, Singh C, Singh A, Singh MP, Singh BK (2020) Mitochondrial dysfunction: a potential therapeutic target to treat Alzheimer’s disease. Mol Neurobiol 57:3075–3088
WHO (2015) World Alzheimer Report 2015 The global impact of dementia an analysis of prevalence, incidence, cost and trends.
GBD (2016) Dementia Collaborators A: Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2019(18):88–106
WHO (2003) World Health Report 2003-Global burden of dementia in year 2000. Geneva
Dorszewska J, Prendecki M, Oczkowska A, Dezor M, Kozubski W (2016) Molecular basis of familial and sporadic Alzheimer’s disease. Curr Alzheimer Res 13:952–963
Huang YA, Zhou B, Wernig M, Sudhof TC (2017) ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and abeta secretion. Cell 168:427–441 e421
Ittner LM, Gotz J (2011) Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci 12:65–72
Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120:545–555
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185
Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH (2006) A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440:352–357
Kim J, Chakrabarty P, Hanna A, March A, Dickson DW, Borchelt DR, Golde T, Janus C (2013) Normal cognition in transgenic BRI2-Abeta mice. Mol Neurodegener 8:15
Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T et al (2007) Abeta40 inhibits amyloid deposition in vivo. J Neurosci 27:627–633
Chetelat G, La Joie R, Villain N, Perrotin A, de La Sayette V, Eustache F, Vandenberghe R (2013) Amyloid imaging in cognitively normal individuals, at-risk populations and preclinical Alzheimer’s disease. Neuroimage Clin 2:356–365
Godyn J, Jonczyk J, Panek D, Malawska B (2016) Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep 68:127–138
Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, Tariot PN, Vellas B et al (2019) Randomized trial of verubecestat for prodromal Alzheimer’s disease. N Engl J Med 380:1408–1420
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS et al (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370:322–333
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R et al (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370:311–321
Loureiro JC, Pais MV, Stella F, Radanovic M, Teixeira AL, Forlenza OV, de Souza LC (2020) Passive antiamyloid immunotherapy for Alzheimer’s disease. Curr Opin Psychiatry 33:284–291
Yang T, Dang Y, Ostaszewski B, Mengel D, Steffen V, Rabe C, Bittner T, Walsh DM et al (2019) Target engagement in an Alzheimer trial: crenezumab lowers amyloid beta oligomers in cerebrospinal fluid. Ann Neurol 86:215–224
Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333:1109–1112
LaFrance R, Brustovetsky N, Sherburne C, Delong D, Dubinsky JM (2005) Age-related changes in regional brain mitochondria from Fischer 344 rats. Aging Cell 4:139–145
Navarro A, Boveris A (2010) Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front Aging Neurosci 2:34
Reddy PH, Manczak M, Yin X (2017) Mitochondria-division inhibitor 1 protects against amyloid-beta induced mitochondrial fragmentation and synaptic damage in Alzheimer’s disease. J Alzheimers Dis 58:147–162
Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF (1802) Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta 2010:122–134
Cai Q, Tammineni P (2017) Mitochondrial aspects of synaptic dysfunction in Alzheimer’s disease. J Alzheimers Dis 57:1087–1103
Guo L, Tian J, Du H (2017) Mitochondrial dysfunction and synaptic transmission failure in Alzheimer’s disease. J Alzheimers Dis 57:1071–1086
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A 107:18670–18675
Pickett EK, Rose J, McCrory C, McKenzie CA, King D, Smith C, Gillingwater TH, Henstridge CM et al (2018) Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer’s disease. Acta Neuropathol 136:747–757
Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD (2009) Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 106:14670–14675
Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, Reddy AP, Shirendeb U, Lo HH et al (2012) Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet 21:2973–2990
Swerdlow RH, Khan SM (2004) A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 63:8–20
Swerdlow RH, Burns JM, Khan SM (1842) The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta 2014:1219–1231
Khan SM, Cassarino DS, Abramova NN, Keeney PM, Borland MK, Trimmer PA, Krebs CT, Bennett JC et al (2000) Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol 48:148–155
Scheffler K, Krohn M, Dunkelmann T, Stenzel J, Miroux B, Ibrahim S, von Bohlen Und Halbach O, Heinze HJ et al (2012) Mitochondrial DNA polymorphisms specifically modify cerebral beta-amyloid proteostasis. Acta Neuropathol 124:199–208
Leuner K, Schutt T, Kurz C, Eckert SH, Schiller C, Occhipinti A, Mai S, Jendrach M et al (2012) Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 16:1421–1433
Kukreja L, Kujoth GC, Prolla TA, Van Leuven F, Vassar R (2014) Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol Neurodegener 9:16
Su B, Wang X, Lee HG, Tabaton M, Perry G, Smith MA, Zhu X (2010) Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci Lett 468:267–271
Pagani L, Eckert A (2011) Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis 2011:925050
Martin-Maestro P, Gargini R, Garcia E, Simon D, Avila J, Garcia-Escudero V (2019) Mitophagy failure in APP and tau overexpression model of Alzheimer’s disease. J Alzheimers Dis 70:525–540
Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet 20:2495–2509
Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA (2009) S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324:102–105
Sanz-Blasco S, Valero RA, Rodriguez-Crespo I, Villalobos C, Nunez L (2008) Mitochondrial Ca2+ overload underlies A beta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS One 3:e2718
Cardoso SM, Santos S, Swerdlow RH, Oliveira CR (2001) Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J 15:1439–1441
Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60:748–766
Mossmann D, Vogtle FN, Taskin AA, Teixeira PF, Ring J, Burkhart JM, Burger N, Pinho CM et al (2014) Amyloid-beta peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab 20:662–669
Todd K, Fossati S, Ghiso J, Rostagno A (1842) Mitochondrial dysfunction induced by a post-translationally modified amyloid linked to a familial mutation in an alternative model of neurodegeneration. Biochim Biophys Acta 2014:2457–2467
Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26:9057–9068
Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L (2010) Tau reduction prevents Abeta-induced defects in axonal transport. Science 330:198
Dixit R, Ross JL, Goldman YE, Holzbaur EL (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319:1086–1089
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Akbar M, Essa MM, Daradkeh G, Abdelmegeed MA, Choi Y, Mahmood L, Song BJ (2016) Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res 1637:34–55
Nakamura T, Cho DH, Lipton SA (2012) Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp Neurol 238:12–21
Mizushima N, Yoshimori T, Levine B (2010) Methods in mammalian autophagy research. Cell 140:313–326
Maday S, Wallace KE, Holzbaur EL (2012) Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J Cell Biol 196:407–417
Lee S, Sato Y, Nixon RA (2011) Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J Neurosci 31:7817–7830
Yue Z (2007) Regulation of neuronal autophagy in axon: implication of autophagy in axonal function and dysfunction/degeneration. Autophagy 3:139–141
Xie R, Nguyen S, McKeehan WL, Liu L (2010) Acetylated microtubules are required for fusion of autophagosomes with lysosomes. BMC Cell Biol 11:89
Cuervo AM (2008) Autophagy and aging: keeping that old broom working. Trends Genet: TIG 24:604–612
Rubinsztein DC, Marino G, Kroemer G (2011) Autophagy and aging. Cell 146:682–695
Glatigny M, Moriceau S, Rivagorda M, Ramos-Brossier M, Nascimbeni AC, Lante F, Shanley MR, Boudarene N et al (2019) Autophagy is required for memory formation and reverses age-related memory decline. Curr Biol 29:435
Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C et al (2010) Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A 107:14164–14169
Chang JT, Kumsta C, Hellman AB, Adams LM, Hansen M (2017) Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. eLife 6:e18459
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19:983–997
Ravikumar B, Duden R, Rubinsztein DC (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11:1107–1117
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441:880–884
Jiang X, Jin T, Zhang H, Miao J, Zhao X, Su Y, Zhang Y (2019) Current progress of mitochondrial quality control pathways underlying the pathogenesis of Parkinson’s disease. Oxidative Med Cell Longev 2019:4578462
Leites EP, Morais VA (2018) Mitochondrial quality control pathways: PINK1 acts as a gatekeeper. Biochem Biophys Res Commun 500:45–50
Hamacher-Brady A, Brady NR (2016) Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci 73:775–795
Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189:211–221
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI et al (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314
Nguyen TN, Padman BS, Lazarou M (2016) Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol 26:733–744
Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S et al (2016) Autophagy maintains stemness by preventing senescence. Nature 529:37–42
Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmstrom KM, Fergusson MM et al (2015) Measuring in vivo mitophagy. Mol Cell 60:685–696
Ye X, Sun X, Starovoytov V, Cai Q (2015) Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum Mol Genet 24:2938–2951
Bordi M, Berg MJ, Mohan PS, Peterhoff CM, Alldred MJ, Che S, Ginsberg SD, Nixon RA (2016) Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12:2467–2483
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64:113–122
Cai Q, Tammineni P (2016) Alterations in mitochondrial quality control in Alzheimer’s disease. Front Cell Neurosci 10:24
Nixon RA, Yang DS (2011) Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol Dis 43:38–45
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M et al (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141:1146–1158
Coffey EE, Beckel JM, Laties AM, Mitchell CH (2014) Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 263:111–124
Ji ZS, Mullendorff K, Cheng IH, Miranda RD, Huang Y, Mahley RW (2006) Reactivity of apolipoprotein E4 and amyloid beta peptide: lysosomal stability and neurodegeneration. J Biol Chem 281:2683–2692
Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG, Wu L, Yan SF et al (2017) PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 140:3233–3251
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R et al (2010) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One 5:e9979
Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y et al (2018) Parkin and PINK1 mitigate STING-induced inflammation. Nature 561:258–262
Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–273
He CC, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–U171
O’Neill C (2013) PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol 48:647–653
Perluigi M, Di Domenico F, Butterfield DA (2015) mTOR signaling in aging and neurodegeneration: at the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol Dis 84:39–49
Vartak RS, Rodin A, Oddo S (2019) Differential activation of the mTOR/autophagy pathway predicts cognitive performance in APP/PS1 mice. Neurobiol Aging 83:105–113
Bove J, Martinez-Vicente M, Vila M (2011) Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 12:437–452
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S (2010) Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem 285:13107–13120
Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA (2015) Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem 133:739–749
Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012) The transcription factor TFEB Links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5:42
Nezich CL, Wang C, Fogel AI, Youle RJ (2015) MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol 210:435–450
Tan S, Yu CY, Sim ZW, Low ZS, Lee B, See F, Min N, Gautam A et al (2019) Pomegranate activates TFEB to promote autophagy-lysosomal fitness and mitophagy. Sci Rep 9:727
Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA et al (2012) PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med 4:142ra197
Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, Swartzlander DB, Palmieri M et al (2014) Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med 6:1142–1160
Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM et al (2014) Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci 34:9607–9620
Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Tripoli DL et al (2015) Neuronal-targeted TFEB accelerates lysosomal degradation of APP, reducing Abeta generation and amyloid plaque pathogenesis. J Neurosci 35:12137–12151
Song JX, Malampati S, Zeng Y, Durairajan SSK, Yang CB, Tong BC, Iyaswamy A, Shang WB et al (2020) A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell 19:e13069
Settembre C, Medina DL (2015) TFEB and the CLEAR network. Methods Cell Biol 126:45–62
Zhang XL, Cheng XP, Yu L, Yang JS, Calvo R, Patnaik S, Hu X, Gao Q et al (2016) MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun 7:12109
Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A et al (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17:288–299
Puertollano R, Ferguson SM, Brugarolas J, Ballabio A (2018) The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J 37:e98804
Sekar P, Huang DY, Hsieh SL, Chang SF, Lin WW (2018) AMPK-dependent and independent actions of P2X7 in regulation of mitochondrial and lysosomal functions in microglia. Cell Commun Signal 16:83
Boland B, Yu WH, Corti O, Mollereau B, Henriques A, Bezard E, Pastores GM, Rubinsztein DC et al (2018) Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov 17:660–688
Ravikumar B, Berger Z, Vacher C, O’Kane CJ, Rubinsztein DC (2006) Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet 15:1209–1216
Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci 28:6926–6937
Napoletano F, Baron O, Vandenabeele P, Mollereau B, Fanto M (2019) Intersections between regulated cell death and autophagy. Trends Cell Biol 29:323–338
Gibson GE, Shi Q (2010) A mitocentric view of Alzheimer’s disease suggests multi-faceted treatments. J Alzheimers Dis 20(Suppl 2):S591–S607
Zhang L, Guo XQ, Chu JF, Zhang X, Yan ZR, Li YZ (2015) Potential hippocampal genes and pathways involved in Alzheimer’s disease: a bioinformatic analysis. Genet Mol Res 14:7218–7232
Sheng ZH, Cai Q (2012) Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci 13:77–93
Dorn GW 2nd (2019) Evolving Concepts of Mitochondrial Dynamics. Annu Rev Physiol 81:1–17
Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20:1013–1022
Chan DC (2012) Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46:265–287
Pryde KR, Smith HL, Chau KY, Schapira AH (2016) PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol 213:163–171
Xian H, Liou YC (2019) Loss of MIEF1/MiD51 confers susceptibility to BAX-mediated cell death and PINK1-PRKN-dependent mitophagy. Autophagy 15:2107–2125
Yamada T, Dawson TM, Yanagawa T, Iijima M, Sesaki H (2019) SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 15:2012–2018
Han HL, Tan JDO, Wang RX, Wan HD, He YH, Yan XX, Guo JF, Gao QT et al (2020) PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep 21:e48686
Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29:9090–9103
Gan XQ, Huang SB, Wu L, Wang YF, Hu G, Li GY, Zhang HJ, Yu HY et al (1842) Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. BBA Mol Basis Dis 2014:220–231
Tyumentsev MA, Stefanova NA, Muraleva NA, Rumyantseva YV, Kiseleva E, Vavilin VA, Kolosova NG (2018) Mitochondrial dysfunction as a predictor and driver of Alzheimer’s disease-like pathology in OXYS rats. J Alzheimers Dis 63:1075–1088
Zhang L, Trushin S, Christensen TA, Bachmeier BV, Gateno B, Schroeder A, Yao J, Itoh K et al (2016) Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s disease. Sci Rep 6:18725
Gan X, Wu L, Huang S, Zhong C, Shi H, Li G, Yu H, Howard Swerdlow R et al (2014) Oxidative stress-mediated activation of extracellular signal-regulated kinase contributes to mild cognitive impairment-related mitochondrial dysfunction. Free Radic Biol Med 75:230–240
Xu LL, Shen Y, Wang X, Wei LF, Wang P, Yang H, Wang CF, Xie ZH et al (2017) Mitochondrial dynamics changes with age in an APPsw/PS1dE9 mouse model of Alzheimer’s disease. Neuroreport 28:222–228
Manczak M, Reddy PH (2012) Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet 21:2538–2547
Reddy PH, Oliver DM (2019) Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 8:488
Briston T, Hicks AR (2018) Mitochondrial dysfunction and neurodegenerative proteinopathies: mechanisms and prospects for therapeutic intervention. Biochem Soc Trans 46:829–842
Wang W, Yin J, Ma X, Zhao F, Siedlak SL, Wang Z, Torres S, Fujioka H et al (2017) Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum Mol Genet 26:4118–4131
Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X (2008) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 105:19318–19323
Baek SH, Park SJ, Jeong JI, Kim SH, Han J, Kyung JW, Baik SH, Choi Y et al (2017) Inhibition of Drp1 ameliorates synaptic depression, Abeta deposition, and cognitive impairment in an Alzheimer’s disease model. J Neurosci 37:5099–5110
LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 3:862–872
Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31:454–463
Mattson MP, Chan SL (2003) Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium 34:385–397
Suzuki K, Sorimachi H, Yoshizawa T, Kinbara K, Ishiura S (1995) Calpain: novel family members, activation, and physiologic function. Biol Chem Hoppe Seyler 376:523–529
Perrin BJ, Huttenlocher A (2002) Calpain. Int J Biochem Cell Biol 34:722–725
Mercken M, Grynspan F, Nixon RA (1995) Differential sensitivity to proteolysis by brain calpain of adult human tau, fetal human tau and PHF-tau. FEBS Lett 368:10–14
Yang LS, Ksiezak-Reding H (1995) Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur J Biochem 233:9–17
Atherton J, Kurbatskaya K, Bondulich M, Croft CL, Garwood CJ, Chhabra R, Wray S, Jeromin A et al (2014) Calpain cleavage and inactivation of the sodium calcium exchanger-3 occur downstream of Abeta in Alzheimer’s disease. Aging Cell 13:49–59
Jin N, Yin X, Yu D, Cao M, Gong CX, Iqbal K, Ding F, Gu X et al (2015) Truncation and activation of GSK-3beta by calpain I: a molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci Rep 5:8187
Saito K, Elce JS, Hamos JE, Nixon RA (1993) Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A 90:2628–2632
Taniguchi S, Fujita Y, Hayashi S, Kakita A, Takahashi H, Murayama S, Saido TC, Hisanaga S et al (2001) Calpain-mediated degradation of p35 to p25 in postmortem human and rat brains. FEBS Lett 489:46–50
Wang W, Zhang F, Li L, Tang F, Siedlak SL, Fujioka H, Liu Y, Su B et al (2015) MFN2 couples glutamate excitotoxicity and mitochondrial dysfunction in motor neurons. J Biol Chem 290:168–182
Jiang S, Shao C, Tang F, Wang W, Zhu X (2019) Dynamin-like protein 1 cleavage by calpain in Alzheimer’s disease. Aging Cell 18:e12912
Prinz M, Priller J, Sisodia SS, Ransohoff RM (2011) Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 14:1227–1235
Lucin KM, Wyss-Coray T (2009) Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 64:110–122
Wyss-Coray T, Mucke L (2002) Inflammation in neurodegenerative disease--a double-edged sword. Neuron 35:419–432
Giunta B, Fernandez F, Nikolic WV, Obregon D, Rrapo E, Town T, Tan J (2008) Inflammaging as a prodrome to Alzheimer’s disease. J Neuroinflammation 5:51
Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S (2017) Inflammaging and ‘Garb-aging’. Trends Endocrinol Metab 28:199–212
Mosher KI, Wyss-Coray T (2014) Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol 88:594–604
Ojala J, Alafuzoff I, Herukka SK, van Groen T, Tanila H, Pirttila T (2009) Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol Aging 30:198–209
Prolla TA (2002) DNA microarray analysis of the aging brain. Chem Senses 27:299–306
Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ et al (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36:401–414
Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S et al (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560:198–203
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D et al (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678
Trautmann A (2009) Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal 2:pe6
Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463–477
Zhong ZY, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D et al (2016) NF-kappa B restricts inflammasome activation via elimination of damaged mitochondria. Cell 164:896–910
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–230
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Yan Y, Finkel T (2017) Autophagy as a regulator of cardiovascular redox homeostasis. Free Radic Biol Med 109:108–113
Radi E, Formichi P, Battisti C, Federico A (2014) Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis 42(Suppl 3):S125–S152
Zhao Y, Zhao B (2013) Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxidative Med Cell Longev 2013:316523
Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X (1842) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 2014:1240–1247
Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP (2017) NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med 214:1351–1370
Sun N, Youle RJ, Finkel T (2016) The mitochondrial basis of aging. Mol Cell 61:654–666
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865
Venegas C, Heneka MT (2019) Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J 33:13075–13084
Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, Vieira-Saecker A, Schwartz S et al (2017) Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 552:355–361
Ising C, Venegas C, Zhang SS, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S et al (2019) NLRP3 inflammasome activation drives tau pathology. Nature 575:669
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225
Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, Horng T (2014) Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci U S A 111:15514–15519
Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS et al (2019) LC3-associated endocytosis facilitates beta-amyloid clearance and mitigates neurodegeneration in murine Alzheimer’s disease. Cell 178:536–551 e514
Dionisio PEA, Oliveira SR, Amaral J, Rodrigues CMP (2019) Loss of microglial parkin inhibits necroptosis and contributes to neuroinflammation. Mol Neurobiol 56:2990–3004
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1:a006189
Caselli RJ, Beach TG, Yaari R, Reiman EM (2006) Alzheimer’s disease a century later. J Clin Psychiatry 67:1784–1800
Cotman CW, Anderson AJ (1995) A potential role for apoptosis in neurodegeneration and Alzheimer’s disease. Mol Neurobiol 10:19–45
Eckert A, Marques CA, Keil U, Schussel K, Muller WE (2003) Increased apoptotic cell death in sporadic and genetic Alzheimer’s disease. Ann N Y Acad Sci 1010:604–609
Rohn TT, Head E, Nesse WH, Cotman CW, Cribbs DH (2001) Activation of caspase-8 in the Alzheimer’s disease brain. Neurobiol Dis 8:1006–1016
Yuan JY, Yankner BA (2000) Apoptosis in the nervous system. Nature 407:802–809
Su JH, Anderson AJ, Cummings BJ, Cotman CW (1994) Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 5:2529–2533
Troncoso JC, Sukhov RR, Kawas CH, Koliatsos VE (1996) In situ labeling of dying cortical neurons in normal aging and in Alzheimer’s disease: correlations with senile plaques and disease progression. J Neuropathol Exp Neurol 55:1134–1142
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ et al (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1:112–119
Yuan J, Amin P, Ofengeim D (2019) Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat Rev Neurosci 20:19–33
Wallach D, Kang TB, Dillon CP, Green DR (2016) Programmed necrosis in inflammation: toward identification of the effector molecules. Science 352:aaf2154
Schenk B, Fulda S (2015) Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene 34:5796–5806
Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, Cai Q, Yang ZH et al (2017) RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 8:14329
Yamada T, Adachi Y, Fukaya M, Iijima M, Sesaki H (2016) Dynamin-related protein 1 deficiency leads to receptor-interacting protein kinase 3-mediated necroptotic neurodegeneration. Am J Pathol 186:2798–2802
Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT et al (2017) Necroptosis activation in Alzheimer’s disease. Nat Neurosci 20:1236
Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou C, Das S, Adiconis X et al (2017) RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc Natl Acad Sci U S A 114:E8788–E8797
Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H et al (2016) RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353:603–608
Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L et al (2014) Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81:1001–1008
Iannielli A, Bido S, Folladori L, Segnali A, Cancellieri C, Maresca A, Massimino L, Rubio A et al (2018) Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep 22:2066–2079
Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X et al (2015) Activation of necroptosis in multiple sclerosis. Cell Rep 10:1836–1849
Yang SH, Lee DK, Shin J, Lee S, Baek S, Kim J, Jung H, Hah JM et al (2017) Nec-1 alleviates cognitive impairment with reduction of Abeta and tau abnormalities in APP/PS1 mice. EMBO Mol Med 9:61–77
Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, Dewan MZ, Lieberman SR et al (2017) Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med 214:3687–3705
Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA et al (2014) Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124:3987–4003
Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M et al (2016) The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell 37:337–349
Funding
This work was supported by the National Natural Science Foundation of China (No. 81673209).
Author information
Authors and Affiliations
Contributions
Mingxue Song is the first author of this work, Xiulan Zhao is the co-author, and Fuyong Song is the corresponding author. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Song, M., Zhao, X. & Song, F. Aging-Dependent Mitophagy Dysfunction in Alzheimer’s Disease. Mol Neurobiol 58, 2362–2378 (2021). https://doi.org/10.1007/s12035-020-02248-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02248-y