
Abstract
DNA damage has been reported to induce cell cycle–related neuronal death. This is significant as aberrant cell cycle re-entry of mature, post-mitotic neurons contributes to neurodegeneration. In this study, we investigate how DNA damage elicited by exposure to the topoisomerase I inhibitor camptothecin (CPT) leads to cycle-related death of cultured cortical neurons and examine the function of E2F1 in this process. CPT treatment induced cell cycle initiation of cortical neurons and elevated the expression of certain cell cycle components (e.g., cyclin D1, CDK4, E2F1) but failed to drive S phase entry or DNA synthesis. The arrest in the cell cycle is explained by the elevated expression of the CDK inhibitor p21Cip1. Though its level was increased after CPT treatment, E2F1 did not drive treated neurons into the G1-S phase transition. E2F1 overexpression led to cell cycle activation and acute neuronal apoptosis without detectable entry of the neurons into S phase. ChIPseq analysis demonstrated that E2F1 predominantly occupies positions on or near the promoters of cell cycle related genes. Instead, in CPT-treated neurons, E2F1 preferentially regulated DNA repair related genes. Our study reveals that the functions of E2F1 in postmitotic neurons are context-dependent and offers novel insights into the role of E2F1 in DNA damage induced cycle-related neuronal death.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As terminally differentiated cells, mature neurons have permanently left the cell cycle. Nevertheless, growing evidence supports the idea that post-mitotic neurons can and do begin a cell cycle like process in response to natural or artificial stimuli. Instead of finishing a normal cell division, however, “cycling” neurons eventually die. The first evidence for linkage between cell cycle and neuron death came from studies on SV40 T-antigen transgenic mice. Cell type–specific expression of this viral oncogene resulted in cell cycle re-entry accompanied by apoptosis of cerebellar Purkinje neurons [1] or retinal photoreceptor neurons [2]. Additional evidence came from the analysis of the nervous system of mice carrying a homozygous mutation that disrupted the Retinoblastoma gene (Rb). These animals exhibited extensive apoptosis of postmitotic neurons associated with aberrant S phase entry [3, 4].
The phenomenon of cycle-related neuronal death has also been explored in neuronal culture systems. Cultured neurons exhibit cell cycle dependent cell death when deprived of nerve growth factor or neuronal activity. The critical nature of the cell cycle process was shown by the finding that the death could be abolished by cyclin dependent kinase (CDK) inhibitors [5,6,7]. Importantly, cell cycle activation is robustly associated with neuronal loss during the pathogenesis of neurodegenerative disorders including Alzheimer’s disease (AD). Toxic β-amyloid (Aβ), one of the biochemical markers of the AD brain, triggers a cell cycle mediated death of cultured mouse cortical neurons [8, 9]. Hyper-phosphorylated tau, another pathological hallmark of AD, appears to be a necessary part of the process that leads to neuronal cell cycle re-entry after Aβ exposure [10]. Other insults, including inflammatory factors, excitotoxicity and genotoxic reagents, can also drive a lethal cell cycle event in neurons [11,12,13].
Studies attempting to explain how the cell cycle is linked to cell death have consistently focused on the pRb/E2F1 complex. In non-dividing cells, the tumor suppressor protein retinoblastoma (pRb) binds to E2F1, keeping it in an inactivated state. Following growth stimulation, pRb is hyperphosphorylated. This blocks the ability of pRb to bind E2F1. Released E2F1 forms a dimer with DP1 and acts as a transcription factor, initiating the transcription of genes needed for cell cycle progression. In other situations (e.g., after DNA damage) E2F1 induces cell death by again acting as a transcription factor, but this time activating apoptosis-related genes. For example, in Rb deficient mouse embryos, nerve cells exhibit abnormal S phase entry and apoptosis in association with increased levels of free E2F1 and cyclin E [4]. E2F1 is sufficient to promote apoptosis of cerebellar granule cells induced by potassium deprivation, and suppressing E2F1 action with a CDK inhibitor can block this effect [14]. E2F1 mediates this effect by modulating the expression of CDC2 [6], as well as other apoptosis-related genes. For example, the pro-apoptotic BH3-only protein, Bim, has been identified as a direct target of E2F1 during cycle related apoptosis of sympathetic neurons deprived of NGF [15, 16]. E2F1 was also reported to control cycle-related death upon DNA damage through de-repressing other pro-apoptotic genes including b-myb and c-myb [17].
Several lines of evidence support the concept that DNA damage induces neuronal cell death through cell cycle re-entry. The first evidence was from the Greene laboratory whose work showed that neuronal death induced by UV exposure could be blocked by overexpression of a CDK inhibitor such as p16. They further showed that genotoxic agents lead to an increase of cyclin D-associated kinase activity [18]. Several years later, Kruman et al. reported that treatment of neuronal cultures with the topoisomerase II inhibitor, etoposide, induced neuronal apoptosis coupled with S phase entry [11]. These findings suggest that S phase entry could be a pivotal step triggering programmed death of neurons, yet the mechanism linking the two processes has never been satisfactorily laid out. One hypothesis is that, unlike cycling cells, neurons respond to DNA damage through re-entering cell cycle for DNA repair. This idea was supported by the finding that cell cycle activation is essential for DNA damage repair in cultured rat cortical neurons treated with H2O2 [19]. It was further shown that cyclin C–dependent G0 exit is required for the activation of non-homologous end joining (NHEJ) after DNA double-strand breaks (DSBs) in postmitotic neurons [20]. Nevertheless, these findings only explain a potential role of cell cycle machinery in neurons bearing repairable DNA damage. Details of the mechanism underlying DNA damage-induced cell cycle related neuron death remain unclear. If as hypothesized, S phase entry is required for apoptosis of the cycling neurons, then E2F1, a necessary factor enabling the G1-S transition, must play a significant role.
In this study, we investigate how DNA damage elicited by exposure to the topoisomerase I inhibitor camptothecin (CPT) leads to the cell cycle related death of cultured cortical neurons and examine the function of E2F1 in this process. We show that CPT treatment of cortical neurons induces an increase in the levels of E2F1 as well as cell cycle initiation (cyclin D1 upregulation) but fails to drive S phase entry or DNA synthesis. Using ChIPseq, we demonstrate that E2F1 predominantly occupies genomic locations near cell cycle genes, but its effects in CPT-treated neurons are more closely associated with its occupancy of DNA repair genes. Overexpression of E2F1 leads to cell cycle activation but not progression and drives neuronal apoptosis. Our findings are the first comprehensive look at the genomic locations of E2F1 in neurons and confirm its context dependent function.
Materials and Methods
Cell Cultures
Embryonic cortical neurons were isolated from E16.5 mice according to standard procedures [21]. Briefly, cerebral cortices were dissociated into single cells by mild trypsinization and trituration. After neutralization, cells were suspended in Neurobasal medium supplemented with B27 and 2 mM GlutaMax (Thermo Fisher Scientific) and then seeded onto coverslips or dishes coated with 0.5 mg/ml of poly-l-lysine (Sigma). Neuronal enrichment of the cell population in culture was performed using the protocol previously reported [21]. We added 1 μM 5-fluoro-2′-deoxyuridine (FdU) (F0503, Sigma) to the culture at DIV4 (4 days in vitro) where it remained for 24 h to kill the proliferating cells. On DIV5, medium containing FdU was replaced with fresh medium without FdU. The resulting enriched cultures of primary neurons were grown for an additional 8 days before any treatment.
Murine Neuro2A cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS for routine passage. To differentiate N2a cells, the growth medium was replaced with DMEM without FBS. Cells were kept in differentiation medium for 24 h before use.
Plasmids and Transfection
pMAX-GFP-E2F1 plasmid was a gift from Dr. Olimpia Meucci (Drexel University, Philadelphia, PA). The pCMV-E2F1, pCMV-E2F1 (E132), and pCMV-E2F1 (1–284) plasmids were provided by Dr. Kristian Helin (Danish Cancer Society, Copenhagen, Denmark). The pCMV-HA-hRB vector was a gift from Dr. Steven Dowdy (University of California, San Diego, CA, USA). The pCMV-Neo-Bam-DP1 vector was a gift from Dr. Jacqueline Lees (The Massachusetts Institute of Technology, Cambridge, MA, USA). Plasmids were transfected into N2a cells and primary neurons using Lipofectamine 2000 and Lipofectamine LTX (Thermo Fisher Scientific) respectively according to manufacturer’s protocols.
Immunofluorescence
Cells were washed with PBS and then fixed in cold 4% PFA for 15 min followed by PBS wash. Cells were blocked with 5% donkey serum in PBS containing 0.3% Triton X-100 at room temperature (RT) for 1 h and then incubated with primary antibodies at 4 °C overnight. After rinsing with PBS, cells were incubated with secondary antibodies at RT for 1 h. Cells were then washed with PBS and counter-stained with DAPI (4′,6-diamidino-2-phenylindole) (Thermo Fisher Scientific) for 5 min. After rinsing, all coverslips were mounted with Hydromount media (National Diagnostics) on glass slides. Images were taken using a Leica TCS SP8 confocal laser scanning microscope or an Olympus DP80 fluorescent microscope.
For bromodeoxyuridine (BrdU) labelling, 10 μM BrdU (Sigma) was added to the culture medium and incubated for 24 h before fixation. Before staining, DNA was hydrolyzed by exposing the cells to 2 N HCl for 10 min. Specimens were then neutralized in 0.1 M sodium borate (pH = 8.5) for 20 min and then rinsed extensively in PBS (three times). Immunostaining was then proceeded as described above.
Co-immunoprecipitation
Primary cortical neurons were lysed with 1× RIPA lysis buffer (Millipore) containing protease and phosphatase inhibitors (Roche). Samples (1 mg) of each protein lysate were incubated with either primary antibodies or normal control IgG at 4 °C overnight. The protein-antibody mixtures were then incubated with magnetic beads (Thermo Fisher Scientific) at 4 °C for 6 h, followed by rinsing with lysis buffer three times. Afterwards, the beads were isolated using a magnet according to the manufacturer’s protocol. The immune complexes were then eluted with 50 μl of 4× loading buffer (0.02% bromophenol blue, 40% glycerol, 250 mM Tris-HCl, 5% β-mercaptoethanol) by aggressive vortexing followed by incubation at room temperature, for 10 min. Further elution was achieved by heating the samples at 95 °C, for 5 min. Eluates were isolated using a magnet and were transferred to micro-centrifuge tubes containing 150 μl of lysis buffer and used for Western blot analysis.
Western Blot Analysis
Cells were homogenized in 1× RIPA lysis buffer (Millipore) containing protease and phosphatase inhibitors (Roche). Cell lysates were then centrifuged at 15,000 rpm at 4 °C for 20 min. After protein assay, samples were diluted with 4× loading buffer (0.02% bromophenol blue, 40% glycerol, 250 mM Tris-HCl, 5% β-mercaptoethanol) and RIPA lysis buffer to the required concentration and then denatured at 95 °C for 5 min. Protein samples were separated by SDS polyacrylamide gel electrophoresis and transferred to a PVDF membrane (Bio-Rad). After blocking with TBST containing 5% bovine serum albumin (BSA) (Sigma) or 5% non-fat milk (Bio-Rad), membranes were incubated with primary antibodies overnight at RT. After washing with TBST, membranes were incubated with HRP-conjugated secondary antibodies (Cell Signaling Technology) for 1 h at RT. Protein bands were visualized using ECL substrates (34075, 34095, or 34580, Thermo Fisher Scientific). The intensities of the bands were quantified by ImageJ (NIH) and normalized to the levels of GAPDH.
Reverse Transcription and Quantitative RT-PCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen) according to the manufacture’s protocol. Equal amounts of RNA were reverse transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Real-time PCR was performed using Roche LightCycler 480 SYBR Green I Master on a LightCycler 480 instrument. The mRNA level of Rpl13 was used as an internal control.
FACS
Primary cortical neurons were treated with DMSO or CPT (s1288, Selleckchem) and then digested with 0.25% Trypsin-EDTA (Thermo Fisher Scientific) for 5 min at 37 °C. After adding the media back, neuron suspensions were centrifuged, washed with 1× PBS, and fixed in cold 4% PFA for 10 min, followed by rinsing in 1× PBS. For BrdU labeling, DNA was hydrolyzed by exposing the cells to 2 N HCl for 15 min. Specimens were then neutralized in 0.1 M sodium borate buffer (pH = 8.5) for 10 min and then rinsed with PBS. Non-specific binding was blocked with 5% donkey serum in PBS containing 0.3% Triton X-100 at RT for 30 min and then incubated with antibodies against MAP2 and cyclin D1 or BrdU at RT for 1 h. After rinsing with PBS, cells were incubated with secondary antibodies at RT for 30 min. Donkey anti-rabbit 488, donkey anti-rat 488 (Thermo Fisher Scientific), and donkey anti-chicken 647 (Jackson ImmunoResearch) were used to detect cyclin D1, BrdU, and MAP2 respectively. After rinsing with 1× PBS, cells were resuspended in fresh medium and transferred to FACS tubes, followed by analysis with a Becton Dickinson FACS Aria III machine according to the manufacturer’s instructions. The results were processed with FlowJo software.
ChIP
Chromatin associated proteins from DMSO or CPT treated cortical neurons were crosslinked in 1% formaldehyde (in PBS) at RT for 10 min. Crosslinking was then quenched by addition of glycine to a final concentration of 125 mM followed by rinsing three times with ice-cold PBS. Cells were then scraped off the surface of the dish and collected in ice-cold PBS with protease inhibitor cocktail (Roche). After centrifugation, the cell pellet was re-suspended and lysed with SDS lysis buffer (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 0.5% SDS). The lysates were sonicated to generate DNA fragments with 500 bp average size. Sheared lysates were then pre-cleared by incubation with unblocked magnetic beads (Thermo Fisher Scientific) at 4 °C for 2 h. For immunoprecipitation, 5 μg of E2F1 antibody (sc-193x, Santa Cruz Biotechnology) or normal rabbit IgG (sc-2027, Santa Cruz Biotechnology) was added to the samples and incubated at 4 °C with gentle agitation overnight. Fresh magnetic beads were simultaneously blocked in dilution buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 2% glycerol) with 5% BSA plus 0.2 mg/ml salmon sperm DNA. The next day, blocked beads were added to the samples and allowed to incubate at 4 °C with agitation for 4 h. The beads were then isolated with a magnet and washed sequentially with ice-cold dilution buffer (5 min × 2), low salt wash buffer (20 mM Tris-HCl, pH 8.0, 0.1% SDS, 150 mM NaCl, 5 mM EDTA, pH 8.0, 1% NP-40) (5 min × 2), high salt wash buffer (20 mM Tris-HCl, pH 8.0, 0.1% SDS, 500 mM NaCl, 5 mM EDTA, pH 8.0, 1% NP-40) (5 min × 2), LiCl wash buffer (20 mM Tris-HCl, pH 8.0, 1% NP-40, 1% sodium deoxycholate, 250 mM LiCl, 5 mM EDTA, pH 8.0) (5 min × 2), and TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, pH 8.0) (5 min × 1). After the last wash, immune complexes were eluted with fresh elution buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, pH 8.0, 1% SDS, 0.1 M Na2CO3) with vigorous vortexing followed by incubation at 65 °C for 15 min. The elution step was repeated once. Crosslinking was reversed for both chromatin immunoprecipitation (ChIP) samples and input by incubation in elution buffer containing 200 mM NaCl at 65 °C overnight. On the following day, the samples were treated with 0.2 mg/ml RNase A (Thermo Fisher Scientific) at 37 °C for 1 h, followed by 0.2 mg/ml proteinase K (Thermo Fisher Scientific) at 62 °C for 2 h. Lastly, DNA was purified using phenol/chloroform extraction (Thermo Fisher Scientific) and eluted with nuclease-free water. ChIP on N2a cells was performed using an identical protocol.
ChIP-seq and Downstream Analysis
Cortical cultures treated with DMSO or CPT were harvested for chromatin immunoprecipitation (ChIP) as described above. ChIP DNA was further sheared to an average size of 300 bp using a Covaris S220 focused-ultrasonicator before library preparation. DNA libraries were constructed using the Ovation Ultralow System V2 kit (NuGEN Technologies). The final purified DNA library pool was sequenced using NextSeq 500/550 High Output Kit v2 (75 cycles) (Illumina, Inc) on an Illumina NextSeq 500 instrument. Quantity control of the reads was performed using NGSQCToolkit_v2.3.3. Reads were aligned to the reference mouse genome: GRCm38/mm10, using Bowtie 2, v2.1.0 [22]. The same file was sorted transferred into a bam file by samtools 1.3.1. Peak calling was done using MACS 2 with parameters “-B -g mm --nomodel --extsize 200 (p < 0.05)” [23]. Differential binding regions were identified by comparing two pileup trucks of two conditions using MACS 2 with parameters “bdgdiff -g 60 -l 120.” Peaks were annotated using R package ChIPseeker [24]. A heatmap of the signal near the TSS (± 3 kb) of all genes was generated using ngs.plot [25]. GO and KEGG pathway annotation were performed using R package clusterProfiler [26].
Statistical Analysis
Statistical analysis was performed using Prism 6 (GraphPad software). Differences between each group were assessed with unpaired Student’s T test. p < 0.05 was considered statistically different. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Camptothecin Induces DNA Double-Strand Breaks and Neuronal Loss
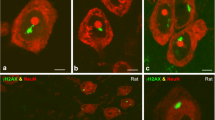
As a DNA topoisomerase I inhibitor, camptothecin (CPT) prevents DNA re-ligation during DNA synthesis and causes DNA breaks leading to apoptosis of mitotic cells [27, 28]. Given this cell cycle restricted mechanism of action, it is somewhat surprising that CPT also induces the apoptosis of postmitotic neurons [29]. The hypothesis is that, in cultured cortical neurons, CPT inhibits topoisomerase I (Top1) resulting in the accumulation of unrepairable DNA damage, which leads to neuronal apoptosis [29]. One explanation is that Top1 inhibition perturbs its function in relieving DNA supercoils generated by RNA polymerases leading to transcription-dependent genome instability [30]. We confirmed that CPT induces DNA double strand breaks (DSBs) in mouse cortical neurons. By immunostaining, we observed the formation of γH2AX foci in the nucleus of primary neurons after a short-term CPT treatment (Fig. 1a and b). γH2AX is regarded as a sensitive and early indicator of DSBs [31] and has been proved useful for the detection of low-level of DNA damage in cells [32, 33]. We also looked at 53BP1, a mediator of DNA repair that has been implicated in the repair of DSBs by non-homologous end joining (NHEJ) during G1 phase [34]. Similar to γH2AX, 53BP1 also formed foci in the neurons treated for a short time with CPT (Fig. 1a). These observations confirmed that postmitotic neurons, just as cycling cells, have a DNA damage response, and that they seem to repair DNA damage by NHEJ. After 24 h of treatment, we observed that around 30% of neurons were killed by CPT (Fig. 1c), revealing that CPT is highly toxic to postmitotic neurons.

Camptothecin induces DNA double strand breaks and neuronal loss in mouse cortical culture. a γH2AX and 53BP1 staining of DIV8 cortical neurons treated with 1 μM CPT. Scale bar = 25 μm. b The percentage of neurons with > 5 γH2AX foci after CPT treatment for 1 or 24 h. c Neuronal numbers (MAP2+ cells) were counted after DMSO or CPT treatment. Data from three independent experiments are represented as mean ± SEM. **p < 0.01; ****p < 0.0001
Camptothecin Triggers Elevated Expression of E2F1
It was reported previously that inhibition of cyclin-dependent kinases (CDKs) and G1/S cell cycle could suppress camptothecin-induced apoptosis of cultured sympathetic and cortical neurons [35]. This finding suggests that pathways controlling cell cycle are involved in neuronal apoptosis induced by CPT. To test this hypothesis, we stained primary neurons with antibody against G1 cell cycle component cyclin D1 and observed that the percentage of cyclin D1-positive neurons was significantly increased after a 24-h CPT treatment, confirming that CPT leads to aberrant activation of the neuronal cell cycle (Fig. 2a and b). Besides cyclin D1, the levels of other cell cycle and death related proteins were examined. Using Western blot analysis, however, we observed that different cell cycle proteins exhibited differential responses to CPT treatment. The proteins involved in cell cycle stages earlier than S phase (cyclin D1, CDK4, E2F1, PCNA) tended to increase their expression levels while other cell cycle proteins showed no significant change (cyclin E, cyclin B1) or decreased (cyclin A2) (Fig. 2c and d). As expected, levels of some apoptosis-related proteins including Bim EL and cleaved caspase-3 were elevated (Fig. 2c and d). By checking the mRNA levels using RT-qPCR, we found that the changes in mRNA were largely consistent with the levels of protein (Fig. 2e). Thus, the observed increases in cell cycle and cell death related proteins probably resulted from changes in gene expression. These data suggest that CPT induces neuronal apoptosis associated with ectopic cell cycle activation.

Camptothecin triggers elevated expression of certain cell cycle components in cultured cortical neurons. a, b Immunostaining of cyclin D1 in DIV8 cortical neurons treated with 1 μM CPT. Data from three independent experiments are represented as mean ± SEM. Scale bar, 50 μm. c, d Western blot of cell cycle and cell death related proteins in cortical neurons treated with 1 μM CPT for 24 h. Data from a minimum of three independent experiments are represented as mean ± SEM. e RT-qPCR to look at the gene expression levels of cell cycle and cell death related proteins after 1 μM CPT treatment for 24 h. Data from a minimum of four independent experiments are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001
Camptothecin Fails to Induce Neuronal S Phase Entry and DNA Synthesis
The pRb/E2F1 axis is a key regulator of the G1/S transition, and in cultured rat cortical neurons, it has been reported that genotoxic compounds lead to cell cycle initiation and DNA synthesis [11]. Yet, after CPT treatment of mouse cortical neurons, even though E2F1 was upregulated, we found that proteins that are required for S phase such as cyclin E and cyclin A2 were not accordingly elevated. Thus, CPT may not induce S phase entry of neurons. To verify this suggestion, we incubated cortical cultures in BrdU during CPT treatment. The cells were then harvested and FACS analysis performed. We found that after CPT treatment, the fraction of cyclin D1 positive neurons increased (Fig. 3a), yet we observed no increase in the percentage of BrdU positive neurons (Fig. 3b). CPT therefore triggers only the earliest events of the cell cycle; it fails to drive S phase entry and DNA replication.

Camptothecin fails to induce S phase entry in cortical neurons. a, b DIV8 cortical neurons treated with DMSO or 1 μM CPT for 24 h were co-labeled with MAP2-Cy7 and cyclin D1 or BrdU antibody followed by FITC-conjugated 2nd antibody and then were analyzed with FACS. c DIV8 cortical neurons treated with DMSO or 1 μM CPT for 24 h were lysed to perform Western blot in order to analyze pRb phosphorylation. d Quantification of the level of p-pRb (S780) and p-pRb (S807) normalized to GAPDH level. Data from four independent experiments are represented as mean ± SEM. **p < 0.01
In a normal cell cycle, hyperphosphorylation of pRb is required to release E2F1, which then forms a heterodimer with DP1 to activate the transcription of genes for S phase. To further confirm the inability of CPT to drive S phase, we examined pRb phosphorylation after CPT treatment. Unlike earlier reports [36], we observed decreased phosphorylation of pRb in neurons treated with CPT (Fig. 3c and d). Taken together, these data indicate that CPT-mediated DNA damage does not induce S phase entry and DNA replication in cultured cortical neurons.
Elevated Expression of p21Cip1 in Neuronal Cells Treated with Camptothecin
We have shown that CPT induces expression of G1 cell cycle components (cyclin D1 and CDK4) but fails to induce hyperphosphorylation of pRb or S-phase markers. We therefore hypothesized that the activity of CDK4 was somehow suppressed. During a typical cell cycle, CDK4 activity can be inhibited by several CKIs (cyclin-dependent kinase inhibitors), such as p16INK4, p19INK4, p21Cip1, and p27Kip1. Using RT-qPCR, we found that in neurons treated with CPT, p21 mRNA was greatly increased, even as the levels of p16 mRNA were downregulated (Fig. 4d). Consistent with this, CPT treatment significantly increased the number of neurons with a strong p21 immunostaining signal (Fig. 4a). Western blot results confirmed these findings (Fig. 4b and c). Next, we used a co-immunoprecipitation assay to determine if cyclin D1 functionally interacted with CDK4 and found that cyclin D1 failed to form a complex with CDK4 in either control or CPT-treated cortical neurons. In order to block the cyclin D1/CDK4 complex and suppress CDK4 kinase activity, p21 interacts with cyclin D1 through the cyclin-binding motif [37]. Significantly, CPT treatment induced the physical interaction of cyclin D1 and p21 (Fig. 4e). Our data suggest that even though CPT drives expression of cyclin D1 and CDK4, they fail to form a functional complex because of the elevated levels of p21.

Camptothecin upregulates the expression of CDK inhibitor p21Cip1. a Immunostaining of p21 in DIV8 primary cortical neurons treated with either DMSO or 1 μM CPT. Scale bar, 50 μm. b, c Western blot of p21 protein levels in neurons treated with DMSO or 1 μM CPT, for 24 h. d RT-qPCR determined mRNA levels of several CKI genes in neurons treated with DMSO or 1 μM CPT, for 24 h. e Lysates of primary neurons treated with either DMSO or 1 μM CPT for 24 h were immunoprecipitated with cyclin D1 antibody followed by Western blot analysis. Co-IP was repeated for three times. Quantitative data from a minimum of three independent experiments are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001
E2F1 Predominantly Binds to the Promotor Regions of Cell Cycle-Related Genes in Primary Cortical Neurons
Despite the elevated expression of cyclin D1 and CDK4 after CPT, no DNA synthesis was observed, even though E2F1 was significantly upregulated. Therefore, E2F1 presumably does not function in its normal role of promoting the G1-S transition. We reasoned that since most of the data on E2F1 function comes from observations made in epithelial cells lines there remained the possibility that the role of E2F1 in postmitotic neurons might be different. To test this, we performed chromatin immunoprecipitation combined with high throughput sequencing (ChIPseq) to uncover the E2F1 target genes in neurons under different physiological conditions. As described above, enriched cortical cultures were treated with CPT for 24 h and then harvested for ChIP-seq analysis. The DMSO-containing buffer without CPT was used as a control. We first analyzed the distribution of E2F1-binding in the vicinity of transcription start sites (TSS), using a 3-kb window. We found that in both control and CPT-treated neurons, the majority of E2F1-binding sites were located within 1.5 kb of the TSS (Fig. 5a). Indeed, in both groups, E2F1 peaks were mostly commonly found in promoter regions (Fig. 5b). Next, we analyzed E2F1-binding motifs using MEME-ChIP. We verified that the most significant E2F1 binding motif was nearly identical to the canonical E2F1-binding motif defined in other cells—TTTSSCGC (S=C or G) (Fig. 5c).

E2F1 predominantly binds to the promoter region of cell cycle-related genes in cultured cortical neurons, and is involved in DNA repair in camptothecin-treated neurons. a Average profile of ChIP peaks within 3 kb of TSS. b E2F1 binding sites were mostly distributed in promoter and distal intergenic regions. c E2F1 binding motifs in primary cortical neurons. d GO biological process of E2F1 enriched regions in primary cortical neurons. e KEGG analysis of E2F1 enriched sites in primary cortical neurons. f Venn diagram shows the numbers of significantly changed genes in both CPT and control (DMSO) and both conditions. g Comparison of respective GO biological process analysis data from CPT and control conditions. h GO biological process analysis of differential binding events between CPT and DMSO samples
To clarify the categories of genes associated with E2F1 binding sites in primary neurons, we performed a gene ontology (GO) analysis. This analysis revealed that the enriched regions in control neurons were most significantly associated with DNA replication and DNA repair (Fig. 5d). KEGG pathway analysis further showed that the enriched regions were mostly significantly associated with pathways involved in cell cycle and DNA replication (Fig. 5e). These data provided the first evidence that in mouse primary cortical neurons, E2F1 predominantly binds to, and presumably regulates, cell cycle and DNA replication-related genes. Unexpectedly, no cell death-related genes appeared in the GO terms, suggesting that in WT primary cortical neurons, E2F1 may not be involved with the expression of cell death genes. Taken together, our data suggest that E2F1 is deeply involved in cell cycle regulation in cultured cortical neurons even though these cells are postmitotic.
E2F1 Is Involved in DNA Repair in Camptothecin-Treated Neurons
We next compared the E2F1 binding regions in CPT-treated neurons with those found in untreated (DMSO) control cells. First, peaks of the two conditions were called separately. A total of 487 enriched regions were identified in the control group while 398 were identified in the CPT group. Significantly, 243 of the genes identified were found in both groups (Fig. 5f). Next, we performed a simple comparison of respective GO analysis data from both conditions. In both control and CPT groups, the enriched E2F1 binding events were most significantly associated with genes responsible for DNA replication (Fig. 5g). Finally, to determine the genes that were more highly enriched in the CPT group compared to the control, we performed call differential binding events to determine where in the genome, the CPT responsive genes were located. GO analysis of these differential binding sites showed that the regions related to DNA repair were more highly enriched in CPT group than in the DMSO group (Fig. 5h). This indicates that even though E2F1 predominantly sits on cell cycle-related genes in both CPT and control conditions, CPT-induced DNA damage enhanced the presence of E2F1 on the promoters of DNA repair-related genes. The implication is that E2F1 is an important part of DNA damage response in primary cortical neurons.
To further examine how E2F1 is involved in DNA repair, we treated the neurons with an E2F1 inhibitor HLM006474. E2F1 inhibition aggravated DNA damage and apoptosis (Figure S1A), suggesting a direct role for E2F1 in the DNA repair process in CPT-treated neurons instead of in the induction of cell cycle and cell death. Non-homologous end joining (NHEJ) is the most prominent form of DSBs repair in postmitotic neurons [38]. The PI3K family member, DNA-PKcs, is an important component of the NHEJ machinery. Consistent with this relationship, if we inhibited DNA-PKcs with NU7441, CPT-induced neuronal apoptosis was significantly aggravated (Figure S1B). This finding suggests that treatment with CPT triggers a DNA-PKcs-mediated repair pathway. Significantly, the CPT-induced elevation of E2F1 was abolished by DNA-PKcs inhibition (Figure S1B), indicating that E2F1 is a part of the DNA-PKcs-mediated repair pathway.
Overexpression of E2F1 Triggers Cell Cycle Activation and Neuronal Apoptosis
While the CPT studies suggest a role for E2F1 in DNA repair, the functional significance of the presence of E2F1 on the promoters of cell cycle-related genes remained unexplained. We therefore turned to overexpression studies in postmitotic neurons. We transfected cultured cortical neurons with a plasmid encoding E2F1 and found that over 80% of the transfected neurons entered into cell cycle, as assessed with staining for Ki67, a general cell cycle marker (Fig. 6a and f). Next, we asked if neurons overexpressing E2F1 can enter S phase and replicate DNA. We found that even though some neurons expressed cyclin A2 (Fig. 6b and f), E2F1 overexpression was unable to stimulate DNA replication as assessed by BrdU incorporation (Fig. 6c and f). Although the transfected neurons could not fully enter into S phase, overexpression of E2F1 did initiate a cell death process. By immunostaining cleaved caspase-3, we observed that these neurons underwent apoptosis, exhibited DNA fragmentation and ultimately died (Fig. 6e and f). By immunostaining the pro-apoptotic protein Bim (which plays an important role in the intrinsic apoptosis pathway and has been identified as a E2F1 target [15]), we found that E2F1 overexpression triggered a typical apoptotic process in cultured cortical neurons (Fig. 6d and f).

Overexpression of E2F1 leads to cell cycle activation and neuronal apoptosis. DIV8 cortical neurons were transfected with pEGFP-N1 or pMax-GFP-E2F1 plasmid and harvested after 24 h. a, b Cells were co-immunostained with the neuronal marker MAP2 and the cell cycle protein Ki67 or cyclin A2. c BrdU incorporation assay was performed to detect replicated DNA. d, e Cells were co-immunostained with MAP2 and the apoptotic protein Bim d or cleaved caspase-3 e. Scale bar, 25 μm. f Quantification of the results illustrated in a–e. Data from a minimum of three independent experiments are represented as mean ± SEM; ***p < 0.001; ****p < 0.0001
Neuronal Cell Cycle–Related Death Induced by E2F1 Overexpression Is Dependent on Its DNA Binding Activity
Overexpression of E2F1 thus induces cell cycle events and apoptosis of cultured cortical neurons. To examine whether this effect was dependent on its classical role as a transcription factor, we repeated our transfection studies using an E2F1 mutant (E2F1 E132) with a mutation that eliminates its DNA binding activity by replacing the sequences encoding amino acids L132 and N133 (CTGAAT) with an EcoRI restriction site sequence (GAATTC) but other domains are not altered [39]. As expected, the DNA binding deficient E2F1 failed to drive cell cycle re-entry of cortical neurons (Fig. 7a and b). Interestingly, a C-terminal truncated E2F1 that loses the transactivation domain but retains the domain needed to bind DNA could drive a significant fraction of the cells to become Ki67 positive (Fig. 7a and b). While highly significant, the effect was not as pronounced as that for wild type E2F1. To determine the effect of mutant E2F1 on neuronal death, we transfected both wild type and mutant E2F1 and incubated the transfected neurons for extended times. We found that wild type and C-terminal truncated E2F1 led to significant neuron death between 24 and 48 h after transfection, but the E132 DNA binding mutant was ineffective even at these later times (Fig. 7c). This observation suggests a consistent effect on cell cycle and death induced by E2F1 in cortical neurons.

Neuronal cell cycle-related death induced by E2F1 overexpression is dependent on its DNA binding activity. a, b DIV8 primary neurons were transfected with pCMV-E2F1, pCMV-E2F1(E132), or pCMV-E2F1(1-284) and fixed after 24 h. Cells were stained with the cell cycle marker Ki67, and the percentage of Ki67-positive neurons was quantified. Scale bar, 25 μm. c Primary neurons were harvested after transfection with different E2F1 mutants for 12, 24, or 48 h, and neuronal numbers were counted. d Chromatin immunoprecipitation of E2F1 in differentiated N2a cells transfected with wildtype and DNA binding mutant E2F1 plasmids, followed by PCR with the primers against the 5′ flanking region of cell cycle and death related genes. e mRNA levels of cell cycle and cell death related genes in differentiated N2a expressing wild type or DNA binding defective E2F1. Rpl13 was used as the internal control. Data from a minimum of three independent experiments are represented as mean ± SEM; *p < 0.05; ***p < 0.001; ****p < 0.0001
With the knowledge that DNA binding activity is required for E2F1 to induce both cell cycle and cell death, we asked if E2F1 directly bound to the genes encoding cell cycle and death-related proteins. We performed ChIP-qPCR on differentiated Neuro2A cells transfected with E2F1. We found a significant enrichment of wild-type E2F1 on the promoter regions of cell cycle and death-related genes such as Mki67, Ccna2, Casp3, and Bcl2l11 (Fig. 7d). By contrast, the DNA binding mutant did not bind to these genes. Using RT-qPCR, we further validated that E2F1 indeed upregulated the mRNA level of some cell cycle and death-related genes, but the DNA binding mutant could not (Fig. 7e). Taken together, our findings indicate that E2F1 regulates cell cycle-related neuronal death by binding to the promoter regions of cell cycle and death-related genes and modulating their transcription.
Discussion
We show here, using a CPT-based model, that DNA damage leads to the cell cycle-related death of cultured cortical neurons. G1 cell cycle components (cyclin D1 and CDK4) are upregulated, but the transition to S phase appears to be blocked as no DNA synthesis can be observed. The block is most likely caused by the elevated expression of the CDK inhibitor p21Cip1. E2F1 is involved in the neuronal CPT response, not as a regulator of the cell cycle, but rather as a part of the DNA repair process. Nonetheless, in keeping with its occupancy of the promoter regions of cell cycle-related genes, E2F1 overexpression can drive cell cycle initiation, but even this cycle is stopped before S phase can begin. Instead, the excess E2F1 drives an apoptotic cell death process. Our findings are very much in keeping with previous observations that the behavior of E2F1 is highly context dependent. The lack of entrance into a formal S-phase, coupled with the rapid loss of neuronal differentiation markers at early stages of the cell death process, has the paradoxical effect of making it difficult to prove that the cells that are entering the cell division process are the ones that die. Nonetheless, all previous work points to this linkage [12, 40, 41].
Growing evidence suggests that ectopic cell cycle reentry leads to neuronal death [1, 3]. Cycle-related neuronal death (CRND) has been regarded as contributing to multiple neurodegenerative disorders including AD [42]. Learning more about the mechanisms underlying CRND, therefore, would help in understanding the pathogenesis of AD and provide potential therapeutic targets for AD treatment. Numerous studies have identified factors that can drive neurons into cell cycle: Aβ, tau, inflammatory stresses, excitotoxicity, genotoxic reagents, and so on [8,9,10,11,12, 18, 43, 44]. To date, investigations linking neuronal re-entrance into the cell cycle with neurodegeneration have focused heavily on the pRb/E2F1 axis. Classically, pRb/E2F1 forms a conserved pathway that controls cell cycle progression in cycling cells. The fact that E2F1 regulates multiple biological processes including cell cycle, apoptosis, and DNA damage responses has led to the hypothesis that E2F1 may work as a link between cell cycle and neuron death.
In cycling cells, when DNA damage is detected, the cell cycle is arrested at one of several checkpoints to allow the DNA repair to be completed [45, 46]. In postmitotic neurons, however, DNA damage reportedly induces cell cycle reentry and apoptosis [11, 18, 35, 43]. Camptothecin (CPT) is a DNA topoisomerase I inhibitor that can induce apoptosis of postmitotic neurons [29]. Seen in light of the finding that inhibition of the G1/S transition activates the neuronal apoptotic pathway [35], we speculate that, as a key regulator for G1/S transition, E2F1 may play a critical role in this process. In this study, we confirm that CPT treatment induces DNA double strand breaks (DSBs) and apoptosis in mouse cortical neurons where it selectively increases the expression of early cell cycle stage proteins—cyclin D1 and CDK4. In contrast, the S phase cyclin, cyclin A2, decreases dramatically and BrdU incorporation was not observed. The simplest interpretation of these findings is that DNA damage induces cell cycle activation, but these cycling neurons cannot engage a true cell cycle; they fail to enter S phase. This block in cell cycle progression is coupled with and possibly explained by the elevated expression of p21Cip1 and inhibition of the interaction between cyclin D1 and CDK4 [37].
Cyclin D1 synthesis and its nuclear localization are critical for cell cycle progression during G1 phase. After DNA damage, however, cyclin D1 is downregulated to allow for DNA repair. For example, in UV-irradiated G1 fibroblasts, cyclin D1 disappears from the nucleus and overexpression of cyclin D1 impairs DNA repair [47]. Given its increased expression after DNA damage in CPT-treated neurons, however, it seems likely that cyclin D1 has unique functions in non-dividing nerve cells. This concept is supported by the finding that cyclin D1-dependent kinases are essential for the apoptosis of neuronal cells [48]. Cyclin D1-associated kinase activity was detected to be significantly increased in cortical neurons treated with CPT [18], but the exact physiological function of endogenous cyclin D1 in neurons remains elusive. One possibility that would be consistent with our findings is that nuclear cyclin D1 directly functions in homologous recombination-mediated DNA repair in cancer cells [49]. This suggests that its expression may be beneficial for the survival of aging (DNA damaged) neurons.
Uncovering target genes is an important approach to investigate the biological functions of a transcription factor. Although there have been previous studies of the genomic targets of E2F1 in other cell types, we are the first to perform a genome-wide analysis of the genomic binding sites of E2F1 in cortical neurons. Our finding that E2F1 consistently binds to the genomic promoter regions of genes related to DNA replication and cell cycle is significant, especially since neurons are supposed to have permanently exited the cell cycle. The fact that E2F1 promotor occupancy in CPT-treated and control neurons differs primarily in the extent to which E2F1 is bound to DNA repair genes further enriches this picture. In the aggregate, the ChIPseq data imply that even though E2F1 mainly binds to cell cycle-related genes in primary cortical neurons, it tends to regulate genes associated with DNA repair in neurons with DNA breaks evoked by CPT treatment.
The finding of an E2F1 response to DNA damage is intriguing. E2F1 levels are elevated in cells treated with DNA damage agents, including etoposide, doxorubicin, UV radiation, and others [50, 51]. In tumor cells treated with genotoxic reagents, E2F1, but not E2F2 or E2F3, is stabilized by phosphorylation by ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and RAD3-related (ATR). E2F1 is also phosphorylated by checkpoint kinase 2 (Chk2) in cancer cells treated with etoposide [52] where it results in cell death, as it does in mouse thymocytes with induced DNA damage [53]. In addition to phosphorylation, E2F1 can be acetylated in response to DNA damage, a modification that is required for apoptosis. ChIP assays have revealed that acetylation shifts E2F1 from cell cycle-related genes to pro-apoptotic genes [51]. In addition to this function in cell death, E2F1 recruits DNA repair factors, such as NBS1, RPA, and Rad51 [54]. E2F1 depletion also impairs nucleotide excision repair (NER) in UV-exposed primary human fibroblasts [55]. ChIP-microarray assays have identified a set of genes involved in DNA repair processes that are directly regulated by E2F1 in primary human fibroblasts, including genes involved in mismatch repair (MSH2, MLH1), base excision repair (UNG), nucleotide excision repair (RPA3), homologous recombination (RAD51 and RAD54), and non-homologous end-joining (DNA-dependent protein kinase) [56]. In the current study, ChIPseq of E2F1 in primary cortical neurons revealed that E2F1 directly targets DNA repair genes, such as Brca1, Brca2, Rpa1, Rpa2, Xrcc2, Pcna, Chk1, and more.
One question that remains unanswered after our work is the nature of the E2F1-dependent DNA repair process used by neurons treated with CPT. As they are postmitotic cells, neurons rely heavily on NHEJ to repair DSBs [38]. After DNA damage induced by camptothecin, inhibition of DNA-PK aggravated neuronal death provoked by CPT, suggesting that neurons highly rely on DNA-PKcs-mediated NHEJ to repair damaged DNA and survive. The observation that E2F1 inhibition aggravates DNA damage and neuronal apoptosis induced by CPT provides further evidence for a potential role of E2F1 in promoting DNA repair in neurons even though the detailed mechanism still remains to be uncovered.
Our results tend to rule out a role for E2F1 in promoting apoptosis during the CPT-induced cycle-related death of primary cortical neurons. Based on our ChIPseq data, E2F1 does not target pro-apoptotic genes in neurons. We speculate that the reason for this is that elevation of the E2F1 expression induced at the concentrations of CPT that we used is not enough to mediate apoptosis. This idea is supported by a recent report that E2F1 expression level is an important determinant of its function in determining the fate of a cell. Low levels of E2F1 promote proliferation, moderate levels of E2F1 induce cell cycle arrest, and very high levels promote apoptosis [57]. Indeed, when we overexpressed E2F1 in cortical neurons, we also observed acute apoptosis of neurons, indicating a pro-apoptotic function of E2F1 in neurons when concentrations are sufficiently high. The neuronal death induced by E2F1 overexpression is coupled with aberrant cell cycle activation, but no S phase entry could be observed, suggesting a predominant role of E2F1 in triggering apoptosis. We also observed increased expression of checkpoint kinase 1 (Chk1) and the DNA damage marker γH2AX in E2F1-overexpressing neurons (Figure S2), which offers an explanation for why the cell cycle of the neurons was arrested in G1 phase. Since we have shown that both cell cycle and cell death activities induced by E2F1 overexpression are dependent on its DNA binding activity, it is likely that E2F1 functions by modulating the transcription of the relevant cell cycle or cell death-related genes.
References
Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT (1992) Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron 9:955–966
al-Ubaidi MR, Hollyfield JG, Overbeek PA, Baehr W (1992) Photoreceptor degeneration induced by the expression of simian virus 40 large tumor antigen in the retina of transgenic mice. Proc Natl Acad Sci U S A 89:1194–1198
Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359:288–294
Macleod KF, Hu Y, Jacks T (1996) Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J 15:6178–6188
Freeman RS, Estus S, Johnson EM Jr (1994) Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of Cyclin D1 during programmed cell death. Neuron 12:343–355
Konishi Y, Bonni A (2003) The E2F-Cdc2 cell-cycle pathway specifically mediates activity deprivation-induced apoptosis of postmitotic neurons. J Neurosci 23:1649–1658
Padmanabhan J, Park DS, Greene LA, Shelanski ML (1999) Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci 19:8747–8756
Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM, Canonico PL, Nicoletti F et al (1999) Mitotic signaling by beta-amyloid causes neuronal death. FASEB J 13:2225–2234
Varvel NH, Bhaskar K, Patil AR, Pimplikar SW, Herrup K, Lamb BT (2008) Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease. J Neurosci 28:10786–10793
Seward ME, Swanson E, Norambuena A, Reimann A, Cochran JN, Li R, Roberson ED, Bloom GS (2013) Amyloid-beta signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J Cell Sci 126:1278–1286
Kruman II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, Emokpae R Jr, Gorospe M et al (2004) Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41:549–561
Wu Q, Combs C, Cannady SB, Geldmacher DS, Herrup K (2000) Beta-amyloid activated microglia induce cell cycling and cell death in cultured cortical neurons. Neurobiol Aging 21:797–806
Park DS, Obeidat A, Giovanni A, Greene LA (2000) Cell cycle regulators in neuronal death evoked by excitotoxic stress: implications for neurodegeneration and its treatment. Neurobiol Aging 21:771–781
O’Hare MJ, Hou ST, Morris EJ, Cregan SP, Xu Q, Slack RS, Park DS (2000) Induction and modulation of cerebellar granule neuron death by E2F-1. J Biol Chem 275:25358–25364
Biswas SC, Liu DX, Greene LA (2005) Bim is a direct target of a neuronal E2F-dependent apoptotic pathway. J Neurosci 25:8349–8358
Biswas SC, Shi Y, Vonsattel JP, Leung CL, Troy CM, Greene LA (2007) Bim is elevated in Alzheimer’s disease neurons and is required for beta-amyloid-induced neuronal apoptosis. J Neurosci 27:893–900
Liu DX, Greene LA (2001) Regulation of neuronal survival and death by E2F-dependent gene repression and derepression. Neuron 32:425–438
Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA (1998) Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol 143:457–467
Schwartz EI, Smilenov LB, Price MA, Osredkar T, Baker RA, Ghosh S, Shi FD, Vollmer TL et al (2007) Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle 6:318–329
Tomashevski A, Webster DR, Grammas P, Gorospe M, Kruman II (2010) Cyclin-C-dependent cell-cycle entry is required for activation of non-homologous end joining DNA repair in postmitotic neurons. Cell Death Differ 17:1189–1198
Hui CW, Zhang Y, Herrup K (2016) Non-neuronal cells are required to mediate the effects of neuroinflammation: results from a neuron-enriched culture system. PLoS One 11:e0147134
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM et al (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137
Yu G, Wang LG, He QY (2015) ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31:2382–2383
Shen L, Shao N, Liu X, Nestler E (2014) ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15:284
Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16:284–287
Ryan AJ, Squires S, Strutt HL, Johnson RT (1991) Camptothecin cytotoxicity in mammalian cells is associated with the induction of persistent double strand breaks in replicating DNA. Nucleic Acids Res 19:3295–3300
Hsiang YH, Hertzberg R, Hecht S, Liu LF (1985) Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260:14873–14878
Morris EJ, Geller HM (1996) Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell cycle-independent toxicity. J Cell Biol 134:757–770
Marinello J, Chillemi G, Bueno S, Manzo SG, Capranico G (2013) Antisense transcripts enhanced by camptothecin at divergent CpG-island promoters associated with bursts of topoisomerase I-DNA cleavage complex and R-loop formation. Nucleic Acids Res 41:10110–10123
Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM (2000) Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 275:9390–9395
Banath JP, Macphail SH, Olive PL (2004) Radiation sensitivity, H2AX phosphorylation, and kinetics of repair of DNA strand breaks in irradiated cervical cancer cell lines. Cancer Res 64:7144–7149
Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC et al (2013) Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat Neurosci 16:613–621
Panier S, Boulton SJ (2014) Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol 15:7–18
Park DS, Morris EJ, Greene LA, Geller HM (1997) G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci 17:1256–1270
Park DS, Morris EJ, Bremner R, Keramaris E, Padmanabhan J, Rosenbaum M, Shelanski ML, Geller HM et al (2000) Involvement of retinoblastoma family members and E2F/DP complexes in the death of neurons evoked by DNA damage. J Neurosci 20:3104–3114
Chen J, Saha P, Kornbluth S, Dynlacht BD, Dutta A (1996) Cyclin-binding motifs are essential for the function of p21CIP1. Mol Cell Biol 16:4673–4682
Chow HM, Herrup K (2015) Genomic integrity and the ageing brain. Nat Rev Neurosci 16:672–684
Johnson DG, Schwarz JK, Cress WD, Nevins JR (1993) Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature 365:349–352
Cicero S, Herrup K (2005) Cyclin-dependent kinase 5 is essential for neuronal cell cycle arrest and differentiation. J Neurosci 25:9658–9668
Zhang J, Herrup K (2011) Nucleocytoplasmic Cdk5 is involved in neuronal cell cycle and death in post-mitotic neurons. Cell Cycle 10:1208–1214
Busser J, Geldmacher DS, Herrup K (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J Neurosci 18:2801–2807
Zhang Y, Qu D, Morris EJ, O’Hare MJ, Callaghan SM, Slack RS, Geller HM, Park DS (2006) The Chk1/Cdc25A pathway as activators of the cell cycle in neuronal death induced by camptothecin. J Neurosci 26:8819–8828
Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P (2005) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 25:5446–5454
Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15:2177–2196
Roos WP, Kaina B (2006) DNA damage-induced cell death by apoptosis. Trends Mol Med 12:440–450
Pagano M, Theodoras AM, Tam SW, Draetta GF (1994) Cyclin D1-mediated inhibition of repair and replicative DNA synthesis in human fibroblasts. Genes Dev 8:1627–1639
Kranenburg O, van der Eb AJ, Zantema A (1996) Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J 15:46–54
Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T et al (2011) A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 474:230–234
Hofferer M, Wirbelauer C, Humar B, Krek W (1999) Increased levels of E2F-1-dependent DNA binding activity after UV- or gamma-irradiation. Nucleic Acids Res 27:491–495
Pediconi N, Ianari A, Costanzo A, Belloni L, Gallo R, Cimino L, Porcellini A, Screpanti I et al (2003) Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat Cell Biol 5:552–558
Stevens C, Smith L, La Thangue NB (2003) Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol 5:401–409
Lin WC, Lin FT, Nevins JR (2001) Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev 15:1833–1844
Chen J, Zhu F, Weaks RL, Biswas AK, Guo R, Li Y, Johnson DG (2011) E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 10:1287–1294
Guo R, Chen J, Zhu F, Biswas AK, Berton TR, Mitchell DL, Johnson DG (2010) E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J Biol Chem 285:19308–19315
Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16:245–256
Shats I, Deng M, Davidovich A, Zhang C, Kwon JS, Manandhar D, Gordan R, Yao G et al (2017) Expression level is a key determinant of E2F1-mediated cell fate. Cell Death Differ 24:626–637
Acknowledgments
We thank Dr. Hiu Tung Cheung and Ms. Pui Shuen Wong for the help with ChIPseq experiment.
Funding
This work is supported by grants from the Research Grants Council (RGC) of the Hong Kong SAR (16101315, 660813, AoE/M-604/16). Additional support was received from the Hong Kong University of Science and Technology (R9321).
Author information
Authors and Affiliations
Contributions
YZ and KH designed the study. YZ and XS conducted experiments. YZ, XS, and KH wrote and edited the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interest.
Ethics Approval
All animal procedures were approved by Animal and Plant Care Facility of Hong Kong University of Science and Technology.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, Y., Song, X. & Herrup, K. Context-Dependent Functions of E2F1: Cell Cycle, Cell Death, and DNA Damage Repair in Cortical Neurons. Mol Neurobiol 57, 2377–2390 (2020). https://doi.org/10.1007/s12035-020-01887-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-01887-5