
Abstract
The complement C1q plays a critical role in microglial phagocytosis of glutamatergic synapses and in the pathogenesis of neuroinflammation in Alzheimer’s disease (AD). We recently reported that upregulation of metabotropic glutamate receptor signaling is associated with increased synaptic C1q production and subsequent microglial phagocytosis of synapses in the rodent models of AD. Here, we explored the role of astrocytic glutamate transporter in the synaptic C1q production and microglial phagocytosis of hippocampal glutamatergic synapses in a rat model of AD. Activation of astrocyte and reduction glutamate transporter 1 (GLT1) were noted after bilateral microinjection of amyloid-beta (Aβ1–40) fibrils into the hippocampal CA1 area of rats. Ceftriaxone is a β-lactam antibiotic that upregulates GLT1 expression. Bilateral microinjection of ceftriaxone recovered GLT1 expression, decreased synaptic C1q production, suppressed microglial phagocytosis of glutamatergic synapses in the hippocampal CA1, and attenuated synaptic and cognitive deficits in rats microinjected with Aβ1–40. In contrast, artificial suppression of GLT1 activity by dl-threo-beta-benzyloxyaspartate (DL-TBOA) in naïve rats induced synaptic C1q expression and microglial phagocytosis of glutamatergic synapses in the hippocampal CA1 area, resulting in synaptic and cognitive dysfunction. These findings demonstrated that impairment of astrocytic glutamate transporter plays a role in the pathogenesis of AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The complement 1q (C1q) is an initiator of the classical pathway of complement activation. C1q is upregulated in neurons and glial cells in different neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease, Huntington’s disease, and frontotemporal dementia [1,2,3,4,5,6,7,8,9,10]. For instance, significantly increased C1q expression was observed in many plaques and neurons in human AD brain [2]. C1q is a ligand for C1qRp, a transmembrane receptor on microglia [11]. C1q-mediated microglial activation plays a pivotal role in synaptic pruning and remodeling that are characteristic of neurodegenerative disorders such as AD [3, 6, 12,13,14].
Metabotropic glutamate receptor 1 (mGluRl) is a member of a large family of G protein–coupled glutamate receptors and is involved in the regulation of synaptic plasticity and spatial and associative learning [15,16,17,18,19]. We have shown that amyloid fibrils increased the activity of mGluRl that resulted in activation of mGluR-protein phosphatase 2A (PP2A) signaling, dephosphorylation of fragile X mental retardation protein (FMRP), and increased local translation of synaptic C1q mRNA [14]. This resulted in C1q upregulation and microglial phagocytosis of glutamatergic synapse [14]. Impairment of synaptic plasticity was noted in mice lacking mGluR1 [16], and overactivation of mGluRl plays an important role in the pathogenesis of synaptic and cognitive deficits in the rodent models of AD [20, 21]. In contrast, suppression of mGluR1 signaling or knockout of mGluR1 receptor attenuated the microglial phagocytosis of hippocampal synapses, and recovered the central synaptic and cognitive function in the rodent models of AD [14]. The present study was aimed to further explore the potential mechanism of extracellular glutamate accumulation and its involvement in the induction of C1q-mediated microglial phagocytosis of the hippocampal glutamatergic synapses.
Several types of glutamate transporters regulate glutamate homeostasis at the synaptic and extrasynaptic sites through active uptake of glutamate in the central nervous system [22, 23]. Among the five subtypes of glutamate transporters in the brain, GLAST (glutamate aspartate transporter/excitatory amino acid transporter-1; EAAT1) and glutamate transporter 1 (GLT1/EAAT2) are primarily located on astrocytes, and EAAT3 is mostly present on neurons [24]. Astrocytes are equipped to remove glutamate from the synaptic cleft, to maintain homeostasis, and convert it to glutamine through the action of glutamine synthetase [25, 26]. Glutamine is then released and taken up by neurons to replenish neurotransmitter pools in both glutamatergic and GABAergic neurons [27]. Astrocytic GLT1 is responsible for ~ 95% of all glutamate uptake [28], and GLAST accounts for ∼ 5% of the total glutamate transport in the adult brain [28, 29]. The expression of astrocytic GLAST and GLT1 in the hippocampus and other brain regions of AD patients appear to be markedly impaired, even in early clinical stages of the disease [30, 31]. In addition, partial loss of GLT1 (GLT1+/−) accelerated the development of cognitive deficits in a transgenic AD mouse model (APPswe/PS1ΔE9 mice) [32]. Considering the critical role of glutamate transporter GLT1 in regulating extracellular glutamate content, we studied the impact of astrocytic GLT1 in C1q-mediated microglial phagocytosis of hippocampal glutamatergic synapses and synaptic and cognitive impairment in a rodent model of AD.
Based on recent work from several groups, including ours [3, 10, 14], we hypothesized that amyloid fibrils induce downregulation of astrocytic glutamate transporter GLT1, which results in upregulation of synaptic C1q and microglial phagocytosis of glutamatergic synapses in the hippocampal CA1 area, leading to impaired glutamatergic transmission and cognitive deficit.
Materials and Methods
Animals
All animal procedures were approved by the Animal Care and Use Committee of Cleveland Clinic. Adult male Sprague–Dawley (250–300 g, 2–3 months of age, Charles River) rats were used, and all experiments were performed during the light cycle. The animals were randomly assigned to different groups with specific treatment, and another party blinded the experimenter to the individual groups. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in the previous publications [33, 34].
Microinjection in the Hippocampal CA1 Area
Rats were anesthetized with sodium pentobarbital (45 mg/kg i.p.) and restrained in a stereotaxic apparatus [35]. Aβ1–40 fibrils were formed as described previously [36]. Aβ1–40 fibrils (10 μg/3 μl) or 3 μl of artificial cerebrospinal fluid (aCSF) was injected stereotaxically and bilaterally into each hippocampus (anteroposterior, − 3.5 mm; mediolateral, ± 2.0 mm; dorsoventral, − 3.0 mm) [37] using a 10-μl Hamilton syringe with a 27-G stainless steel needle at a rate of 0.5 μl/min. This experimental model has been previously used for studying AD [14, 34, 36, 38, 39].
For in vivo treatment by microinjection, a 26-gauge double-guide cannula was inserted into the brain, aimed at the hippocampal CA1 area (the same coordinate as above) [34]. The guide cannula was then cemented in place to the skull and securely capped. The rat was allowed to recover for at least 5 days before subsequent treatment. Specific glutamate transporter inhibitor DL-TBOA (DL-TBOA; 10 nmol/side × 7 days), ceftriaxone (0.1 mg/side × 7 days), or aCSF was delivered daily into the hippocampal CA1 area through a 33-gauge double injector at a rate of 0.5 μl/min for 7 days. Behavioral tests were performed 14 days after amyloid fibril microinjection, when substantial neuroinflammation and synaptic dysfunction is expected to be established in the hippocampal CA1 [34, 40, 41]. Cellular and molecular analyses were performed 1 day after the behavioral test. The injection sites for the hippocampal CA1 were histologically verified afterward by injecting the same dose of ink [14, 40].
Morris Water Maze Test
The Morris water maze test was employed to determine the memory function of rats [36, 40, 42, 43]. The water maze model was performed in a circular tank (diameter 1.8 m) filled with opaque water. A platform (15 cm in diameter) was submerged below the water’s surface in the center of the target quadrant. The swimming path of the animal was recorded by a video camera and analyzed by EthoVision XT software (Noldus Information Technology). Each rat underwent 4 trials per day for 5 days. They underwent four trials a day for 7 days with a 10-min inter-trial interval. For each training session, the animals were placed into the maze consecutively from four random points of the tank, and were allowed to search for the platform. If the animal did not find the platform within 120 s, they were gently guided to it. Animals were allowed to remain on the platform for 20 s. The latency for each trial was recorded for analysis. During the probe trial, the platform was removed from the tank and rats were allowed to swim in the maze for 60 s.
Protein Extraction and Immunoblotting
The protocol for protein extraction and immunoblotting was generally based on the previous reports [35, 44]. The hippocampal CA1 tissues or synaptosomal preparations were collected and lysed in ice-cold lysis buffer containing 50 mM Tris–Cl, 150 mM NaCl, 0.02 mM NaN2, 100 μg/ml phenylmethyl sulfonyl fluoride, 1 μg/ml aprotinin, 1% Triton X-100, and proteinase and phosphatase inhibitor cocktail. The proteins were extracted and subjected to 7.5–15% SDS–PAGE followed by immunoblotting. The blots were incubated overnight at 4 °C with the primary antibodies as follows: monoclonal anti-C1q antibody (1:1000; Abcam, ab71940), polyclonal anti-GLT1 antibody (1:1000; Cell Signaling Technology, #3838), and monoclonal anti-β-actin antibody (1:2000; Santa Cruz Biotechnology, sc-81178). The membranes were washed extensively and then incubated with horseradish peroxidase (HRP)–conjugated anti-mouse and anti-rabbit IgG antibody (1:10,000; Jackson ImmunoResearch Laboratories Inc., West Grove, PA). The immunoreactivity was detected using enhanced chemiluminescence (ECL Advance Kit; Amersham Biosciences). The intensity of the bands was captured digitally and analyzed quantitatively with ImageJ software. The immunoreactivity of all proteins was normalized to that of β-actin.
Immunostaining and 3D Confocal Imaging
Immunostaining on the serial sections containing hippocampal CA1 area in all groups (30 μm, 15–20/rat, n = 5 rats per group) was performed as previously described [35, 40]. Mouse monoclonal antibodies against the microglial marker Iba1 (1:500, Abcam, ab5076), glutamatergic synapse marker PSD95 (1:200, Abcam, ab104898), lysosomal marker CD68 (1:200, Abcam, ab955), monoclonal anti-C1q antibody (1:1000; Abcam, ab71940), and glial fibrillary acidic protein (GFAP, an astrocyte marker) (1:200, Abcam, ab71940) were used. The sections were then incubated with FITC-, Cy3-conjugated secondary antibody (1:500, Jackson ImmunoResearch), or Alex Fluor 633 (1:500, Invitrogen) for 1 h. Mouse IgG isotype (Abcam, ab188776) was applied as control of C1q immunostaining in the same groups of tissues as above. Negative controls were run in parallel and involved omission of one or both primary antibodies and/or inclusion of an irrelevant isotype control antibody. All such controls were devoid of staining. All sections were examined by confocal microscopy, and fluorescent images were acquired using a Leica TCS-SP8-AOBS inverted confocal microscope (Leica Microsystems, GmbH, Wetzlar, Germany). Sections were examined and analyzed in a blinded fashion. Image rendering and analyses were performed using Image-Pro Plus (Media Cybernetics, Inc., Rockville, MD) and Velocity (PerkinElmer, Waltham, MA). The presentation and analysis of internalization of PSD95 in microglial (Iba1) lysosomes (CD68) were adopted from the method described by Schafer et al. [45] with minor modification. Briefly, the internalization of PSD95 in microglial lysosomes was defined by rotating the 3D micrographs to confirm the colocalization of synaptic markers with CD68 immunoreactivity within microglia in hippocampal CA1. The representative 3D images were presented with resolution 1024 × 1024 dpi, z-step size 0.3 μm. Usually, a 200 × 200 × 20 μm neuropil (containing about 9–12 microglia) in each section and 4 sections in each animal were randomly sampled and analyzed in different groups. The volume of PSD95 within lysosome (CD68-positive) and the total volume of these synaptic markers in a neuropil (200 μm × 200 μm × 20 μm) were measured by Velocity (PerkinElmer, Waltham, MA), and the ratio was calculated as the measurement of microglial phagocytosis of synapses in hippocampal sections in all groups using the following formula: PSD95 engulfment (%) = volume of CD68-positive PSD95 puncta (μm3) / volume of total PSD95 (μm3) × 100.
Hippocampal Slice Preparation and Whole-Cell Recordings
Brain slices containing hippocampal CA1 areas were prepared as previously described [35, 46, 47]. The brain was quickly removed and cut on a Vibratome in cold physiological saline to obtain coronal slices (300 μm thick) containing the hippocampus. Whole-cell voltage-clamp recordings from the CA1 area were taken using an Axopatch 200B amplifier (Molecular Devices) with 2–4-MΩ glass electrodes containing the internal solution (mM): K-gluconate or cesium methanesulfonate, 125; NaCl, 5; MgCl2 1; EGTA, 0.5; Mg-ATP, 2; Na3GTP, 0.1; HEPES, 10; pH 7.3; 290–300 mOsmol. A seal resistance of ≥ 2 GΩ and an access resistance of 15–20 MΩ were considered acceptable. The series resistance was optimally compensated by ≥ 70% and constantly monitored throughout the experiments. Schaffer collateral–commissural fibers were stimulated by ultrathin concentric bipolar electrodes (FHC Inc.), and the excitatory postsynaptic currents (EPSCs) were recorded in the CA1 area in the presence of bicuculline (30 μM). The evoked EPSCs were filtered at 2 kHz, digitized at 10 kHz, and acquired and analyzed using Axograph X software. The amplitude of the EPSCs was monitored for a baseline period of at least 15 min. Miniature EPSC (mEPSC) was recorded in the presence of tetrodotoxin (TTX, 1 μM) and bicuculline (10 μM) at a holding potential of − 70 mV. All electrophysiological experiments were performed at room temperature.
Compounds
Aβ peptide consisting of residues 1–40 of the human wild-type sequence (Aβ1–40) was purchased from Bachem (Torrance, CA). Bicuculline and other chemicals were purchased from Sigma–Aldrich (St. Louis, MO) or Tocris (Ellisville, MO).
Statistical Analyses
Normality was tested using the Shapiro–Wilk test. For electrophysiological and behavioral analyses, the data were compared with two-way ANOVA. The data from the histological and Western blot studies were analyzed using one-way ANOVA test or Student’s t test. For non-normally distributed data, we used the Mann–Whitney U test or the Kruskal–Wallis test. Post hoc analyses were performed using the Student–Newman–Keuls test or the Dunn multiple comparisons test as appropriate. All statistical analyses were performed with BMDP statistical software (Statistical Solutions, Saugus, MA). All data were expressed as means ± SEM. For all tests, a two-tailed P < 0.05 was considered statistically significant.
Results
Amyloid Fibrils Induced Astrocyte Activation and Reduction of Glutamate Transporter GLT1
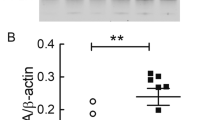
Astrocytes play a pivotal role in synaptic transmission by modulating GluR activity through GluT1-mediated control of synaptic and extra-synaptic glutamate clearance [48]. Following Aβ1–40 administration, we found the increased levels of GFAP immunoreactivity in the hippocampal CA1 area, indicating enhanced astrocyte activation (Fig. 1a). We also found a decreased glutamate transporter GLT1 expression, which was colocalized with GFAP immunoreactivity (Fig. 1b). We also observed a substantial reduction of GLT1 expression in the immunoblotting studies (Fig. 2a).

Reduction of GLT1 in astrocytes in the hippocampal CA1 in rats injected with amyloid fibrils. Increased immunoreactivity of astrocytic marker GFAP was noted in the hippocampal CA1 in the rats injected with Aβ1–40 (a, n = 5 rats in each group, two-tailed t = 4.34, DF = 8, P = 0.0003, scale bar = 100 μ). Decreased GLT1 immunoreactivity that was colocalized with GFAP was noted in the hippocampal CA1 in the rats injected with Aβ1–40 (b, n = 5 rats in each group, Mann–Whitney U statistic < 0.0001, two-tailed P = 0.008, scale bar = 10 μ). **, P < 0.01. Data represent mean ± SEM

Microinjection of ceftriaxone suppressed the synaptic C1q expression in the hippocampal CA1 area in rats injected with amyloid fibrils. Microinjection of ceftriaxone significantly recovered the expression of glutamate transporter GLT1 in the hippocampal CA1 in rats injected with amyloid fibrils (a, n = 7 rats in each group, F3,24 = 7.05, P = 0.0015). b Representative immunostaining images showed the increased the C1q expression, colocalized with the synaptic marker PSD95, in the hippocampal CA1 in rats injected with amyloid fibrils (b, n = 5 rats in each group, F3,16 = 14.95, P < 0.0001), which was significantly attenuated by microinjection with ceftriaxone. Representative immunoblots showing that ceftriaxone significantly attenuated C1q upregulation in the hippocampal CA1 in the modeled rats (c, n = 7 rats in each group, F3,24 = 7.03, P = 0.0015). Scale bar = 10 μ **, P < 0.01. Data represent mean ± SD
Upregulation of GLT1 by Ceftriaxone Suppressed Synaptic C1q Upregulation Induced by Amyloid Fibrils
Loss of GLT1 protein and reduced glutamate uptake are correlated with cognitive decline in AD patients [31, 49, 50]. Beta-lactam antibiotics stimulate GLT1 expression and function [51]. These include ceftriaxone, cefazolin, amoxicillin, ampicillin, and cefoperazone, of which ceftriaxone has been most studied. Considering the involvement of astrocytic glutamate transporter in the regulation of extrasynaptic glutamate hemostasis, we next tested the effect of artificial upregulation of GLT1 by ceftriaxone on the hippocampal C1q production and microglial phagocytosis of glutamatergic synapses. Ceftriaxone (0.1 mg × 7 days) was microinjected into the hippocampal CA1 in the modeled or control rats. Several studies have indicated that ceftriaxone can upregulate GLT1 expression (through GLT1 promoter activation [51]) in stroke [52], amyotrophic lateral sclerosis [51], and Parkinson’s disease [53]. As shown in Fig. 2a, microinjection of ceftriaxone restored the expression of glutamate transporter GLT1 in the hippocampal CA1 in rats injected with amyloid fibrils but did not affect the hippocampal GLT1 expression in the control rats. We also noted an upregulation of C1q expression, which was colocalized with the synaptic marker PSD95 in the modeled rats (Fig. 2b). Furthermore, administration of ceftriaxone attenuated Aβ1–40-induced C1q overproduction (Fig. 2b, c).
Upregulation of GLT1 by Ceftriaxone Attenuated Microglial Phagocytosis of Glutamatergic Synapses and Cognitive Impairment Induced by Amyloid Fibrils
Consistent with our previous report [14], we noted an increased internalization of synaptic marker PSD95 in microglial (CD68-positive) lysosomes, indicating phagocytosis of glutamatergic synapses in the hippocampal CA1 areas injected with amyloid fibrils, which was attenuated by the microinjection of ceftriaxone (Fig. 3). We noted a significant decrease in the amplitude and increase of inter-events interval of excitatory glutamatergic transmission in the hippocampal CA1 neurons in Aβ1–40-microinjected rats, which was recovered after ceftriaxone administration (Fig. 4a). Ceftriaxone also restored the basal glutamatergic strength in the hippocampal CA1 neurons in rats injected with amyloid fibrils (Fig. 4b). In the Morris water maze test, increased escape latency (Fig. 4c) and decreased time spent in the target quadrant (Fig. 4d) during the probe experiment were noted in rats injected with amyloid fibrils, which was alleviated by ceftriaxone (Fig. 4c, d). Taken together, these results demonstrated that recovery of GLT1 function by ceftriaxone attenuated the C1q-mediated microglial phagocytosis of glutamatergic synapses and mitigated the synaptic and cognitive dysfunction in a rodent model of AD.

Microinjection of ceftriaxone suppressed the microglial phagocytosis of glutamatergic synapses in the hippocampal CA1 in rats injected with amyloid fibrils. Significantly increased internalization of synaptic marker PSD95 within lysosomal marker CD68 immunosignal in microglia (Iba1-positive, green) was observed in hippocampal CA1 in the modeled rats, which was attenuated by ceftriaxone (n = 5 rats in each group, F3,16 = 67.2, P < 0.0001). Right micrographs were presented to show the same microglia in which only the lysosomes (CD68, red) and PSD95 (blue) were visualized. ***, P < 0.001. Scale bar = 10 μ. Data represent mean ± SEM

Microinjection of ceftriaxone recovered the hippocampal glutamatergic transmission and cognitive function in rats injected with amyloid fibrils. a Significantly decreased amplitude (a, n = 26 neurons in each group, Kruskal–Wallis statistic = 46.358, P < 0.0001) and extended inter-events interval of mEPSCs were observed in hippocampal CA1 neurons in rats injected with amyloid fibrils, which was attenuated by ceftriaxone. Significantly attenuated input (stimulus intensity)–output (EPSC amplitude) response of evoked EPSCs was also observed in the hippocampal CA1 neurons of rats injected with amyloid fibrils, which was recovered by ceftriaxone (b, n = 10 neurons in each group, effect of group (F3,36 = 7.48, P = 0.0005), effect of strength (F2,36 = 138.5, P < 0.0001), and interaction between group and strength (F6,36 = 2.17, P = 0.06)). Increased escape latency (c, n = 10 rats in each group, effect of group (F3,36 = 8.05, P = 0.0003), effect of time (F4,36 = 202.9, P < 0.0001), interaction between group and time (F12,36 = 0.88, P = 0.56)), and decreased time spent in the target quadrant (d, n = 10 rats in each group, F3,36 = 8.99, P = 0.0001) during the probe trial were noted in rats injected with amyloid fibrils, which was attenuated by ceftriaxone (T, target quadrant; R, right quadrant; O, opposite quadrant; L, left quadrant). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data represent mean ± SEM. For box-and-whiskers plots, the box extends from the 25th to 75th percentiles; a line within the box marks the median. Whiskers (error bars) above and below the box represent the minimum and maximum values
Suppression of GLT1 Function by DL-TBOA Induced Synaptic C1 Upregulation and Microglial Phagocytosis of Glutamatergic Synapse
Next, we test the effect of inhibiting GLT1 function by microinjection of DL-TBOA (10 nmol × 7 days) on the hippocampal C1q expression and microglial phagocytosis of glutamatergic synapse in naïve rats. Microinjection of DL-TBOA increased the expression of C1q in the hippocampal CA1 (Fig. 5a, b), which was largely colocalized with the synaptic marker PSD95 (Fig. 5b). We also found increased internalization of synaptic marker PSD95 within the lysosomes (CD68-positive) in microglia (Iba1 immunoreactivity) in naïve rats treated with DL-TBOA (Fig. 5c). In addition, microinjection of DL-TBOA significantly abolished the effect of ceftriaxone-induced suppression of C1q production in hippocampal CA1 in rats injected with amyloid fibrils (Supplementary Figure). These results indicated that suppression of GLT1 function by DL-TBOA induced synaptic C1q upregulation and microglial phagocytosis of glutamatergic synapses in the hippocampal CA1 in naïve rats.

Suppression of astrocytic GLT1 by DL-TBOA induced the C1q expression and microglial phagocytosis of glutamatergic synapses. Increased C1q expression, colocalized with the synaptic marker PSD95, in the hippocampal CA1 induced by DL-TBOA (a, n = 5 rats in each group, t = 11.95, DF = 8, two-tailed P < 0.0001). DL-TBOA induced C1q expression in the hippocampal CA1 (b, n = 7 rats in each group, t = 2.76, DF = 12, two-tailed P = 0.017). Increased internalization of synaptic marker PSD95 within lysosomal marker Iba1 immunosignal in microglia (CD68-positive, green) was observed in the hippocampal CA1 in rats injected with DL-TBOA (c, n = 5 rats in each group, t = 10.33, DF = 8, two-tailed P < 0.0001). Right micrographs were presented to show the same microglia in which only the lysosomes (CD68, red) and PSD95 (blue) were visualized. ***, P < 0.01. Data represent mean ± SEM
Suppression of GLT1 Function by DL-TBOA Impaired Synaptic and Cognitive Function
We then examined the effect of suppressing GLT1 function by DL-TBOA on the synaptic and cognitive function in naïve rats. As shown in Fig. 6a, repeated microinjection of DL-TBOA decreased the amplitude and increased the inter-events interval of mEPSCs in hippocampal CA1 neurons. It also decreased the strength of basal glutamatergic strength in these neurons (Fig. 6b). Microinjection of DL-TBOA increased escape latency in the Morris water maze test (Fig. 6c) and decreased the time spent in target quadrant in probe trail in the naïve rats (Fig. 6d). Together, these results demonstrated that suppression of GLT1 function by DL-TBOA impaired the synaptic and cognitive function in naïve rats.

Microinjection of DL-TBOA impaired the hippocampal glutamatergic transmission and cognitive function in rats. Significantly decreased amplitude (a, n = 23 neurons, unpaired t test, t = 2.85, DF = 44, two-tailed P = 0.0067) and extended inter-events interval (n = 23 neurons, unpaired t test, t = 2.99, DF = 44, two-tailed P = 0.0046) of mEPSCs were observed in hippocampal CA1 neurons in rats injected with DL-TBOA. Significantly decreased strength of evoked EPSCs in the hippocampal CA1 neurons induced by DL-TBOA (b, n = 11 neurons in each group, effect of group (F1,20 = 15.35, P = 0.0009), effect of EPSCs strength (F2,20 = 58.84, P < 0.0001), interaction between group and EPSCs strength (F2,20 = 3.21, P = 0.051)). Increased escape latency (c, n = 10 rats in each group, effect of group (F1,18 = 20.91, P = 0.0002), effect of time (F4,18 = 60.7, P < 0.0001), interaction between group and time (F4,18 = 0.84, P = 0.50)), and decreased time spent in the target quadrant (d, n = 10 rats in each group, F1,18 = 14.99, P = 0.0011) during the probe trial were noted in rats injected with DL-TBOA (T, target quadrant; R, right quadrant; O, opposite quadrant; L, left quadrant). Data represent mean ± SEM. For box-and-whiskers plots, the box extends from the 25th to 75th percentiles; a line within the box marks the median. Whiskers (error bars) above and below the box represent the minimum and maximum values. *, P < 0.05; **, P < 0.01
Discussion
C1q is normally synthesized in microglia and/or neurons in CNS, but upregulated expression of C1q is seen in normal aging and in neurodegenerative disorders [3, 10, 54]. Complement proteins (C1q-C3) are involved in the microglial synaptic remodeling and phagocytosis that occur normally during development [45]. Increased complement-mediated synaptic loss is seen in different neurological disorders including AD [1, 3, 10, 12, 14, 55, 56]. Astrocyte transforming growth factor β (TGF-β) can induce the C1q expression in synapses [57].
We recently reported that activation of mGluR signaling induces synaptic C1q upregulation and microglial phagocytosis of the hippocampal glutamatergic synapses that resulted in synaptic and cognitive deficiency in the rodent models of AD [14]. Here, we extended our hypothesis by providing evidence that inhibition of astrocytic glutamate transporter GluT1 increased synaptic C1q production and microglial phagocytosis of the hippocampal synapses in the rodent model of AD. Glutamate transporters, through a secondary active transport machinery with Na+/H+/K+ exchange, uptake the extrasynaptic glutamate and maintain the glutamate concentration gradients of 106 across the plasma membrane in the brain under physiological conditions [58]. Dynamic functional adaptation of glutamate transporters contributes to the development of neural circuit and synaptic plasticity in the central neurons [59]. Disruption of astrocytic glutamate transporters is associated with the development of synaptic and cognitive deficits in several neurodegenerative diseases. For example, global or astrocyte-specific knockout of GLT1 induced the fatal hyperactivity and severe epileptic seizures in 2 to 3 weeks after birth [60, 61]. Suppression of astrocytic GLT1 induced increased activity of presynaptic metabotropic glutamate receptors [62]. We noted in the present study that repeated application of glutamate transporter inhibitor DL-TBOA attenuated the basal glutamatergic strength and decreased the amplitude and frequency of mEPSCs in the hippocampal CA1 neurons in naïve rats.
Astrocytic GluT1 prevents accumulation of extracellular glutamate and subsequent overstimulation of GluRs, thus preventing possible neurotoxicity and neuronal death [63]. Astrocytic GLT1 expression is decreased in early stages of neuroinflammation [61, 64]. The expression of astrocytic GLAST and GLT1 in the hippocampus and other brain regions of AD patients was markedly impaired, even in early clinical stages of disease [30, 31], and the loss of EAATs immunoreactivity was particularly obvious near the amyloid plaques [30]. Partial loss of GLT1 accelerated the early occurring cognitive deficits in APP/PS1 mice [32]. Similarly, in the present study, the reduction of GLT1 expression in the hippocampal CA1 in rats injected with amyloid fibrils resulted in impairments in glutamatergic synapse and cognition functions. Suppression of glutamate transporter activity by DL-TBOA inhibited LTP and facilitated long-term depression in the hippocampal CA1 neurons [65]. It is recognized that downregulation of astrocytic glutamate transporters impairs the homeostasis of extracellular glutamate, which leads to excitotoxicity in neurodegenerative diseases [66]. We noted that artificial restoration of GLT1 expression by ceftriaxone attenuated the synaptic C1q production and C1q-mediated microglial phagocytosis of synapses, and ameliorated the synaptic and cognitive dysfunction in a rodent model of AD. Conversely, artificial suppression of glutamate transporter activity promoted C1q-mediated microglial phagocytosis of synapses and induced hippocampal synaptic and cognitive dysfunction in naïve rats. We have shown that activation of metabotropic GluRs induced complement C1q upregulation and microglial phagocytosis of glutamatergic synapses in rodent models of AD [14]. In this study, we attempted to examine the role of GLT1 in modulating synaptic and cognitive function in rodent model of AD. Nevertheless, additional studies are needed to dissect the molecular mechanism underlying the GLT1-involved synaptic pruning in various models of AD.
In conclusion, we report that astrocyte glutamate transporter GLT1 dysfunction contributes to AD pathoetiology. GLT1 modulates synaptic C1q-mediated microglial phagocytosis of synapses, and synaptic and cognitive functions in the setting of AD. Experimental disruption of GLT1 function was neurotoxic, as reflected by increased C1q expression–induced microglial phagocytosis of glutamatergic synapses, and severe synaptic and cognitive impairments.
References
Jiang H, Burdick D, Glabe CG, Cotman CW, Tenner AJ (1994) beta-Amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol 152(10):5050–5059
Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ (1996) Localization and cell association of C1q in Alzheimer’s disease brain. Exp Neurol 138(1):22–32. https://doi.org/10.1006/exnr.1996.0043
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q et al (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. https://doi.org/10.1126/science.aad8373
Depboylu C, Schafer MK, Arias-Carrion O, Oertel WH, Weihe E, Hoglinger GU (2011) Possible involvement of complement factor C1q in the clearance of extracellular neuromelanin from the substantia nigra in Parkinson disease. J Neuropathol Exp Neurol 70(2):125–132. https://doi.org/10.1097/NEN.0b013e31820805b9
Singhrao SK, Neal JW, Morgan BP, Gasque P (1999) Increased complement biosynthesis by microglia and complement activation on neurons in Huntington’s disease. Exp Neurol 159(2):362–376. https://doi.org/10.1006/exnr.1999.7170
Stephan AH, Barres BA, Stevens B (2012) The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35:369–389. https://doi.org/10.1146/annurev-neuro-061010-113810
Rappert A, Bechmann I, Pivneva T, Mahlo J, Biber K, Nolte C, Kovac AD, Gerard C et al (2004) CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci 24(39):8500–8509. https://doi.org/10.1523/JNEUROSCI.2451-04.2004
Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, Tenner AJ (2017) Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflammation 14(1):48. https://doi.org/10.1186/s12974-017-0814-9
Fonseca MI, Kawas CH, Troncoso JC, Tenner AJ (2004) Neuronal localization of C1q in preclinical Alzheimer’s disease. Neurobiol Dis 15(1):40–46. https://doi.org/10.1016/j.nbd.2003.09.004
Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC et al (2016) Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell 165(4):921–935. https://doi.org/10.1016/j.cell.2016.04.001
Korotzer AR, Watt J, Cribbs D, Tenner AJ, Burdick D, Glabe C, Cotman CW (1995) Cultured rat microglia express C1q and receptor for C1q: implications for amyloid effects on microglia. Experimental neurology 134(2):214–221. https://doi.org/10.1006/exnr.1995.1051
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK et al (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131(6):1164–1178. https://doi.org/10.1016/j.cell.2007.10.036
Benoit ME, Tenner AJ (2011) Complement protein C1q-mediated neuroprotection is correlated with regulation of neuronal gene and microRNA expression. J Neurosci 31(9):3459–3469. https://doi.org/10.1523/jneurosci.3932-10.2011
Bie B, Wu J, Foss JF, Naguib M (2019) Activation of mGluR1 mediates C1q-dependent microglial phagocytosis of glutamatergic synapses in Alzheimer’s rodent models. Mol Neurobiol 56(8):5568–5585. https://doi.org/10.1007/s12035-019-1467-8
Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S (1994) Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell 79(2):365–375. https://doi.org/10.1016/0092-8674(94)90204-6
Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A et al (1994) Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature 372(6503):237–243. https://doi.org/10.1038/372237a0
Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ (2001) Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci 21(16):5925–5934
Kishimoto Y, Fujimichi R, Araishi K, Kawahara S, Kano M, Aiba A, Kirino Y (2002) mGluR1 in cerebellar Purkinje cells is required for normal association of temporally contiguous stimuli in classical conditioning. Eur J Neurosci 16(12):2416–2424
Topolnik L, Congar P, Lacaille JC (2005) Differential regulation of metabotropic glutamate receptor- and AMPA receptor-mediated dendritic Ca2+ signals by presynaptic and postsynaptic activity in hippocampal interneurons. J Neurosci 25(4):990–1001. https://doi.org/10.1523/jneurosci.4388-04.2005
Ostapchenko VG, Beraldo FH, Guimaraes AL, Mishra S, Guzman M, Fan J, Martins VR, Prado VF et al (2013) Increased prion protein processing and expression of metabotropic glutamate receptor 1 in a mouse model of Alzheimer’s disease. J Neurochem 127(3):415–425. https://doi.org/10.1111/jnc.12296
Chen X, Lin R, Chang L, Xu S, Wei X, Zhang J, Wang C, Anwyl R et al (2013) Enhancement of long-term depression by soluble amyloid beta protein in rat hippocampus is mediated by metabotropic glutamate receptor and involves activation of p38MAPK, STEP and caspase-3. Neuroscience 253:435–443. https://doi.org/10.1016/j.neuroscience.2013.08.054
Danbolt NC, Furness DN, Zhou Y (2016) Neuronal vs glial glutamate uptake: resolving the conundrum. Neurochem Int 98:29–45. https://doi.org/10.1016/j.neuint.2016.05.009
Ugbode C, Hu Y, Whalley B, Peers C, Rattray M, Dallas ML (2017) Astrocytic transporters in Alzheimer’s disease. Biochem J 474(3):333–355. https://doi.org/10.1042/BCJ20160505
Flores-Mendez M, Mendez-Flores OG, Ortega A (2016) Glia plasma membrane transporters: key players in glutamatergic neurotransmission. Neurochem Int 98:46–55. https://doi.org/10.1016/j.neuint.2016.04.004
Norenberg MD, Martinez-Hernandez A (1979) Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 161(2):303–310. https://doi.org/10.1016/0006-8993(79)90071-4
Schousboe A, Scafidi S, Bak LK, Waagepetersen HS, McKenna MC (2014) Glutamate metabolism in the brain focusing on astrocytes. Advances in neurobiology 11:13–30. https://doi.org/10.1007/978-3-319-08894-5_2
Qureshi T, Sorensen C, Berghuis P, Jensen V, Dobszay MB, Farkas T, Dalen KT, Guo C et al (2019) The glutamine transporter Slc38a1 regulates GABAergic neurotransmission and synaptic plasticity. Cereb Cortex. https://doi.org/10.1093/cercor/bhz055
Vandenberg RJ, Ryan RM (2013) Mechanisms of glutamate transport. Physiol Rev 93(4):1621–1657. https://doi.org/10.1152/physrev.00007.2013
Bjornsen LP, Hadera MG, Zhou Y, Danbolt NC, Sonnewald U (2014) The GLT-1 (EAAT2; slc1a2) glutamate transporter is essential for glutamate homeostasis in the neocortex of the mouse. J Neurochem 128(5):641–649. https://doi.org/10.1111/jnc.12509
Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W et al (2007) Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis 11(1):97–116
Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L (1996) Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol 40(5):759–766. https://doi.org/10.1002/ana.410400512
Mookherjee P, Green PS, Watson GS, Marques MA, Tanaka K, Meeker KD, Meabon JS, Li N et al (2011) GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. J Alzheimers Dis 26(3):447–455. https://doi.org/10.3233/JAD-2011-110503
Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y et al (2009) HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459(7243):55–60. https://doi.org/10.1038/nature07925
Bie B, Wu J, Yang H, Xu JJ, Brown DL, Naguib M (2014) Epigenetic suppression of neuroligin 1 underlies amyloid-induced memory deficiency. Nat Neurosci 17(2):223–231. https://doi.org/10.1038/nn.3618 http://www.nature.com/neuro/journal/vaop/ncurrent/abs/nn.3618.html#supplementary-information
Bie B, Zhang Z, Cai YQ, Zhu W, Zhang Y, Dai J, Lowenstein CJ, Weinman EJ et al (2010) Nerve growth factor-regulated emergence of functional delta-opioid receptors. J Neurosci 30(16):5617–5628. https://doi.org/10.1523/JNEUROSCI.5296-09.2010
Chacon MA, Barria MI, Soto C, Inestrosa NC (2004) Beta-sheet breaker peptide prevents Abeta-induced spatial memory impairments with partial reduction of amyloid deposits. Mol Psychiatry 9(10):953–961. https://doi.org/10.1038/sj.mp.4001516
Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates, vol 1, 4th edn. Academic Press, New York
Shin RW, Ogino K, Kondo A, Saido TC, Trojanowski JQ, Kitamoto T, Tateishi J (1997) Amyloid beta-protein (Abeta) 1-40 but not Abeta1-42 contributes to the experimental formation of Alzheimer disease amyloid fibrils in rat brain. J Neurosci 17(21):8187–8193
Ahmed T, Enam SA, Gilani AH (2010) Curcuminoids enhance memory in an amyloid-infused rat model of Alzheimer’s disease. Neuroscience 169(3):1296–1306. https://doi.org/10.1016/j.neuroscience.2010.05.078
Wu J, Bie B, Yang H, Xu JJ, Brown DL, Naguib M (2013) Activation of the CB(2) receptor system reverses amyloid-induced memory deficiency. Neurobiol Aging 34:791–804. https://doi.org/10.1016/j.neurobiolaging.2012.06.011
Wu J, Bie B, Yang H, Xu JJ, Brown DL, Naguib M (2013) Suppression of central chemokine fractalkine receptor signaling alleviates amyloid-induced memory deficiency. Neurobiol Aging 34(12):2843–2852. https://doi.org/10.1016/j.neurobiolaging.2013.06.003
Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K (2015) Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138(Pt 6):1738–1755. https://doi.org/10.1093/brain/awv081
Wu J, Hocevar M, Foss JF, Bihua Bie B, Naguib M (2017) Activation of CB2 receptor system restores cognitive capacity and hippocampal Sox2 expression in a transgenic mouse model of Alzheimer’s disease. Eur J Pharmacol 811:12–20. https://doi.org/10.1016/j.ejphar.2017.05.044
Bie B, Brown DL, Naguib M (2011) Increased synaptic GluR1 subunits in the anterior cingulate cortex of rats with peripheral inflammation. Eur J Pharmacol 653(1–3):26–31. https://doi.org/10.1016/j.ejphar.2010.11.027
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME et al (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74(4):691–705. https://doi.org/10.1016/j.neuron.2012.03.026
Bie B, Zhu W, Pan ZZ (2009) Rewarding morphine-induced synaptic function of delta-opioid receptors on central glutamate synapses. J Pharmacol Exp Ther 329(1):290–296. https://doi.org/10.1124/jpet.108.148908
Bie B, Zhu W, Pan ZZ (2009) Ethanol-induced delta-opioid receptor modulation of glutamate synaptic transmission and conditioned place preference in central amygdala. Neuroscience 160(2):348–358. https://doi.org/10.1016/j.neuroscience.2009.02.049
Rose CR, Felix L, Zeug A, Dietrich D, Reiner A, Henneberger C (2018) Astroglial glutamate signaling and uptake in the hippocampus. Front Mol Neurosci 10:451–451. https://doi.org/10.3389/fnmol.2017.00451
Scott HA, Gebhardt FM, Mitrovic AD, Vandenberg RJ, Dodd PR (2011) Glutamate transporter variants reduce glutamate uptake in Alzheimer’s disease. Neurobiol Aging 32(3):553.e551–553.e511. https://doi.org/10.1016/j.neurobiolaging.2010.03.008
Kashani A, Lepicard E, Poirel O, Videau C, David JP, Fallet-Bianco C, Simon A, Delacourte A et al (2008) Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging 29(11):1619–1630. https://doi.org/10.1016/j.neurobiolaging.2007.04.010
Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M et al (2005) Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433(7021):73–77. https://doi.org/10.1038/nature03180
Thone-Reineke C, Neumann C, Namsolleck P, Schmerbach K, Krikov M, Schefe JH, Lucht K, Hortnagl H et al (2008) The beta-lactam antibiotic, ceftriaxone, dramatically improves survival, increases glutamate uptake and induces neurotrophins in stroke. J Hypertens 26(12):2426–2435. https://doi.org/10.1097/HJH.0b013e328313e403
Rebec GV (2013) Dysregulation of corticostriatal ascorbate release and glutamate uptake in transgenic models of Huntington’s disease. Antioxid Redox Signal 19(17):2115–2128. https://doi.org/10.1089/ars.2013.5387
Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH et al (2013) A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci 33(33):13460–13474. https://doi.org/10.1523/JNEUROSCI.1333-13.2013
Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L et al (1992) Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 89(21):10016–10020. https://doi.org/10.1073/pnas.89.21.10016
Takano M, Kawabata S, Komaki Y, Shibata S, Hikishima K, Toyama Y, Okano H, Nakamura M (2014) Inflammatory cascades mediate synapse elimination in spinal cord compression. J Neuroinflammation 11:40. https://doi.org/10.1186/1742-2094-11-40
Bialas AR, Stevens B (2013) TGF-[beta] signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16(12):1773–1782. https://doi.org/10.1038/nn.3560 http://www.nature.com/neuro/journal/v16/n12/abs/nn.3560.html#supplementary-information
Zerangue N, Kavanaugh MP (1996) Interaction of L-cysteine with a human excitatory amino acid transporter. J Physiol 493(Pt 2):419–423. https://doi.org/10.1113/jphysiol.1996.sp021393
Sigler A, Oh WC, Imig C, Altas B, Kawabe H, Cooper BH, Kwon HB, Rhee JS et al (2017) Formation and maintenance of functional spines in the absence of presynaptic glutamate release. Neuron 94(2):304–311.e304. https://doi.org/10.1016/j.neuron.2017.03.029
Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA et al (2015) Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci 35(13):5187–5201. https://doi.org/10.1523/JNEUROSCI.4255-14.2015
Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T et al (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276(5319):1699–1702
Oliet SH, Piet R, Poulain DA (2001) Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292(5518):923–926. https://doi.org/10.1126/science.1059162
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA et al (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16(3):675–686
Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K (2012) L-Glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J Neuroinflammation 9:275. https://doi.org/10.1186/1742-2094-9-275
Huang S, Tong H, Lei M, Zhou M, Guo W, Li G, Tang X, Li Z et al (2018) Astrocytic glutamatergic transporters are involved in Abeta-induced synaptic dysfunction. Brain Res 1678:129–137. https://doi.org/10.1016/j.brainres.2017.10.011
Hu YY, Xu J, Zhang M, Wang D, Li L, Li WB (2015) Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. J Neurochem 132(2):194–205. https://doi.org/10.1111/jnc.12958
Acknowledgments
The authors thank John Peterson, Ph.D., Imaging Core, Cleveland Clinic, for his expertise and help provided for imaging analysis.
Funding
Dr. Naguib is financially supported by the National Institute of Aging of the National Institutes of Health under Award Number R56AG051594. This work utilized the Leica SP8 confocal microscope that was purchased with funding from National Institutes of Health SIG grant1S10OD019972-01.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal procedures were approved by the Animal Care and Use Committee of Cleveland Clinic.
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 92 kb)
Rights and permissions
About this article

Cite this article
Wu, J., Bie, B., Foss, J.F. et al. Amyloid Fibril–Induced Astrocytic Glutamate Transporter Disruption Contributes to Complement C1q-Mediated Microglial Pruning of Glutamatergic Synapses. Mol Neurobiol 57, 2290–2300 (2020). https://doi.org/10.1007/s12035-020-01885-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-01885-7