
Abstract
α-Synuclein (α-syn), a metabolite of neurons, induces glial activation and neuroinflammation and participates in pathogenesis of neurodegenerative diseases. This inflammatory response involves activation of toll-like receptors (TLRs) and its neurotoxic outcomes such as cytokine expression and release. However, regulatory role of cytokines on α-syn-induced neurotoxicity is still unclear. In this study, we used interferon (IFN)-γ to costimulate primary astrocytes with wild-type or A53T mutant α-syn, and evaluated inflammatory pathway activation. Four α-syn concentrations (0.5, 2, 8 and 20 μg/mL, 24 h) and four α-syn time-points (3, 12, 24 and 48 h, 2 μg/mL) were chosen to coincubate with one IFN-γ concentration (2 ng/mL). IFN-γ alone upregulated expressions of TLR3 and tumor necrosis factor (TNF)-α (mRNA level), and A53T mutant or wild-type α-syn alone activated the pathway components including TLR2, TLR3, nuclear factor-κB, TNF-α and interleukin (IL)-1β. Additive application of IFN-γ amplified this activation effect except for IL-1β at mRNA and protein levels or TNF-α release, displaying a synergistic effect of α-syn and IFN-γ. Blocking TLR2 other than TLR4 suppressed TLR3, TLR2 and TNF-α expressions induced by α-syn or plus IFN-γ, reflecting an interaction of TLR2 and TLR3 in TNF-α expression. These data collectively showed that IFN-γ potentiated α-syn stimulation and inflammatory outcomes via TLR2, TLR3 and TNF-α other than IL-1β in astrocytes, suggesting that involvement of IFN-γ in α-syn-induced innate immunity may be required for initiation and maintenance of glial activation, a novel neurotoxic mechanism underlying pathogenesis of neurodegenerative diseases.

IFN-γ potentiates α-synuclein (A53T or wild-type)-induced innate immunity, involving expressions of TLR2, TLR3, NF-κB, and TNF-α, other than IL-1β. This effect is suppressed by blockage of TLR2 other than TLR4, reflecting an interaction of TLR2 and TLR3 in TNF-α expression. Thus, involvement of IFN-γ in α-syn-induced neurotoxicity may be required for initiation and maintenance of glial activation, a novel neurotoxic mechanism underlying pathogenesis of neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alpha-synuclein (α-syn), a neuronal 140-amino acid protein, is strongly implicated in pathogenesis of synucleinopathies or neurodegenerative diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and multiple system atrophy (MSA) [1,2,3]. It can be released and aggregated to induce glial activation and innate immune response, leading to neuroinflammatory damage to neuronal function [4,5,6]. This mechanism involves two aspects of α-syn immunogenic attribution: one is activation of canonical inflammatory pathway and neurotoxic outcomes, presented by upregulated expression of toll-like receptors (TLRs) and nuclear factor (NF)-κB and increased release of various cytokines or chemokines; the other is its immune regulatory role by other inflammatory factors [7]. Our previous study [8] demonstrates that α-syn activates innate immunity and downregulates expression of interferon (IFN)-γ, and also reveals a phenomenon that TLR types or cytokine types may interact to modulate inflammatory process. Other studies show that IFN-γ and tumor necrosis factor (TNF)-α can synergistically enhance MHC-II expression [9], and IFN-γ and interleukin (IL)-1β can synergistically stimulate TNF-α expression in rat astrocytes [10]. Likewise, in human microglia from surgical specimens, coincubation of α-syn and IFN-γ (24 h) produces more neurotoxicity for SH-SY5Y cells than any one alone [11], indicating a possible correlation of α-syn with IFN-γ in innate immunity activation. In this study, we used α-syn and IFN-γ to costimulate primary astrocytes, and directly determined their reciprocal regulation in inflammatory response.
Traditionally, α-syn is an exclusively intracellular acidic protein and highly enriched in presynaptic neuronal terminals, and largely localized within neurons in substantia nigra, neocortex, and hippocampus [12]. More research shows that it can be expressed in astrocyte, oligodendroglia [13], and peripheral immune cells such as T cells, B cells, natural killer cells, and monocytes [14], and can be released into biological fluids [15,16,17], displaying a considerable concentration at nanogram/milliliter level or even up to 10–10,000-fold higher in cerebrospinal fluid or blood [18, 19]. These widespread cell sources of α-syn indicate that it serves as an important immune mediator to stimulate innate immunity and inflammatory response. Emerging evidence shows that extracellular existence of α-syn in the brain has been recognized as one of antigen stresses by self-debris danger/damage-associated molecular patterns (DAMPs) [20] or senescence-associated secretory phenotype (SASP) [21], which can stimulate activation of microglia and astrocytes to induce neuroinflammation and neurotoxic damage [7, 22], involving TLRs and pro-inflammatory cytokines such as IFN-γ, TNF-α, and IL-1β [11, 23].
TLRs are a group of type I transmembrane glycoprotein containing an ectodomain, a transmembrane domain, and an intracellular signaling domain, to recognize foreign pathogens such as pathogen-associated molecular patterns (PAMPs), DAMPs, or SASP. Among 12 functional components of TLRs, TLRs 1, 2, 4, 5, and 6 are expressed on cell surface responsible for extracellular recognition and TLRs 3, 7, 8, and 9 are localized on endosomes or lysosomes for detecting intracellular molecules such as viral or bacterial nucleic acids. α-Syn, as a self-antigen of DAMPs, can be recognized by almost all TLRs such as TLRs 1–4, TLR7, and TLR8, to participate in different profiles of inflammatory process [8, 24]. This complex receptor involvement provides a possibility for α-syn to interact with other inflammatory factors, among which IFN-γ is primarily considered as a costimulatory molecule of α-syn based on the following evidence. IFN-γ, as a pro-inflammatory cytokine, is widely expressed in glial cells and blood cells, such as microglia, macrophages, natural killer cells, and T helper cells, and exerts a critical immunomodulatory effect in TLR activation and innate immune response [25, 26]. IFN-γ level in blood plasma is elevated in PD patients [27], and cerebral ventricle injection of IFN-γ in mice can promote neuronal loss and degeneration in nigrostriatal tract and basal ganglia [28]. Likewise, IFN-γ-deficiency in PD mice ameliorates death of dopaminergic neurons by reducing IFN-γ-mediated neurotoxicity in microglia [27]. Moreover, IFN-γ and TNF-α costimulation potentiates MHC-II expression at mRNA level in astrocytes [9], and IFN-γ and IL-1β induce a higher TNF-α expression in rat astrocytes [10]. Also, IFN-γ is proven to be related to the enhanced cell sensitivity to TNF-α cytotoxicity (about 400%) in human glioblastoma cell line [29]. Regarding correlation of IFN-γ with α-syn, an in vivo study shows that coexpression of α-syn and IFN-γ is found in PD substantia nigra, whereas a strong negative correlation between them is detected in the healthy brain, indicating that coexistence of α-syn and IFN-γ may be a cause of PD pathogenesis [30]. In this study, α-syn and IFN-γ were designed to costimulate astrocytes in vitro to evaluate their interaction in innate immunity and their contribution to neurotoxic microenvironment.
Taken together, α-syn, as an abnormal metabolite of neurons, is released into extracellular space to activate glial cells and participate in neuroinflammatory response, reflecting its immunogenic attribution. IFN-γ, as an important regulator of inflammatory response, has a synergistic effect with multiple immune factors such as TNF-α and IL-1β. However, whether IFN-γ regulates α-syn-induced activation of innate immunity in astrocytes has not been fully investigated. In this study, we chose A53T mutant or wild-type (WT) α-syn to costimulate astrocytes with IFN-γ, and on the basis of our previous study that α-syn alone induces innate immunity [8], we further found that IFN-γ enhanced α-syn-induced inflammatory response and neurotoxicity via TLR2, TLR3, NF-κB, and TNF-α other than IL-1β at multiple α-syn doses and time-points in astrocytes. Also, blockage of TLR2 other than TLR4 inhibited activation of the inflammatory components. These data suggest that both persistent accumulation of α-syn and its interplay with cytokine IFN-γ may be required for initiation and maintenance of immune response [26], a novel neurotoxic mechanism underlying pathogenesis of synucleinopathies.
Materials and Methods
Experimental Design
Our aim is to evaluate the regulatory role of IFN-γ in α-syn-induced activation of innate immunity by determining canonical inflammatory pathway and its neurotoxic outcomes. In experimental design, we considered the following issues. (1) Choice of stimulators. IFN-γ is a crucial immune modulator with widespread cell sources. α-Syn (A53T mutant or WT) is of high neurotoxicity. IFN-γ and α-syn are both involved in astrocyte-mediated neuroinflammation; (2) Doses and time-points. α-Syn concentration in human blood (normal, 0.02 pg/mL; PD, 1.56 pg/mL) and in cerebrospinal fluid (PD, 1.6 ng/mL) indicates that α-syn level is supposed to be much higher in inflammatory brain parenchyma, or even up to 10 μg/mL level in whole blood [18, 19]. In previous in vitro studies, a range of α-syn concentrations are used from 0.1 to 10 μg/mL at one time-point (24 h) [31]. In this study, we chose four concentrations of α-syn (0.5, 2, 8, and 20 μg/mL, 24 h) for dose dependency and four time-points (3, 12, 24, 48 h, 2 μg/mL) for time dependency to coincubate with one IFN-γ concentration (2 ng/mL), which is a moderate level compared with previous IFN-γ application (10 ng/mL, equivalent to 200 U/mL, 48 h) in human monocytes [32]; (3) Determination of inflammatory factors. We chose the canonical inflammatory pathway components as indicators of inflammatory neurotoxicity: TLR2 (cell surface receptor), TLR3 (intracellular receptor), NF-κB (core regulator), and effector factors (TNF-α and IL-1β); and (4) Data normalization. According to the manufacturer’s instruction (ThermoFisher Scientific, Waltham, MA, USA) and previous studies [33], we assigned comparison between treatment groups at ΔΔCt level, because the interference of control and GAPDH values is subtracted, and comparison between control and treatment groups at ΔCt level due to all control values indicated at 0 at ΔΔCt level.
Chemicals and Reagents
Cell culture reagents were purchased from Gibco Fisher Scientific (Waltham, MA, USA): Advanced Dulbecco’s modified Eagle medium (DMEM)-nutrient mixture F12 Ham (DMEM-F12), Hank’s Balanced Salt Solution (HBSS), fetal bovine serum (FBS), penicillin/streptomycin, trypsin-EDTA, and TRIzol® Reagent. Cell treatment reagents: WT or A53T mutant α-syn from rPeptide (Bogart, GA, USA), mouse IFN-γ from Cell Signaling Technology (Beverly, MA, USA), mouse IFN-β from R&D Systems (Minneapolis, MN, USA), TLR4-IN-C34 (C34, TLR4 antagonist), CU-CPT22 (CPT22, TLR2 antagonist), polyinosinic-polycytidylic acid (Poly I:C, TLR3 agonist), and lipopolysaccharide (LPS, TLR4 agonist) from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies (rabbit anti-mouse IgG) against: glial fibrillary acidic protein (GFAP), TLR2, and TLR3 from Abcam (Cambridge, MA, USA); TNF-α, IFN-γ, and β-Actin from ThermoFisher Scientific (Waltham, MA, USA). Other reagents: Alexa Fluor® 514 goat anti-rabbit secondary antibody, reagent kits for reverse transcription and quantitative real-time polymerase chain reaction (qRT-PCR), and 4′,6-diamidino-2-phenylindole (DAPI) purchased from ThermoFisher Scientific (Waltham, MA, USA), enzyme-linked immunoabsorbent assay (ELISA) kits from R&D System (Minneapolis, MS, USA), lysis buffer, complete protease inhibitor cocktail and horseradish peroxidase (HRP)-conjugated secondary antibody from Cell Signaling Technology (Beverly, MA, USA), IgG from rabbit serum (negative control), and Fluoromount™ Aqueous Mounting Medium from Sigma-Aldrich (St. Louis, MO, USA).
Animals
Male and female C57BL/6 mice were purchased from Jackson Laboratories (Sacramento, CA) and were bred in animal facilities of Johns Hopkins University School of Medicine. All experimental procedures were approved by Institution’s Animal Care Committee and performed in accordance with the guidelines by Institution of Laboratory Resources, National Research Council (Department of Health and Human Service, National Institutes of Health, Bethesda, MD).
Preparation of Primary Astrocytes and Cell Culture
Mixed glial cells were prepared from 9 to 10 cortices per experiment of newborn (3–4 days) C57BL/6 mouse pups and astrocytes were separated by differential adhesion of cell types according to previously described method [34]. Briefly, cerebral cortex was removed from the brain and homogenized into single-cell suspension by mincing, trypsinizing, and passaging through nylon mesh. Primary glial cells were washed and cultured for 16 h in poly-D-lysine-coated 75-cm2 flasks with complete DMEM-F12 containing 10% FBS (heat-inactivated), 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified 5% CO2, 95% air atmosphere at 37 °C. In 16 h incubation, horizontal shakes and HBSS washes were performed to remove oligodendrocytes and microglia, and astrocytes were adhered for further culture. Purity of astrocytes was determined by immunostaining for GFAP (a specific marker for astrocytes) and immunofluorescence microscopy. All astrocytes were cultured for a total of 7–10 days, and then shaken and washed again before they were detached from plates by trypsinization (0.05% trypsin EDTA), resuspended and plated in complete DMEM-F12 medium for planned experiments.
Immunofluorescence Microscopy
Astrocytes were seeded and cultured on coverslips until confluent, and fixed in ice-cold solution of methanol and acetone (1:1) for 5 min. After they were rinsed twice in phosphate-buffered saline (PBS, pH 7.4), the main steps below were performed with PBS washes of 10–15 min in between: (1) Glycine (2.25%, 20 min) was put to remove endogenous fluorescent; (2) Blocker buffer containing 1% bovine serum albumin, 0.3% Triton X-100, and 10% goat serum in PBS was made for blocking nonspecific staining at room temperature for 1 h; (3) Primary antibody for GFAP (rabbit anti-mouse, 1:1000) and IgG from rabbit serum (negative control) were used at 4 °C overnight; (4) Alexa Fluor® 514 fluorescein-conjugated goat-anti-rabbit antibody (1:1000) was added at room temperature for 1 h; (5) DAPI was used for nuclear and chromosome counterstaining at 1 μg/mL for 10 min. After final wash in PBS, coverslips were mounted with Fluoromount™ Aqueous Mounting Medium and examined under fluorescence microscope (Nikon Model: ECLIPSE Ni-U, Tokyo, Japan).
Astrocyte Treatment
Astrocytes were divided into multiple treatment groups in 6-well plates and cultured to about 90% confluency. Culture medium was replaced with fresh complete DMEM-F12 containing appropriate final concentration of individual drugs or their combinations. A53T mutant or WT α-syn was set at 0.5, 2, 8, and 20 μg/mL (24 h) for dose dependency, and 0, 3, 12, 24, and 48 h (2 μg/mL α-syn) for time dependency; For combination treatment, each dose or time group of α-syn was coincubated with one concentration of individual drugs, including IFN-γ and IFN-β at 2 ng/mL, CPT22 and C34 at 5 μg/mL, Poly I:C at 10 μg/mL, and LPS at 0.2 μg/mL. These treatment groups were cultured in a humidified 5% CO2, 95% air atmosphere at 37 °C for specified hours. Supernatants from each group were collected and stored at − 80 °C, and astrocytes were harvested with TRIzol Reagent (0.5 mL/well) or lysis buffer (0.2 mL/well) and properly stored according to the instruction on qRT-PCR or western blot analysis.
Determination of TLRs and Cytokine Gene Expression
Expression of genes for TLRs 1–4, NF-κB, TNF-α, IFN-γ, and glyceraldehyde phosphate dehydrogenase (GAPDH, a loading control) was determined by using two-step qRT-PCR method. Total RNA was extracted from astrocytes of each group with TRIzol Reagent; RNA concentration was measured with NanoDrop™ 2000 Spectrophotometer from Biotek (Winooski, VT, USA); RNA (2 μg) was reverse-transcribed with High Capacity cDNA Reverse Transcription Kit according to the manufacturer’s protocol; and qRT-PCR was performed with TaqMan® Fast Advanced Master Mix and TaqMan primer and probe sets by using Stratagene Mx3000P Real Time PCR Systems from Bio-Rad (Hercules, CA, USA) according to the manufacturer’s protocol. Multiple transcripts prepared on a 96-well PCR plate were analyzed simultaneously under standardized thermal cycling conditions: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. Target transcripts were measured with TaqMan probes for TLR1 (Mm01208874_m1), TLR2 (Mm00442346_m1), TLR3 (Mm00628112_m1), TLR4 (Mm00445273_m1), NF-κB (Mm 00476361_m1), TNF-α (Mm00443258_m1), and IL-1β (Mm01336189_m1), and their relative abundance was normalized with respect to the expression of GAPDH (Mm03302249_g1). Changes in gene expression levels were determined by using ΔCt (cycle threshold) value taking off GAPDH (internal control), and fold differences in expression level of individual gene under each treatment condition over untreated control were calculated by formula of 2-ΔΔCt in the ΔΔCt method (Livak and Schmittgen, 2001). All qRT-PCR reactions were run in duplicate and their average values were used in data analysis.
Western Blotting
Protein expression of TLR2, TLR3, and TNF-α was determined by western blotting with β-actin as internal control. Briefly, astrocytes (1 × 106 cells/well) treated in a 6-well plate were washed once with ice-cold PBS and lysed in lysis buffer supplemented with complete protease inhibitor cocktail and 1 mM phenylmethanesulfonyl fluoride (PMSF, before use). Cell debris was removed by centrifugation at 13,000×g, 4 °C for 30 min, and protein concentrations were determined by Bradford reagent (Bio-Rad, Hercules, CA). A total of 30 μg lysate protein for one lane was added to appropriate denaturing sample buffer and heated at 95 °C for 5 min, and then was resolved by Mini-PROTEIN TGX Precast Gels from Bio-Rad and transferred to 0.45 μm polyvinylidenedifluoride (PVDF) membrane (Millipore, USA) by using Bio-Rad Trans-Blot Semi-dry Transfer Cell and System. After blocked in 5% milk at room temperature for 1 h, the membrane was probed with appropriately diluted primary antibodies (TLR2 and TLR3 at 1:300, TNF-α at 1:800, β-actin at 1:2000) in 5% milk at 4 °C overnight and then hybridized with appropriate HRP-conjugated secondary antibody (1:2000) at room temperature for 1 h. Results were visualized by using Enhanced Chemiluminescence (ECL) Substrate Kit (Bio-Rad, Hercules, CA, USA) and films were exposed and developed in dark room according to the manufacturer’s instruction. To confirm equal loading of proteins in each lane, membrane was reprobed for β-actin following removal of background bands by incubation at 37 °C for 30 min with Gentle Review™ Stripping Buffer (Amresco-inc, Solon, USA).
Cytokine Release Measurement
Supernatants collected from each group were centrifuged at 300×g at 4 °C for 10 min, and then stored at − 80 °C or used for measurement of cytokine release. TNF-α was chosen as a representative of pro-inflammatory cytokines to assess neurotoxicity by astrocytes. TNF-α protein level in each supernatant was measured by using specific sandwich ELISA kits according to the manufacturer’s instruction, and the normalized concentrations were automatically generated through 460 nm filters plus 570 nm subtraction on microplate reader (Bio-Rad, Hercules, CA, USA).
Data Analysis
Data are expressed as means ± standard deviation and scatter charts, displaying sample distribution, sample number (n), means (−), significant differences (* or #), and p values in group comparison, and statistically analyzed on ΔΔCt values between treatment groups or on ΔCt values between control and treatment groups. Fold difference in means was determined by 2-ΔΔCt calculation. Comparisons between single and combination treatment groups and between single treatment groups were performed by two-way ANOVA methods combined with margin means. Normality and homogeneity of variance were examined by Shapiro-Wilk test and Levene test at a 0.05 significance level, respectively. All statistical analyses were conducted using SAS software (SAS 9.1.3, SAS Institute Inc., Carey, NC), and statistically significant differences were defined as p < 0.05, with p values shown in the text.
Results
Identification of Primary Astrocytes
Astrocytes were identified by immunofluorescence analysis for GFAP. The GFAP-positive cells showed a heterogeneous population as described previously [34], consisting of small-, medium- and large-sized cells, with multiple primary and secondary branches that extended to outer space in a long distance (Fig. 1a). This morphological feature may reflect multiple cell division phases of astrocytes with 7–10 day culture, and is different from microglia (1–2 short branches). Purity of astrocytes was determined by calculating the proportion of GFAP-positive cells in the numerated cells with DAPI-positive nuclei staining within five randomly selected areas under microscope, showing that approximately 94% (93.96 ± 4.03%) of DAPI-positive cells were GFAP-positive. Immunostaining for IgG from rabbit serum, the equivalent to GFAP antibody, was used as negative control and no GFAP-positive cells were detected (Fig. 1b).

Expression profiles of TLR2, TLR3, and NF-κB induced by A53T mutant or wild-type (WT) α-syn plus IFN-γ. a A representative image showing GFAP-immunolabeled astrocytes (93.96 ± 4.03%) with a heterogeneous cell population. Scale bar, 50 μm. b Paralleling immunostaining for IgG equivalent to GFAP antibody, showing no GFAP-positive cells detected. Scale bar, 50 μm. c–f Additive application of IFN-γ significantly potentiated mRNA expressions of TLR3 (c, d) and TLR2 (e, f) in all α-syn treatment groups, indicating an amplification effect of IFN-γ on TLR2 or TLR3 expression. g, h IFN-γ significantly enhanced NF-κB expression induced by A53T mutant or WT α-syn at high doses (8 or 20 μg/mL) and early time-points (3 or 12 h). The data are presented by scatter charts showing sample distribution, sample number (n), and means (−), with p values (in the text) specified at p < 0.05 (ANOVA, margin means). Asterisk (*) represents significant difference at ΔΔCt values between single and combination groups, with pound sign (#) between the indicated groups at ΔCt values. (Dose treatment at 2 μg/mL, 24 h is the same condition as time treatment at 24 h, 2 μg/mL)
IFN-γ Amplified α-Syn-Induced Activation of TLR2, TLR3, and NF-κB
To explore the regulatory effect of IFN-γ on α-syn-induced innate immunity, we examined expression profiles of upstream molecules TLR2, TLR3, and NF-κB, as well as TLR1 and TLR4, which reflect the induction status of inflammatory response. Our previous study demonstrates that α-syn (A53T mutant or WT) upregulates mRNA expressions of TLR2 and TLR3 in a positive dose-effect correlation (PDEC) or a negative time-effect correlation (NTEC) pattern [8]. In this study, we used IFN-γ to coincubate with α-syn of multiple doses and time-points (Fig. 1c), and compare induction effects of α-syn with and without IFN-γ at mRNA and protein levels.
Dose or time dependency showed that (1) IFN-γ alone (2 ng/mL, 24 h) significantly upregulated expression of TLR3 (ΔCt, 5.26 ± 0.67, n = 7) other than TLR2 compared with control group (7.35 ± 0.77, n = 13, p = 0.0284) (Fig. 1c), indicating a selective stimulation effect on TLR3. (2) IFN-γ significantly potentiated expressions of TLR2, TLR3, and NF-κB induced by A53T mutant or WT α-syn in almost all treatment groups. TLR3 (Fig. 1c,d): A53T-2/+IFN-γ (− 0.68 ± 0.22, n = 11/− 2.71 ± 0.43, n = 8, p = 0.0002); A53T-8/+IFN-γ (− 1.08 ± 0.09, n = 5/− 2.51 ± 0.42, n = 4, p = < 0.0001); A53T 12 h/+IFN-γ (− 2.08 ± 0.50, n = 6/− 3.47 ± 0.26, n = 4, p = 0.0005); A53T 48 h/+IFN-γ (− 0.75 ± 0.20, n = 5/− 2.07 ± 0.42, n = 4, p = 0.002); WT-2/+IFN-γ (− 0.27 ± 0.18, n = 4/− 1.98 ± 0.25, n = 4, p = 0.0107); WT-8/+IFN-γ (− 0.61 ± 0.15, n = 4/− 1.92 ± 0.40, n = 4, p = 0.0186); WT 12 h/+IFN-γ (− 1.03 ± 0.25, n = 4/− 3.16 ± 0.26, n = 4, p = 0.0007); WT 48 h/+IFN-γ (− 0.18 ± 0.12, n = 4/− 1.11 ± 0.28, n = 4, p = 0.0015);TLR2 (Fig. 1e,f): A53T-2/+IFN-γ (− 1.41 ± 0.27, n = 9/− 3.52 ± 0.26, n = 7, p = < 0.0001); A53T-8/+IFN-γ (− 1.97 ± 0.21, n = 5/− 4.09 ± 0.35, n = 4, p = < 0.0001); A53T 12 h/+IFN-γ (− 2.59 ± 0.23, n = 4/− 3.85 ± 0.52, n = 4, p = < 0.0001); A53T 48 h/+IFN-γ (− 1.54 ± 0.29, n = 5/− 3.00 ± 0.46, n = 4, p = < 0.0001); WT-2/+IFN-γ (− 0.80 ± 0.44, n = 4/− 3.30 ± 0.17, n = 4, p = 0.0006); WT-8/+IFN-γ (− 0.85 ± 0.61, n = 4/− 3.87 ± 0.30, n = 4, p = 0.0019); WT 12 h/+IFN-γ (− 2.16 ± 0.63, n = 4/− 3.30 ± 0.17, n = 4, p = 0.0013); NF-κB (Fig. 1g,h): A53T-2/+IFN-γ (− 0.49 ± 0.39, n = 8/− 1.02 ± 0.36, n = 7, p = 0.004); A53T-8/+IFN-γ (− 1.26 ± 0.38, n = 4/− 1.91 ± 0.16, n = 4, p = 0.0419); WT-8/+IFN-γ (− 0.66 ± 0.46, n = 4/− 1.32 ± 0.28, n = 4, p = 0.0108); WT-20/+IFN-γ (− 0.51 ± 0.10, n = 4/− 1.84 ± 0.29, n = 4, p = 0.0117); (3) Additive application of IFN-γ antibody (Ab, 5 μg/mL) significantly reduced mRNA expression of TLR2 or TLR3, compared with A53T mutant α-syn (2 μg/mL) plus IFN-γ (2 ng/mL) (Fig. 2a), supporting the amplification effect of IFN-γ on the α-syn induction. TLR2 (Fig. 2a, left): A53T-2/IFN-γ/+IFN-γAb (− 3.52 ± 0.26, n = 7/− 2.12 ± 0.265, n = 5, p = 0.0007); TLR3 (Fig. 2a, right): A53T-2/IFN-γ/+IFN-γAb (− 2.71 ± 0.43, n = 8/− 1.51 ± 0.301, n = 5, p = 0.0169).

Other evidence to support synergistic effect of α-syn and IFN-γ. a IFN-γ antibody (5 μg/mL) significantly reduced expression of TLR2 and TLR3 induced by A53T mutant α-syn plus IFN-γ. b IFN-β significantly upregulated expression of TLR2 and TLR3 induced by A53T mutant α-syn, with no induction effect by IFN-β alone. c Protein expression profiles of TLR2 (140 kDa), TLR3 (104 kDa), and β-actin (45 kDa, reference control) by western blotting analysis, showing protein signal size, or density as identified by manufacturers. d, e Densitometric analyses for western blotting by ImageJ software, indicating that expression level (gray scales) of TLR2 or TLR3 was significantly enhanced by A53T mutant α-syn plus IFN-γ, compared with the control or anyone alone. f, g Expression of TLR1 and TLR4 was significantly potentiated by additive application of IFN-γ, to support TLR2 and TLR3 expression profile. The data are presented as sample distribution, number (n), means (−), and significant difference (*) at a level of p < 0.05 (ANOVA, margin means), with p values shown in the text
To determine whether IFN-γ impact on α-syn induction is specific, we replaced IFN-γ (2 ng/mL) with IFN-β (2 ng/mL), Poly I:C (10 μg/mL) or LPS (0.2 μg/mL) to costimulate with A53T mutant α-syn (2 μg/mL). IFN-β significantly upregulated A53T-induced expression of TLR2 or TLR3 (Fig. 2b), indicating that IFN-β has a similar amplification effect to IFN-γ in α-syn induction. TLR2: A53T-2/+IFN-β (− 1.41 ± 0.27, n = 9/− 2.69 ± 0.28, n = 6, p = 0.0057); TLR3: IFN-β/+A53T-2 (− 2.48 ± 0.27, n = 6/− 3.175 ± 0.22, n = 7, p = 0.0318); Poly I:C or LPS application had no significant effect on A53T mutant-induced expression of TLR2 or TLR3.
To evaluate protein expression alteration of TLRs induced by IFN-γ, we determined TLR2 and TLR3 signals by western blotting analysis (Fig. 2c). The gray scale data showed that (1) IFN-γ alone significantly elevated protein level of TLR3 (0.78 ± 0.10, n = 3) other than TLR2 compared with control group (0.39 ± 0.08, n = 3, p = 0.0353) (Fig. 2d); (2) A53T mutant α-syn alone enhanced expression of TLR3 (1.08 ± 0.15, n = 3) other than TLR2 compared with control group (0.39 ± 0.08, n = 3, p = 0.026) (Fig. 2d), indicating that TLR3 is much more sensitive to α-syn stimulation at its protein level; (3) Additive application of IFN-γ (2 ng/mL) significantly enhanced protein level of TLR2 or TLR3 compared with control group or anyone alone (Fig. 2d,e). TLR3: IFN-γ/+A53T-2 (0.78 ± 0.10, n = 3/1.76 ± 0.16, n = 3, p = 0.0165); A53T-2/+IFN-γ (1.08 ± 0.15, n = 3/1.76 ± 0.16, n = 3, p = 0.0427); TLR2: IFN-γ/+A53T-2 (0.52 ± 0.20, n = 3/1.59 ± 0.24, n = 3, p = 0.0364); A53T-2/+IFN-γ (0.27 ± 0.16, n = 3/1.59 ± 0.24, n = 3, p = 0.0260); (4) IFN-γ synergistically upregulated expression of TLR1 or TLR4 induced by A53T mutant α-syn (2 μg/mL, 24 h) (Fig. 2f,g), providing an extra support to TLR2 and TLR3 data.
Collectively, IFN-γ or α-syn alone significantly upregulated expression of TLR3 other than TLR2, and IFN-γ application substantially enhanced α-syn-induced expression of TLR2, TLR3, or NF-κB at mRNA level, with no enhanced TLR2 expression at protein level. These data strongly indicate an amplification effect of IFN-γ and a functional difference of TLR3 and TLR2 in α-syn-induced innate immunity. Additionally, this amplification effect was also observed with IFN-β other than Poly I:C or LPS.
IFN-γ Potentiated α-Syn-Induced TNF-α Expression and Release
Previous studies show that IFN-γ has a synergistic effect with other cytokines such as TNF-α and IL-1β in neuroinflammatory response. In this study, to reveal neurotoxic mechanism in α-synucleinopathy, we first examined the expression pattern of TNF-α or IL-1β induced by α-syn (A53T mutant or WT) in dose or time dependency, and then observed the expression pattern alteration by additive application of IFN-γ, involving mRNA expression or protein release of cytokines TNF-α and IL-1β.
Dose or time dependency showed that α-syn (A53T mutant or WT) alone significantly upregulated TNF-α mRNA expression in almost all treatment groups (Fig. 3a,b, solid circle), with the maximal level at high doses (8 or 20 μg/mL) and at early time-point (3 h), displaying a PDEC or a NTEC pattern, as evidenced with IL-1β expression in our previous study [8]. TNF-α: A53T-2/A53T-8 (− 2.35 ± 0.31, n = 8/− 3.48 ± 0.68, n = 4, p = 0.0021); A53T 12 h/A53T 24 h (− 4.40 ± 0.60, n = 6/− 2.35 ± 0.31, n = 8, p < 0.0001); WT-0.5/WT-2 (− 0.69 ± 0.22, n = 4/− 2.04 ± 0.41, n = 4, p = 0.0098); WT 12 h/WT 24 h (− 3.29 ± 0.27, n = 4/− 2.04 ± 0.41, n = 4, p = 0.0050).

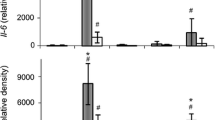
Expression profiles of cytokines TNF-α and IL-1β induced by A53T mutant or wild-type (WT) α-syn plus IFN-γ. a, b TNF-α mRNA expression was significantly upregulated by A53T mutant or WT α-syn, showing a positive dose-effect correlation (PDEC) or a negative time-effect correlation (NTEC) pattern, as shown in TLR2 or TLR3 expression. This induction effect was significantly enhanced by additive application of IFN-γ in almost all α-syn treatment groups. c, d IL-1β mRNA expression was reduced by additive application of IFN-γ at α-syn high doses or early time-points. e IFN-γ antibody (5 μg/mL) significantly inhibited TNF-α expression induced by A53T α-syn plus IFN-γ groups. f Additive application of IFN-β (2 or 5 ng/mL) significantly potentiated TNF-α expression induced by A53T mutant α-syn, a similar amplification effect to IFN-γ. The data are presented by scatter charts showing sample distribution, number (n), means (−), and p values (in the text) at a level of p < 0.05 (ANOVA, margin means). Asterisk (*) represents significant difference at ΔΔCt values between treatment groups, with pound sign (#) between the indicated groups at ΔCt values
Combination application of α-syn and IFN-γ (2 ng/mL) showed that (1) IFN-γ alone significantly upregulated TNF-α mRNA expression (ΔCt, 8.62 ± 0.91; n = 7) (Fig. 3a, b) other than IL-1β (Fig. 3c), compared with control group (10.62 ± 0.89, n = 10, p = 0.0330), but the upregulation amplitude is low, indicating a weak stimulation effect of IFN-γ on TNF-α; (2) IFN-γ significantly potentiated TNF-α expression induced by A53T mutant or WT α-syn in most treatment groups (Fig. 3a,b), whereas IL-1β expression was downregulated by IFN-γ at high doses or early time-point (12 h) of α-syn (Fig. 3c,d), with no synergy in induced IL-1β expression. Additionally, IFN-γ application had no significant effect on endogenous IFN-γ expression under stimulation of α-syn. It’s noteworthy that IFN-γ induced a considerably high amplitude of TNF-α expression in almost all WT α-syn groups (Fig. 3b), even comparable with those in A53T mutant groups, indicating that IFN-γ-mediated increment of WT α-syn cytotoxicity may be endowed with more pathological implication due to the fact that A53T mutant α-syn only exists in a limited human population. TNF-α (Fig. 3a,b): A53T-0.5/+IFN-γ (− 1.43 ± 0.45, n = 5/− 3.39 ± 0.64, n = 4, p = 0.0040); A53T-2/+IFN-γ (− 2.35 ± 0.31, n = 8/− 3.99 ± 0.52, n = 7, p = 0.0081); A53T 48 h/+IFN-γ (− 2.46 ± 0.57, n = 6/− 4.60 ± 0.49, n = 4, p = 0.0046); WT-0.5/+IFN-γ (− 0.69 ± 0.22, n = 4/− 2.48 ± 0.28, n = 4, p = 0.0064); WT-2/+IFN-γ (− 2.04 ± 0.41, n = 4/− 3.68 ± 0.23, n = 4, p = 0.0019); WT 3 h/+IFN-γ (− 4.34 ± 0.39, n = 4/− 5.23 ± 0.12, n = 4, p < 0.0001); WT 12 h/+IFN-γ (− 3.29 ± 0.27, n = 4/− 4.16 ± 0.15, n = 4, p = 0.0085); IL-1β (Fig. 3c,d): A53T 3 h/+IFN-γ (− 9.54 ± 0.33, n = 6/− 9.01 ± 0.16, n = 4, p = 0.0219); A53T 12 h/+IFN-γ (− 9.47 ± 0.56, n = 6/− 7.48 ± 0.29, n = 4, p = 0.0032); WT-8/+IFN-γ (− 9.09 ± 0.44, n = 4/− 8.44 ± 0.14, n = 4, p = 0.0361); WT-20/+IFN-γ (− 9.20 ± 0.41, n = 4/− 8.38 ± 0.17, n = 4, p = 0.0233); WT 12 h/+IFN-γ (− 9.38 ± 0.36, n = 4/− 7.50 ± 0.16, n = 4, p < 0.0001); (3) Additive application of IFN-γ antibody (Ab, 5 μg/mL) significantly reduced TNF-α mRNA expression (− 2.77 ± 0.26, n = 4), compared with A53T mutant α-syn (2 μg/mL) plus IFN-γ (2 ng/mL) (− 3.80 ± 0.33, n = 7, p = 0.0098) (Fig. 3e), supporting the amplification effect of IFN-γ in α-syn-induced TNF-α expression.
To determine the specificity of IFN-γ impact on α-syn induction, we replaced IFN-γ (2 ng/mL) with IFN-β (2 ng/mL) or LPS (0.2 μg/mL) to coincubate with A53T mutant α-syn (2 μg/mL). IFN-β significantly upregulated TNF-α expression induced by A53T, and when IFN-β dose was increased to 5 ng/mL, a more stimulation effect was not observed (Fig. 3f), indicating that both IFN-β and IFN-γ had a synergistic effect with α-syn. TNF-α: A53T-2/+IFN-β2 (− 2.35 ± 0.31, n = 8/− 3.27 ± 0.36, n = 4, p = 0.0195); A53T-2/+IFN-β5 (− 2.35 ± 0.31, n = 8/− 3.40 ± 0.20, n = 5, p = 0.0162); Application of LPS had no effect on A53T-induced TNF-α expression.
Western blotting analysis showed that (1) A53T mutant α-syn alone enhanced protein level of TNF-α at 3 h and 48 h, but in dose dependency, TNF-α levels were gradually downregulated compared with control level (Fig. 4a), an induction pattern different from the PDEC at its mRNA level. This inconsistent phenomenon may be attributed to the inherent immune response capacity of astrocytes, which is preferentially motivated by α-syn stimulation, leading to exhaustive release of effector TNF-α, as no endotoxin or intrinsic toxicity of A53T mutant is detected by manufacturer. TNF-α (Fig. 4b, gray scale): control/A53T 3 h (0.81 ± 0.13, n = 3/1.81 ± 0.19, n = 3, p = 0.0154); control/A53T 48 h (0.51 ± 0.13, n = 3/1.28 ± 0.17, n = 3, p = 0.035); (2) IFN-γ alone (2 ng/mL) did not significantly enhance TNF-α protein level compared with control group (Fig. 4c), differently from its low-level upregulated mRNA expression, indicating that IFN-γ itself is not a strong stimulator to TNF-α production in astrocytes; (3) IFN-γ and A53T mutant α-syn conjointly enhanced TNF-α protein expression (1.076 ± 0.214, n = 3) compared with A53T mutant alone (0.30 ± 0.074, n = 3, p = 0.00967) or IFN-γ alone (0.53 ± 0.13, n = 3, p = 0.0362) (Fig. 4d).

Protein expression and release profiles of TNF-α induced by A53T mutant or wild-type (WT) α-syn plus IFN-γ. a, b Protein expression profiles of TNF-α (75 kDa) and β-actin (45 kDa, reference control) by western blotting analysis and its densitometric analysis by ImageJ software. The dose and time dependency showed that A53T mutant α-syn significantly enhanced TNF-α expression (gray scales) at 0.5 μg/mL and 3 h (or 48 h) ("at 0.5 μg/mL and 3 h (or 48 h)" is replaced with "at 3 hour and 48 hour") compared with control group, with a reduced expression at high doses of α-syn. c, d Protein expression profiles of TNF-α and β-actin. IFN-γ significantly enhanced TNF-α expression induced by A53T mutant α-syn, compared with control group and anyone alone, but IFN-γ alone did not enhance TNF-α expression compared with control group. e, f Protein release profiles of TNF-α by ELISA. A53T or WT α-syn significantly enhanced TNF-α release level in a dose- or time-dependent manner, and this effect was significantly potentiated by IFN-γ at high doses (2, 8, or 20 μg/mL) of A53T mutant α-syn. The data are presented by scatter charts showing sample distribution, number (n), means (−), and significant difference (*) at a level of p < 0.05 (ANOVA, margin means), with p values shown in the text
ELISA data showed that (1) α-Syn (A53T mutant or WT) alone significantly upregulated TNF-α release in a PDEC pattern or a positive time-effect correlation (PTEC) pattern (Fig. 4e,f, solid circle). TNF-α: A53T-0.5/A53T-2 (5.87 ± 2.35, n = 7/23.42 ± 7.37, n = 10, p = 0.0014); A53T 3 h/A53T 24 h (10.785 ± 3.361, n = 7/23.42 ± 7.37, n = 10, p = 0.0381); WT-0.5/WT-2 (3.30 ± 1.11, n = 4/21.87 ± 5.28, n = 4, p = 0.0028); WT 3 h/WT 24 h (6.97 ± 3.14, n = 3/21.87 ± 5.28, n = 4, p = 0.0381); (2) Additive application of IFN-γ significantly enhanced TNF-α release compared with A53T α-syn (2, 8 or 20 μg/mL) (Fig. 4e): A53T-2/+IFN-γ (23.42 ± 7.37, n = 10/26.93 ± 7.48, n = 8, p = 0.0388); A53T-8/+IFN-γ (55.99 ± 12.23, n = 9/77.15 ± 9.75, n = 4, p = 0.0249); (3) In WT α-syn treatment groups, additive application of IFN-γ did not significantly modulate TNF-α release level (Fig. 4f, right).
Collectively, α-syn (A53T mutant or WT) alone upregulated TNF-α mRNA expression in a PDEC or NTEC pattern, and TNF-α release in a PDEC or a PTEC pattern, and IFN-γ potentiated TNF-α expression induced by A53T mutant at mRNA, protein and protein release levels, and by WT α-syn at mRNA level, indicating a neurotoxic amplification mechanism of α-syn and IFN-γ in TNF-α expression.
TLR2 Blockage Suppressed Synergistic Effect of α-Syn and IFN-γ
To further explore the differential role of TLRs in α-syn-induced neurotoxicity, we chose TLR2 antagonist (CPT22, 5 μg/mL) and TLR4 antagonist (C34, 5 μg/mL) to coincubate with A53T mutant α-syn or plus IFN-γ (2 ng/mL) at multiple time-points, and then determined expression profiles of TLR2, TLR3, and TNF-α.
Time dependency showed that (1) CPT22 significantly downregulated TLR3 mRNA expression in almost all treatment groups (Fig. 5a) and TNF-α mRNA expression at later time-points (24 and 48 h), but upregulated TNF-α expression at early time-points (3 and 12 h) (Fig. 5b). The result indicates that TLR2 blockage strongly inhibits TLR3 function, a possible receptor interaction mechanism: TLR2 is involved in α-syn endocytosis, which effectively stimulates TLR3 expression. Also, the inconsistency of TNF-α expression at early and later time-points shows a transient reactivity increase of α-syn neurotoxicity and then a prolonged reactivity decrease due to TLR2 blockage, revealing a complex regulatory mechanism by TLR2 in innate immune response. TLR3 (Fig. 5a): A53T-2/+CPT22 (− 0.679 ± 0.223, n = 11/− 0.207 ± 0.09, n = 6, p = 0.0009); A53T-2/IFN-γ/+CPT22 (− 2.71 ± 0.43, n = 8/− 1.703 ± 0.34, n = 6, p = 0.0006); A53T 3 h/+CPT22 (− 0.145 ± 0.288, n = 6/0.735 ± 0.24, n = 4, p = 0.0007); A53T 12 h/+CPT22 (− 2.076 ± 0.504, n = 6/− 0.5425 ± 0.17, n = 4, p < 0.0001); A53T 48 h/+CPT22 (− 0.752 ± 0.197, n = 6/1.025 ± 0.25, n = 4, p < 0.0001); TNF-α (Fig. 5b): A53T-2/+CPT22 (− 2.35 ± 0.31, n = 8/− 1.95 ± 0.817, n = 7, p = 0.0473); A53T-2/IFN-γ/+CPT22 (− 3.99 ± 0.52, n = 7/− 2.288 ± 0.532, n = 5, p = 0.0157); A53T 3 h/+CPT22 (− 5.73 ± 0.14, n = 6/− 7.1 ± 0.393, n = 4, p = 0.0283); A53T 12 h/+CPT22 (− 4.40 ± 0.60, n = 6/−5.738 ± 0.55, n = 4, p = 0.0394); A53T 48 h/+CPT22 (− 2.46 ± 0.57, n = 6/0.7525 ± 0.316, n = 4, p < 0.0001); (2) CPT22 significantly downregulated TLR2 mRNA expression induced by A53T mutant at 48 h (not shown in the figures), the maximal time-point in our study, with no effect at 3, 12, and 24 h, indicating that persistent TLR2 blockage may inhibit expression and function of TLR2 itself. TLR2: A53T 48 h/+CPT22 (− 1.54 ± 0.29, n = 5/0.03 ± 0.32, n = 3, p < 0.0001); (3) Additive application of C34 had no significant effect on expression of TLR3 (Fig. 5c), TLR2 (not shown) and TNF-α (Fig. 5d), even in costimulation of A53T mutant and IFN-γ, indicating a functional difference between TLR2 and TLR4 in α-syn induction process. In addition, we observed that NF-κB expression was not significantly regulated by CPT22 or C34 in A53T mutant α-syn and IFN-γ costimulation.

Impact of TLR2 or TLR4 blockage on expression of TLR3 or TNF-α. a CPT22 (TLR2 antagonist, 5 μg/mL) significantly downregulated TLR3 expression induced by A53T mutant α-syn in almost all treatment groups, indicating that TLR2 has a potent regulatory effect on TLR3. b CPT22 significantly downregulated TNF-α expression induced by A53T mutant α-syn at later time-points (24 or 48 h) or in α-syn plus IFN-γ group, with the TNF-α expression upregulated at early time-points (3 and 12 h). c, d C34 (TLR4 antagonist, 5 μg/mL) had no significant effect on TLR3 and TNF-α expressions in all treatment groups. e, f Protein release profile of TNF-α by ELISA. CPT22 significantly inhibited TNF-α release induced by A53T mutant α-syn in almost all groups, whereas C34 did not do so. The data are presented by scatter charts, showing sample distribution, number (n), means (−), and significant difference (*) at a level of p < 0.05 (ANOVA, margin means), with p values shown in the text
ELISA data showed that CPT22 significantly downregulated TNF-α release in all treatment groups (Fig. 5e), whereas C34 application only displayed a tendency of TNF-α downregulation (Fig. 5f), indicating that TLR2 plays a crucial role in TNF-α release or α-syn neurotoxicity. TNF-α: A53T-2/+CPT22 (23.42 ± 7.374, n = 10/10.58 ± 4.04, n = 5, p = 0.0361); A53T-2/IFN-γ/+CPT22 (26.93 ± 7.48, n = 8/7.82 ± 1.14, n = 5, p = 0.0082); A53T 3 h/+CPT22 (10.785 ± 3.36, n = 7/4.175 ± 1.14, n = 4, p = 0.0367); A53T 12 h/+CPT22 (20.96 ± 4.71, n = 8/8.2 ± 2.19, n = 4, p = 0.0217); A53T 48 h/+CPT22 (30.25 ± 6.34, n = 8/17.47 ± 2.66, n = 4, p = 0.0359).
Taken together, blockage of TLR2 other than TLR4 downregulated mRNA expressions of TLR3 (all time-points), TLR2 (later time-point) and TNF-α (later time-points), and TNF-α protein release, a suppressive effect on the innate immunity induced by α-syn or plus IFN-γ, suggesting that TLR2 and TLR3 may jointly play a critical role in inflammatory response and neurotoxicity, and a novel interaction mechanism of α-syn, TLRs and cytokines may underlie neurotoxic pathogenesis of neurodegenerative diseases. However, this hypothesis needs to be in-depth investigated elsewhere.
Discussion
Glial activation and concomitant neuroinflammation are a crucial characteristic in pathogenesis of neurodegenerative diseases, and involvement of additional inflammatory factors could be a pivotal risk to exacerbate such a neuroinflammatory process [35]. It has been confirmed that α-syn and its mutant forms (A30P, E46K, and A53T) can potently stimulate microglia and astrocytes to initiate innate immunity [7, 31, 36], involving immune recognition, TLRs activation and release of pro-inflammatory factors such as cytokines and chemokines [37]. On the other side, the self-antigenic attribution of α-syn may be regulated by other inflammatory factors of various origins, to control innate immune response. However, the interaction mechanism of α-syn with other factors has not been fully elucidated. In this study, we chose A53T mutant or WT α-syn to costimulate primary astrocytes with IFN-γ in a dose- or time-dependent manner, and then evaluate their interaction effect on canonical inflammatory pathway components such as TLR2, TLR3, NF-κB, TNF-α, and IL-1β. The results showed that A53T mutant or WT α-syn upregulated expressions of these inflammatory components, and this induction effect except for IL-1β was significantly amplified by additive application of IFN-γ at mRNA, protein or protein release level, suggesting that α-syn and IFN-γ synergistically potentiate innate immunity and neurotoxic outcomes via TLR2, TLR3, and TNF-α, a novel molecular interaction mechanism underlying neuroinflammatory pathogenesis.
TLRs, as a group of antigen recognizing receptors, have a close association with α-syn-induced inflammation. In primary microglia, TLR agonists Pam3Csk4 (TLR2/1) and single-stranded RNA40 (TLR7) differentially affect WT α-syn-induced inflammatory response [31], and misfolded α-syn (50 ng/mL, 24 h) upregulates TLR2 and TLR3 expression, with no effect on NF-κB and TNF-α expression [20]. In memory impairment model in vivo, α-syn oligomers impair memory through TLR2-mediated glial cell activation [24], and in PD brain, TLR2 expression is significantly increased in neurons and shows a strong colocalization with α-syn in Lewy bodies [38]. In neuronal and glial cell culture, TLR2 activation increases release of endogenous α-syn and pro-inflammatory cytokines, which in turn leads to increment of autophagy/lysosomal pathway marker p62 [38], indicating an interplay of TLR2 and TLR3 because lysosomes or endosomes are known as an effective stimulator of TLR3 [39]. Our study directly demonstrated that A53T mutant or WT α-syn stimulated expression of TLR2 and TLR3 in a PDEC pattern and a NTEC pattern, and TLR2 blockage led to downregulation of TLR3 and TLR2 expression, reflecting a strong interaction between TLR2 and TLR3 in α-syn-induced neuroinflammation. Moreover, this α-syn induction effect was significantly amplified by additive application of IFN-γ. Therefore, TLR2 and TLR3 both play a critical role in α-syn-induced activation of inflammatory pathway or even in synergistic neurotoxicity of α-syn and IFN-γ [38].
Cytokines are a group of immune effector factors, and their levels determine status of inflammatory outcomes, involving a range of pro-inflammatory molecules such as TNF-α, IFN-γ, IL-6, IL-1β, nitric oxide, reactive oxygen species, and excitatory amino acids, and several anti-inflammatory cytokines such as IL-10, IL-13, IL-14, and transforming growth factor-β [40]. α-Syn aggregation in the brain is a core feature of PD and leads to microglia/astrocyte activation, inflammatory cytokine and chemokine production, and ultimately to neurodegeneration, reflecting a correlation of α-syn stimulation and cytokine production.
Previous evidence shows that intracellular α-syn by astrocyte endocytosis induces gene expression of pro-inflammatory cytokines and chemokines, displaying a positive correlation with the extent of α-syn accumulation [22]; in mutant α-syn transgenic mice, striatal mRNA levels of cytokines such as TNF-α, IL-1β, and IL-6 are upregulated at 1, 6, and 12 month, with the maximal at 1 month and the minimal at 12 month [41], displaying a similar induction pattern to the NTEC in our in vitro study; in microglia culture, WT α-syn (10, 50, and 250 ng/mL, 24 h) significantly upregulates TNF-α expression in a dose-dependent manner at RNA and protein release levels [42]. Nevertheless, our in vitro study further demonstrated that A53T mutant or WT α-syn upregulated expression of TNF-α and IL-1β in a PDEC or a NTEC pattern, suggesting that α-syn accumulation and persistent release may be required for priming of glial cells and maintenance of neurotoxic microenvironment [43, 44].
IFN-γ, as an immunoregulatory cytokine, is not only able to potently activate microglia and astrocytes, but also modulate immune function of other inflammatory factors. In this study, IFN-γ alone significantly stimulated mRNA expressions of TLR3 and TNF-α other than TLR2, NF-κB, and IL-1β in astrocytes, further supporting a role of IFN-γ in astrocyte activation via TLR3 and TNF-α, despite TNF-α protein enhancement not detected in this study and others [10]. In previous studies on costimulation of IFN-γ with TNF-α, IL-1β, or LPS, IFN-γ has a neurotoxic enhancement attribution by astrocytes and microglia to exacerbate pathology of neurodegenerative diseases, along with IFN-γ receptor upregulation [45]. However, in neonatal rat glial cells, IL-1β other than TNF-α inhibits IFN-γ signaling in astrocytes and production of nitric oxide in microglia, suggesting a negative interplay of IFN-γ and IL-1β [46], as evidenced in our study. Regarding the synergy of IFN-γ and α-syn, previous research only provides a limited evidence. In PD patient sera, an increase in α-syn autoantibody titer is associated with boosted level of IL-6 or TNF-α and decreased level of IFN-γ, indicating a functional difference of TNF-α and IFN-γ in α-syn induction reaction [47]. In glial cell culture, combined application of IL-6 and IFN-γ leads to a sustained protein increase and dispersal of α-syn and Tau compared with their single treatment, along with disruption of normal perinuclear tau and peripheral α-syn patterns [48], indicating that IFN-γ and α-syn or IL-6 synergistically increases neurotoxicity, but it’s noted that astrocyte response induced by IFN-γ suppresses microglia activation mediated by IL-6 [49]. A53T mutant α-syn delivered into rat substantia nigra leads to an inflammatory response in striatum but not in substantia nigra by increasing expression of TNF-α, IFN-γ, and IL-1β [50], indicating a causal correlation of α-syn with TNF-α, IFN-γ, or IL-1β expression. Interestingly, in microglia from surgical specimens, α-syn and IFN-γ induce more toxicity for SH-SY5Y cells than any one alone [11]. Also in PD monkey, a sustained increment of IFN-γ and TNF-α other than IL-1β and IL-6 is detected in blood serum, and high expression of IFN-γ is mainly localized in microglia of substantia nigra, whereas TNF-α is highly expressed in astrocytes but not microglia, suggesting a functional difference of microglia and astrocytes in production or release of cytokines and a potential glial interaction mechanism [23, 25]. Collectively, these previous studies point to a hypothesis that IFN-γ has a synergistic effect with α-syn and other cytokines in glial cell activation, and our results directly demonstrated that exogenous IFN-γ amplified α-syn-induced inflammatory response and neurotoxicity in astrocytes by upregulating TLR2, TLR3, and TNF-α other than IL-1β. This synergistic effect may be mechanistically related to coreceptors or complex intracellular signal connection of TLRs such as toll-IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) or TRIF-related adaptor molecule (TRAM) [51]. Therefore, we may well raise a possible signal amplification pathway in glial reciprocal activation, i.e., α-syn/IFN-γ/TLR2/TLR3/NF-κB/TNF-α, by which α-syn (neurons) orchestrates with IFN-γ and TNF-α released from different cell sources (glia or immune cells) to maintain a sustained inflammatory state at an appropriate level [23, 25, 45].
It is noteworthy that we unexpectedly found a special synergistic effect between IFN-γ and WT α-syn: IFN-γ substantially enhanced mRNA expressions of TLR2, TLR3, and TNF-α induced by WT α-syn in an amplitude comparable with or even more than A53T mutant/IFN-γ induction amplitude. This result indicates that less cytotoxicity of WT α-syn may be greatly enhanced due to IFN-γ involvement in a pro-inflammatory microenvironment, a potential neurotoxicity enhancing mechanism of WT α-syn, which may present a more important inflammatory pathogenesis of neurodegenerative diseases than A53T mutant α-syn, despite lack of the relevant evidence at protein or protein release level in this study.
Conclusions
Our data collectively demonstrated that A53T mutant or WT α-syn alone significantly upregulated mRNA or protein expression of TLR2, TLR3, NF-κB, TNF-α, and IL-1β in a PDEC pattern and a NTEC pattern, and this effect except for IL-1β was significantly amplified by additive application of IFN-γ, as well as IFN-β, displaying a synergistic effect of α-syn and IFN-γ. Moreover, TLR2 blockage other than TLR4 blockage significantly suppressed expression of TLR3, TLR2, and TNF-α in α-syn or plus IFN-γ stimulation, indicating a strong interaction between TLR2 and TLR3 and a critical role of TLR2 in TNF-α expression and release. Therefore, we concluded that α-syn (A53T mutant or WT) activated canonical inflammatory pathway in a positive dose- or a negative time-correlation pattern, and IFN-γ synergistically enhanced α-syn-induced neurotoxicity via TLR2, TLR3, and TNF-α in astrocytes. These findings may unravel a potential neurotoxic mechanism in synucleinopathies, that is, the molecular interaction between α-syn, TLR2/3, IFN-γ, and TNF-α modulates innate immunity and inflammatory outcomes, and their coexistence in inflammatory microenvironment may be required for initiation and maintenance of innate immunity activation, a novel immune amplification mechanism underlying pathogenesis of neurodegenerative diseases.
Data Accessibility Statement
Information about the animal model, experimental methods, and data described in this article are available to scientific and medical communities for review, verification, and research studies; Data supporting the conclusions of this article are all publicly reachable in Pubmed database. For data inquiry, please contact Dr. Jintang Wang at jintangwang@hotmail.com or Dr. Sean Leng at sleng1@jhmi.edu.
Abbreviations
- AD:
-
Alzheimer’s disease
- ANOVA:
-
analysis of variance;
- C34:
-
TLR4-IN-C34
- CPT22:
-
CU-CPT22
- DAMPs:
-
damage-associated molecular patterns
- DAPI:
-
4′,6-diamidino-2-phenylindole
- ELISA:
-
enzyme-linked immunoabsorbent assay
- FBS:
-
fetal bovine serum
- GAPDH:
-
glyceraldehyde phosphate dehydrogenase
- GFAP:
-
glial fibrillary acidic protein
- IFN-γ:
-
interferon-γ
- IL-1β:
-
interleukin-1β
- LPS:
-
lipopolysaccharide
- LSD:
-
fisher’s least significant differences
- NF-κB:
-
nuclear factor-kappa B
- PBS:
-
phosphate-buffered saline
- PD:
-
Parkinson’s disease
- Poly I:C:
-
polyinosinic-polycytidylic acid
- qRT-PCR:
-
quantitative real-time polymerase chain reaction
- TLR:
-
toll-like receptor
- TNF-α:
-
tumor necrosis factor-α
References
Norris EH, Giasson BI, Lee VM (2004) Alpha-synuclein: normal function and role in neurodegenerative diseases. Curr Top Dev Biol 60:17–54. https://doi.org/10.1016/S0070-2153(04)60002-0
Lawand NB, Saadé NE, El-Agnaf OM, Safieh-Garabedian B (2015) Targeting α-synuclein as a therapeutic strategy for Parkinson’s disease. Expert Opin Ther Targets 19(10):1351–1360. https://doi.org/10.1517/14728222.2015.1062877
Woerman AL, Kazmi SA, Patel S, Aoyagi A, Oehler A, Widjaja K, Mordes DA, Olson SH et al (2018) Familial Parkinson’s point mutation abolishes multiple system atrophy prion replication. Proc Natl Acad Sci U S A 115(2):409–414. https://doi.org/10.1073/pnas.1719369115
Lee HJ, Kim C, Lee SJ (2010) Alpha-synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxidative Med Cell Longev 3(4):283–287. https://doi.org/10.4161/oxim.3.4.12809
Alvarez-Erviti L, Couch Y, Richardson J, Cooper JM, Wood MJ (2011) Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci Res 69(4):337–342. https://doi.org/10.1016/j.neures.2010.12.020
Ma D, Jin S, Li E, Doi Y, Parajuli B, Noda M, Sonobe Y, Mizuno T et al (2013) The neurotoxic effect of astrocytes activated with toll-like receptor ligands. J Neuroimmunol 254(1–2):10–18. https://doi.org/10.1016/j.jneuroim.2012.08.010
Rivest S (2009) Regulation of innate immune responses in the brain. Nat Rev Immunol 9(6):429–439. https://doi.org/10.1038/nri2565
Wang J, Chen Z, Walston JD, Gao P, Gao M, Leng SX (2018) α-Synuclein activates innate immunity but suppresses interferon-γ expression in murine astrocytes. Eur J Neurosci 48(1):1583–1599. https://doi.org/10.1111/ejn.13956
Vidovic M, Sparacio SM, Elovitz M, Benveniste EN (1990) Induction and regulation of class II major histocompatibility complex mRNA expression in astrocytes by interferon-gamma and tumor necrosis factor-alpha. J Neuroimmunol 30(2–3):189–200
Chung IY, Norris JG, Benveniste EN (1991) Differential tumor necrosis factor alpha expression by astrocytes from experimental allergic encephalomyelitis-susceptible and -resistant rat strains. J Exp Med 173(4):801–811
Klegeris A, Pelech S, Giasson BI, Maguire J, Zhang H, McGeer EG, McGeer PL (2008) Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol Aging 29(5):739–752. https://doi.org/10.1016/j.neurobiolaging.2006.11.013
Kim S, Seo JH, Suh YH (2004) Alpha-synuclein, Parkinson’s disease, and Alzheimer’s disease. Parkinsonism Relat Disord 10(suppl 1):S9–S13. https://doi.org/10.1016/j.parkreldis.2003.11.005
Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K (2002) Demonstration of alpha-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp Neurol 176(1):98–104
Shin EC, Cho SE, Lee DK, Hur MW, Paik SR, Park JH, Kim J (2000) Expression patterns of alpha-synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol Cell 10(1):65–70
El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA et al (2003) Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J 17(13):1945–1947. https://doi.org/10.1096/fj.03-0098fje
Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25(25):6016–6024. https://doi.org/10.1523/jneurosci.0692-05.2005
Sui YT, Bullock KM, Erickson MA, Zhang J, Banks WA (2014) Alpha-synuclein is transported into and out of the brain by the blood-brain barrier. Peptides 62:197–202. https://doi.org/10.1016/j.peptides.2014.09.018
Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Döring F, Trenkwalder C, Schlossmacher MG (2011) α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. Lancet Neurol 10(3):30–40. https://doi.org/10.1016/S1474-4422(11)70014-X
Lin CH, Yang SY, Horng HE, Yang CC, Chieh JJ, Chen HH, Liu BH, Chiu MJ (2017) Plasma α-synuclein predicts cognitive decline in Parkinson’s disease. J Neurol Neurosurg Psychiatry 88(10):818–824. https://doi.org/10.1136/jnnp-2016-314857
Béraud D, Twomey M, Bloom B, Mittereder A, Ton V, Neitzke K, Chasovskikh S, Mhyre TR et al (2011) α-Synuclein alters toll-like receptor expression. Front Neurosci 5:80. https://doi.org/10.3389/fnins.2011.00080
Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517(7534):311–320. https://doi.org/10.1038/nature14191
Farina C, Aloisi F, Meinl E (2007) Astrocytes are active players in cerebral innate immunity. Trends Immunol 28(3):138–145. https://doi.org/10.1016/j.it.2007.01.005
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE et al (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541(7638):481–487. https://doi.org/10.1038/nature21029
La Vitola P, Balducci C, Cerovic M, Santamaria G, Brandi E, Grandi F, Caldinelli L, Colombo L et al (2018) Alpha-synuclein oligomers impair memory through glial cell activation and via toll-like receptor 2. Brain Behav Immun 69:591–602. https://doi.org/10.1016/j.bbi.2018.02.012
Barcia C, Ros CM, Annese V, Gómez A, Ros-Bernal F, Aguado-Llera D, Martínez-Pagán ME, de Pablos V et al (2012) IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis 3:e379. https://doi.org/10.1038/cddis.2012.123
Papageorgiou IE, Lewen A, Galow LV, Cesetti T, Scheffel J, Regen T, Hanisch UK, Kann O (2016) TLR4-activated microglia require IFN-γ to induce severe neuronal dysfunction and death in situ. Proc Natl Acad Sci U S A 113(1):212–217. https://doi.org/10.1073/pnas.1513853113
Mount MP, Lira A, Grimes D, Smith PD, Faucher S, Slack R, Anisman H, Hayley S et al (2007) Involvement of interferon-gamma in microglial-mediated loss of dopaminergic neurons. J Neurosci 27(12):3328–3337. https://doi.org/10.1523/jneurosci.5321-06.2007
Chakrabarty P, Ceballos-Diaz C, Lin WL, Beccard A, Jansen-West K, McFarland NR, Janus C, Dickson D et al (2011) Interferon-γ induces progressive nigrostriatal degeneration and basal ganglia calcification. Nat Neurosci 14(6):694–696. https://doi.org/10.1038/nn.2829
Cimoli G, Parodi S, Russo P (1996) Interferon-gamma enhances TNF sensitivity in A172 human glioblastoma cell line. Oncol Rep 3(2):369–370
Liscovitch N, French L (2014) Differential co-expression between α-synuclein and IFN-γ signaling genes across development and in Parkinson’s disease. PLoS One 9(12):e115029. https://doi.org/10.1371/journal.pone.0115029
Roodveldt C, Labrador-Garrido A, Gonzalez-Rey E, Lachaud CC, Guilliams T, Fernandez-Montesinos R, Benitez-Rondan A, Robledo G et al (2013) Preconditioning of microglia by α-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS One 8(11):e79160. https://doi.org/10.1371/journal.pone.0079160
Springall R, Amezcua-Guerra LM, Gonzalez-Pacheco H, Furuzawa-Carballeda J, Gomez-Garcia L, Marquez-Velasco R, Mejía-Domínguez AM, Cossío-Aranda J et al (2013) Interferon-gamma increases the ratio of matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 in peripheral monocytes from patients with coronary artery disease. PLoS One 8(8):e72291. https://doi.org/10.1371/journal.pone.0072291
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Smith GM, Rutishauser U, Silver J, Miller RH (1990) Maturation of astrocytes in vitro alters the extent and molecular basis of neurite outgrowth. Dev Biol 138(2):377–390
Wang J, Song Y, Chen Z, Leng SX (2018a) Connection between systemic inflammation and neuroinflammation underlies neuroprotective mechanism of several phytochemicals in neurodegenerative diseases. Oxidative Med Cell Longev 2018:1–16. https://doi.org/10.1155/2018/1972714
Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E et al (2013) Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4:1562. https://doi.org/10.1038/ncomms2534
González-Reyes RE, Nava-Mesa MO, Vargas-Sánchez K, Ariza-Salamanca D, Mora-Muñoz L (2017) Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci 10:427. https://doi.org/10.3389/fnmol.2017.00427
Dzamko N, Gysbers A, Perera G, Bahar A, Shankar A, Gao J, Fu Y, Halliday GM (2017) Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol 133(2):303–319. https://doi.org/10.1007/s00401-016-1648-8
Liu J, Zhou Y, Wang Y, Fong H, Murray TM, Zhang J (2007) Identification of proteins involved in microglial endocytosis of alpha-synuclein. J Proteome Res 6(9):3614–3627. https://doi.org/10.1021/pr0701512
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91(2):461–553. https://doi.org/10.1152/physrev.00011.2010
Su X, Federoff HJ, Maguire-Zeiss KA (2009) Mutant alpha-synuclein overexpression mediates early proinflammatory activity. Neurotox Res 16(3):238–254. https://doi.org/10.1007/s12640-009-9053-x
Su X, Maguire-Zeiss KA, Giuliano R, Prifti L, Venkatesh K, Federoff HJ (2008) Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging 29(11):1690–1701. https://doi.org/10.1016/j.neurobiolaging.2007.04.006
Couch Y, Alvarez-Erviti L, Sibson NR, Wood MJ, Anthony DC (2011) The acute inflammatory response to intranigral α-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation 8:166. https://doi.org/10.1186/1742-2094-8-166
Khandelwal PJ, Herman AM, Moussa CE (2011) Inflammation in the early stages of neurodegenerative pathology. J Neuroimmunol 238(1–2):1–11. https://doi.org/10.1016/j.jneuroim.2011.07.002
Hashioka S, Klegeris A, Schwab C, McGeer PL (2009) Interferon-gamma-dependent cytotoxic activation of human astrocytes and astrocytoma cells. Neurobiol Aging 30(12):1924–1935. https://doi.org/10.1016/j.neurobiolaging.2008.02.019
Tichauer J, Saud K, von Bernhardi R (2007) Modulation by astrocytes of microglial cell-mediated neuroinflammation: effect on the activation of microglial signaling pathways. Neuroimmunomodulation 14:168–174. https://doi.org/10.1159/000110642
Gruden MA, Yanamandra K, Kucheryanu VG, Bocharova OR, Sherstnev VV, Morozova-Roche LA, Sewell RD (2012) Correlation between protective immunity to α-synuclein aggregates, oxidative stress and inflammation. Neuroimmunomodulation 19(6):334–342. https://doi.org/10.1159/000341400
Bick RJ, Poindexter BJ, Kott MM, Liang YA, Dinh K, Kaur B, Bick DL, Doursout MF et al (2008) Cytokines disrupt intracellular patterns of Parkinson’s disease-associated proteins alpha-synuclein, tau and ubiquitin in cultured glial cells. Brain Res 1217:203–212. https://doi.org/10.1016/j.brainres.2008.03.081
Savarin C, Hinton DR, Valentin-Torres A, Chen Z, Trapp BD, Bergmann CC, Stohlman SA (2015) Astrocyte response to IFN-γ limits IL-6-mediated microglia activation and progressive autoimmune encephalomyelitis. J Neuroinflammation 12:79. https://doi.org/10.1186/s12974-015-0293-9
Chung CY, Koprich JB, Siddiqi H, Isacson O (2009) Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci 29(11):3365–3373. https://doi.org/10.1523/jneurosci.5427-08.2009
Hu W, Jain A, Gao Y, Dozmorov IM, Mandraju R, Wakeland EK, Pasare C (2015) Differential outcome of TRIF-mediated signaling in TLR4 and TLR3 induced DC maturation. Proc Natl Acad Sci U S A 112(45):13994–13999. https://doi.org/10.1073/pnas.1510760112
Funding
This work was supported by the Irma and Paul Milstein Program for Senior Health fellowship from the Milstein Medical Asian American Partnership (MMAAP) Foundation (http://www.mmaapf.org) (JT Wang: 1005352440), and in part by NIH grants R21-AG-043874, R01AI108907 (SX Leng).
Author information
Authors and Affiliations
Contributions
J.T.W designed, performed the research, and wrote the manuscript; Z.C. helped design and put the research into effect; J.W. and P.S.G. provided experiment facilities, techniques, and mice use; M.L.G. performed the statistical analysis and its interpretation; S.X.L. designed research, analyzed data, reviewed overall findings, and wrote the manuscript; and all authors approved the final draft of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Ethics Approval
All animal procedures performed in this study were reviewed and approved by the Institution’s Animal Care Committee, and performed in accordance with the guidelines by Institution of Laboratory Resources, National Research Council (Department of Health and Human Service, National Institutes of Health, Bethesda, MD).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wang, J., Chen, Z., Walston, J.D. et al. Interferon-γ Potentiates α-Synuclein-induced Neurotoxicity Linked to Toll-like Receptors 2 and 3 and Tumor Necrosis Factor-α in Murine Astrocytes. Mol Neurobiol 56, 7664–7679 (2019). https://doi.org/10.1007/s12035-019-1567-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-1567-5