
Abstract
Dopamine (DA) neurons in the ventral tegmental area (VTA) are well-known components of the brain involved in reward-related behaviors and participate in the generation of new memories. Much attention has been focused to understand how DA neurons integrate a diversity of afferent signals with local excitatory and inhibitory influences regulated by somatodendritic release of dopamine. However, the mechanisms that actively forget rewarding information are still terra incognita. Using rodents in the conditioned place preference (CPP) behavioral task, we show that during acquisition D1-type DA receptors (D1R) in the VTA are crucial components of a neural circuit involving the hippocampus that induces active forgetting of cocaine-associated long-term memory, while VTA and nucleus accumbens (NAc) D1R are required for its formation. Inhibition of VTA D1R results in increased activation of VTA ERK1/2 and in prolonging memory storage of cocaine-place association in an ERK-dependent manner. Moreover, intra-VTA infusion of a specific D1 agonist induces forgetting of cocaine-associated consolidated memory. In contrast, D1R in the NAc shell, medial prefrontal cortex, or amygdala appear not to participate in the maintenance of cocaine-associated memory. Our present results suggest that at the moment of learning D1R-mediated neurotransmission in the VTA actively participates in at least two processes affecting the fate of appetitive memory: its consolidation involving NAc shell DA neurotransmission and its forgetting via DA activation of the hippocampus.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
VTA DA-releasing neurons are thought to play an important role in motivation, incentive and stimulus salience, reinforcing rewarding and aversive behaviors, and memory processing [1,2,3,4]. These neurons release DA not only in their terminal fields but also in the VTA region. The origin of DA released in the VTA is from collaterals arising on substantia nigra DA neurons as well as from somata and dendrites of VTA DA neurons. The so-called somatodendritic release of DA modifies the firing of DA neurons, and therefore, the release of DA in downstream terminal fields affecting behavior [2, 5, 6].
Cocaine increases extracellular DA at the level of the dendrites in the VTA [7]; DA acts on D1R that comprises D1 and D5 subtypes [8] located on GABA and glutamate axon terminals originating in other regions of the brain and impinging on VTA DA and non-DA neurons [9, 10]. As a consequence, somatodendritic DA release via D1R modifies GABA and glutamate release in the VTA which in turn regulates DA neuronal firing [11]. Therefore, DA in the VTA could play a role in cocaine-associated memories in particular, and in positive valence memories in general. In this context, we recently demonstrated that D1R in the dorsal CA1 region of the hippocampus, a target of dopaminergic fibers originating in the VTA, is critical for the persistent storage but not the formation of a cocaine-associated memory [12]. To test the hypothesis that somatodendritic DA in the VTA is important for the maintenance of rewarding-associated memory, we blocked D1R of the VTA immediately after place conditioning with cocaine or after a food preference learning task. Our present findings support the hypothesis that D1R-mediated neurotransmission in the VTA promotes the forgetting of positive valence memories. In cocaine-associated memory, this effect is via a late postconditioning D1R activation in the VTA and in the dorsal hippocampus. In addition, VTA and nucleus accumbens (NAc) D1R modulate memory formation but not memory maintenance of single-trial cocaine-place conditioning. These results indicate that VTA-NAc DA pathway regulates memory consolidation, whereas VTA-dorsal hippocampus DA inputs control forgetting of cocaine-place conditioning.
Material and Methods
Animals
A total of 556 Sprague-Dawley rats were used in these studies (Faculty of Veterinary Sciences, Argentina). Groups of five rats (weighting around 200 g upon arrival at the laboratory) were housed in an animal vivarium maintained on a reversed 12-h light-dark cycle (lights off at 0700 h) at a constant temperature of 21 °C. Experimental procedures followed the guidelines and were approved by the Animal Care and Use Committees of the University of Buenos Aires (CICUAL). Each experiment involves an independent group of animals and was performed during the dark phase of the diurnal cycle.
Drugs
Cocaine hydrochloride (20 mg/ml/kg, Laboratorios Verardo y Cia., Argentina); the dopamine D1R antagonist SCH 23390 hydrochloride (1.5 μg/μl, Sigma-Aldrich, Germany); and GABAA receptor agonist muscimol (0.1 μg/μl, Sigma-Aldrich, Germany) were all dissolved in sterile 0.9% physiological saline. The dopamine D1R agonist SKF 38393 hydrochloride (12.5 μg/μl, Sigma-Aldrich, Germany); SKF 83822 (1 μg/μl, Tocris Bioscience, UK); and selective inhibitor of MEK U0126 (0.8 μg/μl, Sigma-Aldrich, Germany) were dissolved in sterile 0.9% physiological saline supplemented with DMSO (10% final concentration, Ernesto van Rossum y Cia., Buenos Aires, Argentina). The doses used were determined based on previous studies showing the effect of each compound on learning or behavioral performance [12, 13].
Surgical and Intracerebral Infusion Procedures
Each rat was anesthetized with a mix of ketamine and xylazine administered intraperitoneally (i.p.) and placed in a stereotaxic frame. The skull was exposed and leveled (flat skull, lambda, and bregma at the same elevation degree). 22-G guide cannulae (measuring 1 cm length) for intracerebral infusions were bilaterally implanted aimed at different structures. The stereotaxic coordinates used were as follows for the different structures according to [14]: for VTA: AP, + 5.3 mm/L ± 1 mm/ DV, − 8.2 mm; for mPFC: AP, + 3.20 mm/ L, ± 0.75 mm/ DV, − 3.20 mm; for NAc: AP, + 1.5 mm/ L, ± 1.2 mm/ DV, − 7.4 mm; for hippocampus (HP): AP, + 3.90 mm/ L, ± 3.00 mm/ DV, − 3.00 mm; and for basolateral amygdala (BLA): AP − 2.8 mm/ L, ± 4.6 mm/ DV, − 6.6 mm from bregma (Fig. S1). Cannulae were fixed to the skull with acrylic cement. After surgery, animals were injected with a single dose of meloxicam (0.2 mg/kg) as analgesic and animals were left on their homecage to recover for 1 week. For intracerebral infusions, 30-G needles of 1.1 cm connected to Hamilton syringes were used. The infusions were always bilateral with 1 μl per side as volume infusions for hippocampus and 0.5 μl for the other structures (injection rate, 1 μl/30 s). The needle was left in place for an additional minute after infusion to allow diffusion and to prevent reflux. At the end of each experiment, cannulae placement was verified by infusions of 1 μl of 4% methylene blue in saline for hippocampus and 0.5 μl for the other structures. Animals were killed after 15 min by decapitation, and histological localization of the infusion site was established. The extension of the dye infused was taken as indicative of the presumable diffusion of the drugs previously given to each animal. Infusions spread with a radius of ranging from 1 to 1.5 mm3 depending on the volume infused. Animals with both cannulae in the correct place were included in the analysis.
Behavioral Paradigm
Place conditioning experiments were carried out using a three-compartment apparatus [12]; the central compartment was a short-connecting passageway between two other compartments. One of them had black walls, white square patterns and grid floor, whereas the other one had white walls, black lines pattern, and perforated floor. All the experiments were independent and carried out with different groups of animals, but they shared part of the protocol, consisted on three phases (Fig. 1a): a pretest phase in which the animals were allowed to explore the entire apparatus freely for 15 min and the preference for each compartment was determined measuring time spent in each compartment, a conditioning phase 24 h after the pretest in which they were restricted to one of the compartments (black compartment) immediately after saline i.p. injections and, on the next day, confined to the other compartment (white compartment) immediately after saline/cocaine i.p injections. Immediately or 12 h after removing them from this compartment, animals were infused with SCH 23390, SKF 38393, SKF 83822, or muscimol into the VTA, mPFC, hippocampus, NAc, or BLA according to the experiment. Results were analyzed using the score corresponding to time spent in the compartment that was followed by a drug infusion in the brain minus time spent in that compartment on the pretest. For the food-CPP experiment, the first conditioning session was made with nothing on the compartment (no food) while the second session was made with 50 fruitloop cereals on the compartment. For a multiple trial protocol, animals were conditioned three or four times on subsequent days, for cocaine or food protocol, respectively. As the animals receive saline or no food in the black compartment 1 day, cocaine or food in the white compartment on the following day the multi-trial protocol lasts 6 or 8 days in total depending on the conditioned stimulus used. Exploration time was measured with timers by a blind subject seated near the apparatus at a distance where rats cannot see it directly. The intracerebral injections were performed on a separate table, away from the CPP apparatus, while there were no animals being conditioning at that moment. Conditioning drug and modulating drug are the two statistical factors with a 2 × 2 design. Finally, the test phase was made at 24 h or 7 days, in which the animals were allowed again to explore freely the entire apparatus for 15 min in a drug-free state, where time spent in each compartment was also measured. During the pretest phase, animals that spend less than 90 s in any of the compartments were excluded from the study. Four animals out of 520 were excluded for this reason.

D1R signaling in the VTA induces active forgetting of cocaine-associated memory. a Schema of the protocol used. b Animals conditioned with a weak cocaine training protocol show memory at 24 h but not at 7 days (n = 18 and 10, respectively for each group). c Infusion of D1R antagonist SCH 23390 in the VTA immediately after conditioning promotes a cocaine-associated weak memory at 7 days (n = 8 per group). d Animals conditioned with a weak food training show memory at 24 h but not at 7 days (n = 7 per group). e Infusion of D1R antagonist SCH 23390 in the VTA immediately after conditioning promotes a weak food memory at 7 days (n = 10 per group). f Infusion of D1R antagonist SCH 23390 in the VTA immediately after conditioning, impairs memory expression at 24 h (n = 9 per group). g Stimulation of VTA Gs-coupled D1R by the specific agonist SKF 83822 immediately after conditioning induces attenuation of cocaine-associated memory durability (n = 9 per group). Asterisks indicate significant statistical differences between groups (Tukey, *p < 0.05; **p < 0.01; ***p < 0.001). COC, cocaine; SAL, saline
Immunoblots Assays
Animals were divided into three groups: naïve, cocaine conditioned with vehicle infusion or cocaine conditioned with SCH infusion. For the experiments in which we analyzed protein expression at time 0, animals were killed 15 min after drug infusion. For the analysis of the hippocampus at 12 h, animals were killed 12 h after drug infusion. For both cases, tissue was processed and stored immediately after the sacrifice. Tissue from VTA and hippocampus was homogenized in ice-chilled buffer; 7.5 μg of proteins for hippocampus or 12 μg for VTA (determined by BCA assay, ThermoFisher Scientific, USA) were subjected to SDS-PAGE (polyacrylamide 10%) [15]. Proteins were transferred to polyvinylidene (PVDF) membranes overnight at 4 °C. Membranes were incubated first with anti-p-ERK1/2 antibody (1:9000, Cell Signaling, MA, USA), then stripped and incubated with anti-ERK1/2 antibody (1:9000, Cell Signaling, MA, USA). Antigen-antibody complexes were visualized by a fluorescent method using ECF substrate (GE Healthcare Life Sciences, UK) and fluorescence-measuring equipment (STORM scanner, GE Healthcare Life Sciences, UK). Densitometry analysis was performed using ImageQuant Analyzer (GE Healthcare Life Sciences, UK).
Statistical Analysis
Data is presented as a score in seconds (s) calculated as the time spent in the cocaine-associated compartment minus time spent in the to-be cocaine-associated compartment during the pretest. Results were presented as mean ± SEM. Data was analyzed using two-way analysis of variance (ANOVA) or Student’s t test according to the case. In cases of significant interaction, post hoc analysis was made with Tukey test when one or two factors p value was significant. In cases with nonsignificant interaction, a main effect ANOVA was made. A result was considered significant when p < 0.05. All data was analyzed using Graphpad and InfoStat software.
Results
Single pairing of cocaine-place conditioning induces a weak memory lasting 1 day, but neither three [12] nor 7 days (Fig. 1b, Student’s t test, COC vs. SAL, p = 0.0008 for 24 h and p = 0.2868 for 7 days). Given that cocaine exposure resulted in changes in extracellular DA levels in the VTA and the activation of VTA D1R modulates the firing rate of DA-releasing neurons [11], we infused into the VTA the D1R antagonist SCH 23390 (0.5 μl per side) immediately after single pairing of cocaine-place association. As shown in Fig. 1c, the inhibition of VTA D1R prolongs memory duration to 7 days (Tukey post hoc analysis after two-way ANOVA(1,18), F(conditioned drug) = 8.216, p = 0.0103, F(modulating drug) = 6.413, p = 0.0209, F(interaction) = 5.501, p = 0.0307, n = 8). To substantiate the hypothesis that dopamine transmission in the VTA controls the duration of positive valence memories and that the prolonging action of SCH 23390 on memory storage is not only a cocaine-dependent effect, we developed a food preference task using fruitloops as rewarding agent to get a short-lasting appetitive LTM. Therefore, using a 4-pairings protocol memory is evident at 24 h but not at 7 days after the last conditioning session (Fig. 1d, Student’s t test no food vs. food for 24 h, p = 0.0382; no food vs. food for 7 days, p = 0.9332, n = 7). The intra-VTA infusion of D1R antagonist immediately after each pairing induces an increase in the persistence of food-place association (Fig. 1e, Tukey post hoc analysis after two-way ANOVA(1,29), F(conditioned drug) = 11.17, p = 0.0023, F(modulating drug) = 1.313, p = 0.2612, F(interaction) = 6.373, p = 0.0173, n = 10) which is consistent with the findings obtained using cocaine as rewarding agent. Considering that cocaine-place association protocol is a much simple and reliable learning paradigm than food-place association task, we decided to use it in the next experiments. In addition, we were able to develop our food-CPP protocol that generates a weak short-lasting LTM at 24 h, but we were not able to develop a protocol to get a strong long-lasting memory. Two main possibilities may explain our current findings: (1) D1R in the VTA is necessary for active forgetting or (2) D1R activation provokes an attenuation of memory consolidation. Thus, we assessed whether the effect of SCH 23390 observed at 7 days is due to the blockade of an intrinsic forgetting mechanism in the VTA or to an increased consolidation or expression of cocaine-associated LTM. We trained animals using a single-pairing protocol and infused the D1R antagonist in the VTA and tested at 24 h after conditioning. An impairment of memory expression was found (Fig. 1f, Tukey post hoc analysis after two-way ANOVA(1,31), F(conditioning drug) = 2.252, p = 0.1436, F(modulating drug) = 9.777, p = 0.0038, F(interaction) = 6.060, p = 0.0196, n = 9), indicating that the blockade of D1R in the VTA around training results in two main effects: (1) downregulation of recent cocaine-place LTM expression; (2) upregulation of its durability, transforming a short-lasting LTM into a long-lasting LTM, ruling out the possibility that VTA D1R neurotransmission is constraining LTM consolidation.
To give further support to the idea that an active forgetting mechanism underlies the enhanced durability of cocaine-associated LTM induced by the blockade of VTA D1R, we infused the Gs-coupled D1R agonist SKF 83822, which specifically activates D1 but not D5 subtype [16], into the VTA immediately after each pairing of 3-pairings cocaine-place conditioning. This multiple pairing protocol gives a persistent memory lasting at least 21 days [12]. We speculate that stimulating this subtype of D1R would result in marked reduction on the duration of memory storage. Figure 1g shows that stimulation of VTA Gs-coupled D1R immediately after conditioning abolished the persistent LTM because there is no cocaine-associated memory observed at a 7-day test (Tukey post hoc analysis after two-way ANOVA(1,29), F(conditioning drug) = 2.357, p = 0.1356, F(modulating drug) = 2.124, p = 0.1557, F(interaction) = 5.234, p = 0.0296, n = 9), endorsing the hypothesis that dopamine neurotransmission in the VTA is crucial for active forgetting of consolidated cocaine-place association.
The increase in the durability of cocaine-associated memory induced by the intra-VTA infusion of SCH 23390 is associated with an increase in the phosphorylation state of VTA ERK1/2 (Fig. 2a, b, Tukey post hoc analysis after one-way ANOVA(2,24), F = 3.716 p = 0.0393, naïve vs. SCH, VEH vs. SCH; for p42: Tukey post hoc analysis after one-way ANOVA(2,22), F = 4.815 p = 0.0184, VEH vs. SCH, n = 4). In pilots experiments, the blockade of VTA ERK activity by the local infusion of the MEK inhibitor U0126 (0.8 μg/μl, 0.5 μl per side) immediately after cocaine conditioning did not alter CPP performance at 7 days (mean ± SEM, VEH: 24 ± 8.1 s, U0126: 44.1 ± 18.5 s, Student’s t test, VEH vs. U0126, p = 0.303, n = 6). The blockade of ERK1/2 activation by U0126 co-infused immediately after training prevents the effect of SCH 23390 on the persistence of cocaine-place association (Fig. 2c, Tukey post hoc analysis after one-way ANOVA(2,9), F(treatment) = 4.542, p = 0.0433, SCH vs. SCH + U0126, n = 9). Our findings suggest that ERK1/2 signaling cascade in the VTA is required for the increased durability of cocaine-associated memory storage induced by D1R blockade, which is consistent with previous findings showing the participation of ERK1/2 in memory maintenance of learning tasks in rodents and Drosophila [17, 18].

D1R signaling in the VTA induces active forgetting of cocaine-associated memory in an ERK1/2-dependent manner. a–b p44/42 MAP kinases are augmented in the VTA 15 min after SCH 23390 infusion in cocaine conditioned animals (n = 4 per group). c Memory promotion induced by D1R antagonist SCH 23390 infusion is prevented when ERK1/2 inhibitor U0126 is infused previously in the VTA (n = 9 per group). Asterisk indicates significant statistical differences between groups (Tukey, *p < 0.05)
Next, we examined whether D1R in regions receiving VTA DA inputs contribute to maintaining memory storage of cocaine-place association. We bilaterally infused SCH 23390 immediately after a single pairing of conditioning in the mPFC, BLA, or dorsal hippocampus and tested CPP 7 days after. In contrast to what it is observed in the VTA (Fig. 1c), no changes were found in memory retention scores 7 days after training when the antagonist of D1R was microinjected in any of the structures studied (Fig. 3a–c, two-way ANOVA, F(conditioned drug) = 4.233, p = 0.0602, F(modulating drug) = 1.440, p = 0.2414, F(interaction) = 0.1633, p = 0.6895, n = 8, for mPFC; F(conditioned drug) = 0.1419, p = 0.7121, F(modulating drug) = 0.1811, p = 0.6769, F(interaction) = 0.8936, p = 0.3605, n = 5, for hippocampus; F(conditioned drug) = 0.2524, p = 0.6227, F(modulating drug) = 0.00193, p = 0.9655, F(interaction) = 0.01739, p = 0.8968, n = 5, for BLA).

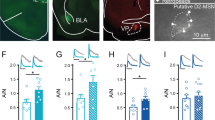
D1R inhibition has no effect on a weak cocaine CPP memory when infused in other limbic or cortical structures. D1R antagonist SCH 23390 has no effect at 7 days when it is infused immediately after a cocaine conditioning in the mPFC (b), hippocampus (c), or BLA d (n = 8 for mPFC, n = 5 for BLA and hippocampus). COC, cocaine; SAL, saline
A single dose of cocaine, as the one used in this study, results in synaptic plasticity changes in VTA DA neurons lasting many hours to few days [19] and a single pairing of cocaine-place association is accompanied by two surges of DA in the hippocampus, an early increase lasting 4–5 h and a late increase starting 13 h after conditioning that is associated with the control of memory duration ([12] and Fig. 4a, Tukey post hoc analysis after two-way ANOVA(1,22), F(conditioned drug) = 6.180, p = 0.0210, F(modulating drug) = 5.822, p = 0.0246, F(interaction) = 12.42, p = 0.0019, n = 7). Therefore, to further determine the role of VTA D1R on cocaine-associated memory durability, we infused SCH 23390 12 h after a single-pairing protocol and found that the blockade of D1R activity late after training also prolonged memory duration (Fig. 4b, Tukey post hoc analysis after two-way ANOVA(1,27), F(conditioned drug) = 9.109, p = 0.0056, F(modulating drug) = 1.199, p = 0.2837, F(interaction) = 4.639, p = 0.0407, n = 8). This finding is consistent with the idea that VTA controls memory duration at two time points, immediately during acquisition and a dozen of hours later, to modulate what is being stored [3]. Consequently, we studied the VTA-dorsal hippocampus relationship measuring changes in ERK1/2 activation, which has been involved as a marker of persistent memories within the hippocampus [17, 18]. We made two experiments in which we trained animals with cocaine, we infused SCH 23390 or VEH immediately after the conditioning session or 12 h after in the VTA, and we extracted the hippocampus to analyze ERK1/2 phosphorylation. No significant differences were found in the activation of ERK1/2 when the infusion of SCH 23390 was made immediately after conditioning (Fig. 4c, d, one-way ANOVA(2,13), F(treatment) = 1.011, p = 0.3907, n = 4 for p44; F(treatment) = 0.9831, p = 0.4024, n = 4 for p42), but blockade of D1R in the VTA 12 h thereafter increases the phosphorylation state of hippocampal ERK1/2 suggesting a functional link between these two structures at this late time point (Fig. 4e, f, for p44:Duncan post hoc after two-way ANOVA(2,23), F = 3.52, p(treatment) = 0.0464, n = 4; For p42: Duncan post hoc after two-way ANOVA(2,24), F = 8.27, p(treatment) = 0.0019, n = 4).

D1R signaling regulates active forgetting 12 h after conditioning of weak cocaine CPP memory. a D1R antagonist SCH 23390 infusion in the HP 12 h after single-pairing cocaine conditioning promotes a long-lasting cocaine-associated memory tested at 7 days (n = 8 per group). b Similarly, D1R antagonist SCH 23390 infusion in the VTA 12 h after cocaine conditioning promotes a long-term persistent memory at 7 days (n = 8 per group). c–d p44/p42 MAP kinases showed no changes in the HP 12 h after D1R antagonist SCH 23390 or VEH infusion in the VTA immediately after cocaine conditioning (n = 4 per group). e–f p44/42 MAP kinases are increased in the HP immediately after D1R antagonist SCH 23390 infusion in the VTA 12 h after cocaine conditioning (n = 4 per group). Asterisks indicate significant statistical differences between groups (Tukey, *p < 0.05; **p < 0.01). COC, cocaine; SAL, saline
Inactivation of VTA neural activity by infusing the GABAA receptor agonist muscimol (0.5 μl per side) immediately after single pairing conditioning blocked the formation of cocaine-place LTM (Fig. 5a, Tukey post hoc analysis after two-way ANOVA(1,31), F(conditioning drug) = 2.560, p = 0.1197, F(modulating drug) = 6.043, p = 0.0198, F(interaction) = 5.011, p = 0.0325, n = 9), extending the importance of VTA to one-trial cocaine-associated memory. In contrast to what we found with the infusion of SCH 23390, muscimol infused in the VTA was not able to prolong memory at 7 days (Fig. 5b, two-way ANOVA(1,13), F(conditioning drug) = 0.0020, p = 0.9658, F(modulating drug) = 0.8956, p = 0.3612, n = 5). In addition and consistent with previous findings on the role of NAc shell D1R on memory formation of classical conditioned place preference [20, 21], we observed that the infusion of SCH 23390 into NAc shell immediately after single-pairing conditioning totally blocked cocaine-associated memory tested at 24 h thereafter (Fig. 5c; Tukey post hoc analysis after two-way ANOVA(3,34), F(conditioning drug) = 7.83, p = 0.0084, F(modulating drug) = 7.91, p = 0.0081, F(interaction) = 13.75, p = 0.0007,. n = 10), having no changes when the animals were tested 7 days after (Fig. 5d, two-way ANOVA(1,29), F(conditioning drug) = 0.9965, p = 0.3264, F(modulating drug) = 0.00827, p = 0.9281, n = 9). The infusion of a D1R agonist (SKF 38393, 0.5 μl per side) into NAc shell resulted in no effects on memory duration (Fig. 5e, two-way ANOVA(1,21), F(conditioning drug) = 0.5721, p = 0.4578, F(modulating drug) = 0.7960, p = 0.3824, n = 8). Altogether, these findings suggest that VTA-NAc pathway is important for appetitive memories formation and that NAc shell D1R plays no important role in controlling the duration of cocaine-associated memory.

D1R modulation in the NAc and neural activity in the VTA are necessary for cocaine-memories formation. a–b Reversible inactivation of the VTA by infusing muscimol immediately after cocaine conditioning impaired memory formation as shown at 24 h (n = 9 per group) and 7 days (n = 5 per group). c Infusion of SCH 23390 in the NAc shell immediately after conditioning impaired the memory at 24 h (n = 10 per group). d SCH 23390 has no effect at 7 days when it is infused immediately after a cocaine conditioning in the NAc shell (n = 9 per group). e D1R agonist SKF 38393 has no effect at 7 days when it is infused immediately after cocaine conditioning in the NAc shell (n = 8 per group). Asterisks indicate significant statistical differences between groups (Tukey, *p < 0.05; ***p < 0.001). COC, cocaine; SAL, saline
Discussion
The main finding of the present study is that DA neurotransmission in the VTA is critical for controlling the durability of rewarding memories. Given that blocking VTA D1R prolongs two consolidated rewarding memories and that stimulation of D1R by a Gs-coupled D1R agonist abolishes the maintenance of cocaine-associated memory storage, our findings indicate that an active forgetting mechanism involving D1R activation is present in the VTA.
Two hypotheses dominated the field of forgetting during the last 50 years. One postulates that forgetting is mainly caused by interference of different steps in memory processing [22]. The other poses that forgetting is due to the passive or active decay of the memory trace [23, 24]. In the last few years, some neurobiological findings involving memory durability have endorsed the idea that active forgetting plays an important role in eroding memory storage [24,25,26]. Active or intrinsic forgetting is referred to the mechanism or the sequence of events required for and sufficient to erase the substrate of a memory [26]. Both requirements are fulfilled by the present experiments.
Intrinsic forgetting has been demonstrated in Drosophila mainly for nonconsolidated aversive memories [27, 28] and also for memories lasting 6 h or more [28, 29]. Activation of DA neurons in the fly triggers the activation of the DA receptor DAMB (a receptor coupled to Gs and Gq signaling pathways; [30]) which is different to the dDA1—an homolog of the mammalian D1 subtype—that modulates actin dynamics in dendritic spines via several proteins (Rac1, Pak, cofilin, Cdc42) leading to the erasure of the memory trace [28, 31, 32]. It appears that while the dDA1 is involved in memory acquisition and consolidation [30, 33], DA receptor DAMB is required for intrinsic forgetting in the fly [28]. The delayed effect of intra-VTA infusion of SCH 23390 in prolonging cocaine-associated memory storage parallels that found when the antagonist of D1R was given in the dorsal hippocampus (Fig. 4a and [12]). Therefore, during conditioning VTA DA neurotransmission via activation of the Gs-coupled D1 receptor subtype, together with a late postconditioning (12 h) D1R activation in the VTA and dorsal hippocampus, play a crucial role in the mechanism of intrinsic forgetting of cocaine-associated memory. It is important to consider that the VTA is not the only dopaminergic source that innervates the hippocampus [34], and hippocampal activity might be also regulated by dopaminergic innervations from the Locus Coeruleus. This brain region has been involved in the enhancement of spatial memory, and recent studies have identified the noradrenergic fibers originating in this structure as an additional source of neurotransmitter acting on dopaminergic receptors in the hippocampus [35].
Considering that SCH 23390 might also act as an agonist at serotonin 5HT2C receptors [36], it may be possible that the prolonging effect of SCH 23390 on memory storage is partially due to a serotoninergic effect. However, it has been shown that the activation of these receptors by its specific agonist lorcaserin is efficacious in decreasing multiple aspects of cocaine motivation and reward [37]. In particular, this agonist impairs food intake and cocaine-self administration when it binds to the 5HT2C receptor [38]. These results indicate that our present findings might not be explained by the activation of 5HT2C receptors in the VTA.
Somatodendritic release of DA in the VTA may modulate D1 subtype present in glutamate and GABA afferents to the VTA [9, 10, 37]. Thus, it can affect local glutamate and GABA concentrations, which control DA neurons [11, 38]. Given the known role of these DA cells in cocaine reward [2, 6, 39], any effect on their activity will affect cocaine-place association. In this context, it has been demonstrated that a single passive injection of cocaine, like that used in the present study, increased calcium-permeable AMPARs and reduced NMDAR EPSCs in VTA DA neurons projecting to NAc shell for several days [19, 40]. The blockade of VTA D1R by SCH 23390 inhibited cocaine-induced synaptic plasticity changes [19, 41] and consistent with previous findings [39, 42] impaired cocaine-induced memory measured 24 h after conditioning (Fig. 1f). Several groups have demonstrated similar effects: SCH 23390 infusions in the VTA impaired dopaminergic signaling on the NAc and blocked cocaine sensitization [43]. In accordance with these results, same infusions blocked acute motor-response to cocaine [44]. NAc shell infusion of D1R antagonist during conditioning also impaired memory consolidation (Fig. 5c), without altering memory persistence (Fig. 5d, e), and blocked accumbal ERK1/2 activation induced by cocaine-place association [45]. Therefore, VTA and NAc shell DA neurotransmission via activation of D1R are required for the formation of cocaine-associated LTM tested 24 h after learning.
Several studies suggest that different functions of the VTA are mediated by diverse subpopulations of VTA DA neurons that are associated with distinct neuronal networks [46]. As it has been reported, VTA DA neurons are heterogeneous in their afferent but also efferent connectivity [47]. The present study indicates that while VTA-NAc pathway regulates cocaine-memories formation, VTA-hippocampus regulates its forgetting. Taken together, our results suggest different mechanisms regulating appetitive memory processing, and those differences might be explained by VTA neural diversity.
We also found that VTA ERK1/2 signaling cascade is activated when VTA D1R is blocked during conditioning leading to persistent cocaine-associated memory. Cocaine-place conditioning did not alter ERK1/2 activation in VTA which is consistent with previous reports [44, 48]. More importantly, the blockade of ERK1/2 activation by the infusion of the specific MEK inhibitor U0126 prevents the establishment of a persistent memory induced by VTA D1R inhibition. A delayed ERK1/2 activation in the hippocampus accompanied the promotion of a persistent cocaine-induced memory when the antagonist of D1R was infused in the VTA many hours (12 h) after conditioning (Fig. 4e–f). These findings support the idea that ERK activation in selected brain regions is important for maintaining memory storage [17, 18].
DA neurotransmission in VTA and dorsal hippocampus is one of several molecular events associated with intrinsic forgetting. For instance, it has also been demonstrated that forgetting is regulated in the rat hippocampus by NMDA receptors and calcineurin [49]. In addition, similar to the findings obtained in Drosophila, Liu and colleagues [50] demonstrated that Rac1 is part of an intrinsic forgetting mechanism in the mouse hippocampus. Inhibition of Rac1 activity in hippocampal neurons prolonged object recognition memory for 5 days, whereas stimulation of Rac1 induced forgetting of object recognition LTM. However, this mechanism of active forgetting seems to be absent in two others hippocampus-dependent learning tasks: contextual fear conditioning and trace fear conditioning [50]. These findings suggest that Rac1-mediated forgetting is not a general mechanism of time-dependent memory loss. Also, we cannot rule out the possibility of a response regulated by dendritic remodeling. In developing neurons, the activation state of Rac1 influences lamellipodial extensions of growth cones, while in adult neurons it influences the size and shape of synaptic spines by regulating actin polymerization [51]. Another candidate that could regulate memory decay is GluA2 receptor; recently, it has been suggest that receptor endocytosis or AMPA receptor trafficking may play an important role on forgetting [52].
In conclusion, we demonstrated the existence of intrinsic time-dependent forgetting mechanisms involving VTA D1R and ERK1/2 signaling that provides proper control for the physiological expression of rewarding experiences. At the time that the memory of cocaine-place association is being formed [53], an active mechanism is triggered that promotes the late postconditioning eroding of the memory trace. An imbalance in these mechanisms or a failure in VTA active forgetting mechanisms will generate aberrant, persistent drug-associated memories which might be crucial in the transitional process between the recreational consumption of the drug of abuse and its compulsive use. In this sense, we propose that appetitive memories need a biological control in order to prevent addictive behaviors that are self-defeating and cause a negative impact on humans and other animals.
References
Schultz RT, Klin A, Lombroso (2002) Genetics of childhood disorders: XLIII. Autism, Part 2: Neural foundations. J Am Acad Child Adolesc Psychiatry 41:1259–1262. https://doi.org/10.1097/00004583-200210000-00018
Wise RA (2004) Rewards wanted: molecular mechanisms of motivation. Discov Med 4:180–186
Rossato JI, Bevilaqua LRM, Izquierdo I, Medina JH, Cammarota M (2009) Dopamine controls persistence of long-term memory storage. Science (80- ) 325:1017–1020. https://doi.org/10.1126/science.1172545
Salamone JD, Correa M (2012) The mysterious motivational functions of mesolimbic dopamine. Neuron 76:470–485. https://doi.org/10.1016/j.neuron.2012.10.021
Adell A, Artigas F (2004) The somatodendritic release of dopamine in the ventral tegmental area and its regulation by afferent transmitter systems. Neurosci Biobehav Rev 28:415–431. https://doi.org/10.1016/j.neubiorev.2004.05.001
Rice ME, Patel JC (2015) Somatodendritic dopamine release: recent mechanistic insights. Philos Trans R Soc B Biol Sci 370:1–14. https://doi.org/10.1098/rstb.2014.0185
Kalivas PW, Duffy P (1989) Similar effects of daily cocaine and stress on mesocorticolimbic dopamine neurotransmission in the rat. Biol Psychiatry 25:913–928. https://doi.org/10.1016/0006-3223(89)90271-0
Sunahara RK, Guan HC, O’Dowd BF, Seeman P, Laurier LG, Ng G, George SR et al. (1991) Cloning of the gene for a human dopamine D5receptor with higher affinity for dopamine than D1. Nature 350:614–619. https://doi.org/10.1038/350614a0
Lu XY, Churchill L, Kalivas PW (1997) Expression of D1 receptor mRNA in projections from the forebrain to the ventral tegmental area. Synapse 25:205–214. https://doi.org/10.1002/(SICI)1098-2396(199702)25:2<205::AID-SYN11>3.0.CO;2-X
Harrison MB, Wiley RG, Wooten GF (1990) Selective localization of striatal D1receptors to striatonigral neurons. Brain Res 528:317–322. https://doi.org/10.1016/0006-8993(90)91674-6
Kalivas PW, Duffy P (1995) D1 receptors modulate tegmental area glutamate transmission in the ventral tegmental area. J Neurosci 15:5379–5388
Kramar CP, Chefer VI, Wise RA, Medina JH, Barbano MF (2014) Dopamine in the dorsal hippocampus impairs the late consolidation of cocaine-associated memory. Neuropsychopharmacology 39:1645–1653. https://doi.org/10.1038/npp.2014.11
Majchrzak M, Di Scala G (2000) Gaba and muscimol as reversible inactivation tools in learning and memory. [Review] [75 refs]. Neural Plast 7:19–29
Paxinos G, Watson C (2005) The rat brain in stereotaxic coordinates, 6th edn. Academic Press, New York
Katche C, Medina JH (2017) Requirement of an early activation of BDNF/c-Fos cascade in the retrosplenial cortex for the persistence of a long-lasting aversive memory. Cereb Cortex 27:1060–1067. https://doi.org/10.1093/cercor/bhv284
Undieh AS (2010) Pharmacology of signaling induced by dopamine D1-like receptor activation. Pharmacol Ther 128:37–60. https://doi.org/10.1016/j.pharmthera.2010.05.003
Bekinschtein P, Cammarota M, Katche C, Slipczuk L, Rossato JI, Goldin A, Izquierdo I, Medina JH (2008) BDNF is essential to promote persistence of long-term memory storage. Proc Natl Acad Sci 105:2711–2716. https://doi.org/10.1073/pnas.0711863105
Eckel-mahan KL, Phan T, Han S, Wang H, Chan GC, Scheiner ZS, Storm DR (2009) Circadian oscillation of hippocampal MAPK activity and cAmp: implications for memory persistence. 11:1074–1082
Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411:583–587. https://doi.org/10.1038/35079077
Di Chiara G (2002) Nucleus accumbens shell and core dopamine: Differential role in behavior and addiction. Behav Brain Res 137:75–114. https://doi.org/10.1016/S0166-4328(02)00286-3
Wise RA (2004) Dopamine, learning and motivation. Nat Rev Neurosci 5:483–494. https://doi.org/10.1038/nrn1406
Wixted JT (2004) The psychology and neuroscience of forgetting. Annu Rev Psychol 55:235–269. https://doi.org/10.1146/annurev.psych.55.090902.141555
Hardt O, Nader K, Nadel L (2013) Decay happens: the role of active forgetting in memory. Trends Cogn Sci 17:111–120. https://doi.org/10.1016/j.tics.2013.01.001
Davis RL, Zhong Y (2017) The biology of forgetting—a perspective. Neuron 95:490–503. https://doi.org/10.1016/j.neuron.2017.05.039
Frankland PW, Stefan K, Josselyn S (2013) Hippocampal neurogenesis and forgetting. Hippocampus from Cells to Syst Struct Connect Funct Contrib to Mem Flex Cogn 95–121. https://doi.org/10.1007/978-3-319-50406-3_4
Medina JH (2018) Neural, cellular and molecular mechanisms of active forgetting. Front Syst Neurosci 12:1–10. https://doi.org/10.3389/fnsys.2018.00003
Shuai Y, Lu B, Hu Y, Wang L, Sun K, Zhong Y (2010) Forgetting is regulated through Rac activity in Drosophila. Cell 140:579–589. https://doi.org/10.1016/j.cell.2009.12.044
Berry JA, Cervantes-Sandoval I, Nicholas EP, Davis RL (2012) Dopamine is required for learning and forgetting in Drosophila. Neuron 74:530–542. https://doi.org/10.1016/j.neuron.2012.04.007
Zhang X, Li Q, Wang L, Liu ZJ, Zhong Y (2016) Cdc42-dependent forgetting regulates repetition effect in prolonging memory retention. Cell Rep 16:817–825. https://doi.org/10.1016/j.celrep.2016.06.041
Himmelreich S, Masuho I, Berry JA, MacMullen C, Skamangas NK, Martemyanov KA, Davis RL (2017) Dopamine receptor DAMB signals via Gq to mediate forgetting in Drosophila. Cell Rep 21:2074–2081. https://doi.org/10.1016/j.celrep.2017.10.108
Shuai Y, Hirokawa A, Ai Y, Zhang M, Li W, Zhong Y (2015) Dissecting neural pathways for forgetting in Drosophila olfactory aversive memory. Proc Natl Acad Sci 112:E6663–E6672. https://doi.org/10.1073/pnas.1512792112
Cervantes-Sandoval I, Chakraborty M, MacMullen C, Davis RL (2016) Scribble scaffolds a signalosome for active forgetting. Neuron 90:1230–1242. https://doi.org/10.1016/j.neuron.2016.05.010
Kim Y-C, Lee H-G, Han K-A (2007) D1 dopamine receptor dDA1 is required in the mushroom body neurons for aversive and appetitive learning in Drosophila. J Neurosci 27:7640–7647. https://doi.org/10.1523/JNEUROSCI.1167-07.2007
Takeuchi T, Duszkiewicz AJ, Sonneborn A, Spooner PA, Yamasaki M, Watanabe M, Smith CC, Fernández G et al. (2016) Locus coeruleus and dopaminergic consolidation of everyday memory. 537:357–362. https://doi.org/10.1038/nature19325.Locus
Mcnamara CG, Dupret D (2017) Two sources of dopamine for the hippocampus. Trends Neurosci 40:383–384. https://doi.org/10.1016/j.tins.2017.05.005
Ramos M, Goñi-Allo B, Aguirre N (2005) Administration of SCH 23390 into the medial prefrontal cortex blocks the expression of MDMA-induced behavioral sensitization in rats: An effect mediated by 5-HT2C receptor stimulation and not by D1 receptor blockade. Neuropsychopharmacology 30:2180–2191. https://doi.org/10.1038/sj.npp.1300735
Higgins GA, Sellers EM, Fletcher PJ (2013) From obesity to substance abuse: therapeutic opportunities for 5-HT2Creceptor agonists. Trends Pharmacol Sci 34:560–570. https://doi.org/10.1016/j.tips.2013.08.001
Harvey-Lewis C, Li Z, Higgins GA, Fletcher PJ (2016) The 5-HT2Creceptor agonist lorcaserin reduces cocaine self-administration, reinstatement of cocaine-seeking and cocaine induced locomotor activity. Neuropharmacology 101:237–245. https://doi.org/10.1016/j.neuropharm.2015.09.028
Herkenham M, Lynn AB, Decosta BR, Richfield EK (1991) Neuronal localizatin of cannabinoid receptors in the basal ganglia of the rat. Brain Res 547:267–274
Michaeli A, Yaka R (2010) Dopamine inhibits GABAA currents in ventral tegmental area dopamine neurons via activation of presynaptic G-protein coupled inwardly-rectifying potassium channels. Neuroscience 165:1159–1169. https://doi.org/10.1016/j.neuroscience.2009.11.045
Galaj E, Manuszak M, Arastehmanesh D, Ranaldi R (2014) Microinjections of a dopamine D1 receptor antagonist into the ventral tegmental area block the expression of cocaine conditioned place preference in rats. Behav Brain Res 272:279–285. https://doi.org/10.1016/j.bbr.2014.07.008
Bellone C, Lüscher C (2006) Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci 9:636–641. https://doi.org/10.1038/nn1682
Steketee JD (1998) Repeated injection of GBR 12909 , but not cocaine or WIN 35 , 065-2 , into the ventral tegmental area induces behavioral sensitization. Behav Brain Res 97:39–48. https://doi.org/10.1016/S0166-4328
Steketee JD, Braswell B (1997) Injection of SCH 23390, but not 7-hydroxy-DPAT, into the ventral tegmental area blocks the acute motor-stimulant response to cocaine. Behav Pharmacol 8:58–64. https://doi.org/10.1097/00008877
Valjent E, Maldonado R (2000) A behavioural model to reveal place preference to delta 9-tetrahydrocannabinol in mice. Psychopharmacology 147:436–438. https://doi.org/10.1007/s002130050013
Morales M, Root DH (2014) Glutamate neurons within the midbrain dopamine regions. Neuroscience 282:60–68. https://doi.org/10.1016/j.neuroscience.2014.05.032
Morales M, Margolis EB (2017) Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci 18:73–85. https://doi.org/10.1038/nrn.2016.165
Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC (2004) Cocaine-induced potentiation of synaptic strength in dopamine neurons: Behavioral correlates in GluRA(-/-) mice. Proc Natl Acad Sci 101:14282–14287. https://doi.org/10.1073/pnas.0401553101
Sachser RM, Santana F, Crestani AP, Lunardi P, Pedraza LK, Quillfeldt JA, Hardt O, De Oliveira Alvares L (2016) Forgetting of long-term memory requires activation of NMDA receptors, L-type voltage-dependent Ca2+ channels, and calcineurin. Sci Rep 6:1–9. https://doi.org/10.1038/srep22771
Liu Y, Du S, Lv L, Lei B, Shi W, Tang Y, Wang L, Zhong Y (2016) Hippocampal activation of Rac1 regulates the forgetting of object recognition memory. Curr Biol 26:2351–2357. https://doi.org/10.1016/j.cub.2016.06.056
Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases : new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9:690–701. https://doi.org/10.1038/nrm2476
Migues PV, Liu L, Archbold GEB, Einarsson EO, Wong J, Bonasia XK, Ko SH, Wang YT et al (2016) Blocking synaptic removal of GluA2-containing AMPA receptors prevents the natural forgetting of long-term memories. J Neurosci 36:3481–3494. https://doi.org/10.1523/JNEUROSCI.3333-15.2016
Ranaldi R, Wise RA (2001) Blockade of D1 dopamine receptors in the ventral tegmental area decreases cocaine reward: possible role for dendritically released dopamine. J Neurosci 21:5841–5846
Acknowledgments
We thank Dr. Cynthia Katche, Ana Belén de Landeta and Magdalena Pereyra for their help with the biochemistry experiments and daily work in general.
Funding
This study was supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, Argentina) to JHM; nos. 2013–0335 and 2016-0034; Universidad de Buenos Aires (UBACyT, Argentina) 2014–2017 and the Argentina National Research Council (Consejo Nacional de Investigaciones Científicas y Técnicas, CONICET).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Figure S1
Histological analysis of cannulae placement. Schematic illustration of the injections sites in the intended areas. Top: VTA, hippocampus and mPFC; Bottom: NAc and BLA. Coordinates were measured from Bregma taken from the atlas of Paxinos and Watson (2005). (PNG 717 kb)
Rights and permissions
About this article

Cite this article
Castillo Díaz, F., Hernandez, M.A., Capellá, T. et al. Dopamine Neurotransmission in the Ventral Tegmental Area Promotes Active Forgetting of Cocaine-Associated Memory. Mol Neurobiol 56, 6206–6217 (2019). https://doi.org/10.1007/s12035-019-1516-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-019-1516-3