
Abstract
Subarachnoid hemorrhage (SAH) is a devastating disease. Cerebral vasospasm is still an important cause of post-SAH poor outcomes, but its mechanisms remain unveiled. Activation of epidermal growth factor receptor (EGFR) is suggested to cause vasoconstriction in vitro, but no report has demonstrated the involvement of EGFR in vasospasm development after SAH in vivo. Cross-talk of EGFR and vascular endothelial growth factor (VEGF) receptor, which may affect post-SAH vasospasm, was also reported in cancer cells, but has not been demonstrated in post-SAH vasospasm. The aim of this study was to investigate whether EGFR as well as EGFR-VEGF receptor cross-talk engage in the development of cerebral vasospasm in a mouse SAH model. C57BL6 mice underwent endovascular perforation SAH or sham modeling. At 30 min post-modeling, mice were randomly administrated vehicle or 2 doses of selective EGFR inhibitors intracerebroventricularly. A higher dose of the inhibitor significantly prevented post-SAH neurological impairments at 72 h and vasospasm at 24 h associated with suppression of post-SAH activation of EGFR and extracellular signal-regulated kinase (ERK) 1/2 in the cerebral artery wall, especially in the smooth muscle cell layers. Anti-EGFR neutralizing antibody also showed similar effects. However, neither expression levels of VEGF nor activation levels of a major receptor of VEGF, VEGF receptor-2, were affected by SAH and two kinds of EGFR inactivation. Thus, this study first showed that EGFR-ERK1/2 pathways may be involved in post-SAH vasospasm development, and that EGFR-VEGF receptor cross-talk may not play a significant role in the development of vasospasm in mice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebral vasospasm remains an important prognostic factor after aneurysmal subarachnoid hemorrhage (SAH) [1, 2]. The mechanisms are still not well unveiled, though many researchers have revealed multiple factors, which may be involved in vasospasm [1, 2]. In a clinical setting, some growth factors such as platelet-derived growth factor [3] and vascular endothelial growth factor (VEGF) [3, 4] were reported to increase in an acute phase of aneurysmal SAH, and were suggested to cause cerebral vasospasm and proliferative angiopathy in experimental SAH [3, 5]. As far as we know, the possible involvement of epidermal growth factors in cerebral vasospasm has never been reported, but a matricellular protein tenascin-C, which is one of epidermal growth factor receptor (EGFR) ligands [6], was reported to increase in the cerebrospinal fluid of SAH patients [7]. Our previous study showed that recombinant tenascin-C administration brought cerebral artery constriction, and that an EGFR inhibitor improved the vasoconstriction in healthy rats [8]. In addition, EGFR activation was implicated in a potential spasmogen endothelin-1-induced arterial contraction [9]. Thus, EGFR is supposed to be involved in vasospasm development after SAH, but no studies have examined the possible involvement of EGFR in post-SAH vasospasm in vivo. On the other hand, our previous study showed that a major receptor of VEGF, VEGF receptor (VEGFR)-2 (VEGFR2), was activated after experimental SAH, and that specific VEGFR2 blockage suppressed post-SAH blood-brain barrier disruption in mice [10]. Several studies also reported VEGFR involvement in vasoconstriction under hypoxic conditions [11, 12]: hyper-contraction of pulmonary artery to phenylephrine was associated with increased expression of VEGF/VEGFR2 in a dog heart failure-induced pulmonary hypertension model [11], and hypoxic increases in VEGF played a non-angiogenic role such as arterial remodeling by altering contractile protein abundances through VEGFR-dependent mechanisms in fetal ovine carotid arteries [12]. These findings suggest a possibility that VEGF(R) is involved in the pathophysiologies not only in brain parenchyma but also in cerebral arteries after SAH. In addition, as EGFR-VEGFR cross-talk was demonstrated in cancer [13] and non-neoplastic diseases such as chronic hydrocephalus after SAH [14, 15], we hypothesized that such a linkage might also contribute to post-SAH vasospasm development. Thus, this study was conducted to investigate whether EGFR activation is involved in post-SAH vasospasm, and if so, to reveal if the EGFR-VEGFR cross-talk contributes to signaling pathways downstream of EGFR in a mouse SAH model.
Materials and Methods
All procedures were approved by the Animal Ethics Review Committee of Mie University, and were carried out in accordance with the institution’s Guidelines for Animal Experiments and ARRIVE guidelines. C57BL/6 male adult mice (age, 10–12 weeks; weight, 25–30 g; SLC, Hamamatsu, Japan) were used for this study. Because this study was intended to clarify the pathophysiology rather than therapeutic intent, we used only male animals. After SAH induction, animals were randomly assigned to one of the treatment or control groups by drawing lots. Data collection and analyses were performed by a researcher blinded with respect to the treatment group.
Study Protocol
In experiment 1, 107 mice underwent SAH modeling (n = 87) or sham operation (n = 20). At 30 min after modeling, mice were randomly administrated vehicle (2.0 μL of 1.101 mg/μL dimethyl sulfoxide [DMSO]) or two dosages of a specific EGFR inhibitor AG1478 (126.4 ng or 632.0 ng in 2.0 μL of 1.101 mg/μL DMSO; 10010244, Cayman, Ann Arbor, MI) intracerebroventricularly. An injection of 126.4 ng of AG1478 was equivalent to a working concentration of 10 μmol/L in the cerebrospinal fluid, which was enough to inhibit tenascin-C-induced vasoconstriction via EGFR activation in healthy rats in our previous study [8]. The 5-time higher dosage was selected as a higher dosage in this study, because it was reported that 3160 ng (=5 mmol/L, 2 μL) of AG1478 could be administered intracerebroventricularly to mice [16]. After assessing neurological scores, mice were sacrificed at 24 or 72 h after surgery, and then SAH grading, Western blotting, and immunohistochemistry were performed (Fig. 1a).

Experimental designs. a Experiment 1 was designed to examine effects of epidermal growth factor receptor (EGFR) inhibitor (AG1478) on cerebral vasospasm after SAH. b Experiment 2 was designed to examine effects of EGFR inhibitor (cetuximab) on cerebral vasospasm after SAH. DMSO, dimethyl sulfoxide; IgG, immunoglobulin G; IHC, immunohistochemistry; LR/WT, lumen radius/wall thickness ratio; PBS, phosphate-buffered saline; SAH, subarachnoid hemorrhage; WB, Western blotting
In experiment 2, 32 mice underwent SAH modeling (n = 22) or sham operation (n = 10). An intracerebroventricular administration of vehicle (2.0 μL phosphate-buffered saline [PBS]) or mouse anti-EGFR monoclonal neutralizing antibody cetuximab (2.0 μg in 2.0 μL PBS; Merck Serono, Tokyo, Japan) was randomly performed at 30 min post-modeling. Dosages of drug were determined according to our pilot study and a previous study [17], the latter of which reported that cetuximab’s concentrations producing 50% survival inhibition were 1.4–3.5 μmol/L or higher in human cell lines that do not harbor EGFR mutations [17]. In our pilot study, 1.0, 2.0, or 10.0 μg cetuximab (n = 2–3, respectively) was intracerebroventricularly administered to SAH mice to obtain working concentrations of 0.16, 0.33, or 1.65 μmol/L, respectively, in the cerebrospinal fluid. As 2.0 μg cetuximab-treated SAH mice showed the best neurological scores at 24 h post-SAH, the dosage was selected for the study (Online Resource, Table S1). At 24 h after modeling, neurological scores were assessed, and then mice were sacrificed for immunohistochemistry. An intracerebroventricular administration of non-specific mouse immunoglobulin G (IgG; 2.0 μg/2 μL PBS; ab188776, Abcam, Cambridge, MA) was also performed to exclude non-specific responses of intraventricular IgG injection (n = 6; Fig. 1b).
Endovascular Perforation SAH Model in Mice
Mice underwent endovascular perforation SAH or sham modeling as previously described [18]. Briefly, mice were anesthetized with an intraperitoneal injection of Avertin® (2,2,2-tribromoethanol; 250 μg/g body weight) solution, positioned supinely, and skin incision was made at the midline of the neck to expose the left carotid arteries. A 4-0 monofilament with a sharpened tip was inserted from the left external carotid artery (ECA) stump into the left internal carotid artery (ICA) and pushed further to perforate the bifurcation of the left anterior cerebral artery (ACA) and the left middle cerebral artery (MCA). Then the filament was withdrawn and the stump of ECA was coagulated. The wound was sutured. The sham group underwent the same procedure as described above except for perforating the artery. During operation, blood pressure and heart rate were measured via the tail (BP-98A; Softron, Tokyo, Japan). Rectal temperature was kept at 37 °C during surgery.
Intracerebroventricular Injection
An intracerebroventricular injection of vehicle or drug was performed as previously described [18]. Mouse was set on the stereotactic head holder, and using a surgical microscope (Zeiss, Oberkochen, Germany), a midline fronto-parietal skin incision was performed. A burr hole was perforated at 0.2 mm caudal and 1.0 mm lateral (left) to the bregma. The needle of Hamilton syringe (Hamilton Company, Reno, Nev) was inserted 2.2 mm below the horizontal plane of the bregma, 2 μL of vehicle or drug diluent was injected intracerebroventricularly at 0.5 μL/min, and the wound was quickly sutured 10 min later.
Neurobehavioral Test
Neurological impairments were blindly evaluated as previously described [18]. Briefly, neurological scores (3–18) were assessed by summing up six test scores (spontaneous activity; spontaneous movement of four limbs; forepaw outstretching; climbing; body proprioception; and response to whisker stimulation). Higher scores indicate better neurological status.
SAH Grading
SAH grading was performed in a blind fashion as previously described [18]. The basal cistern was divided into six segments, and each segment was allotted a grade from 0 to 3 depending on the amount of SAH. A total score ranging from 0 to 18 was determined by summing the scores. Mice with moderate SAH grades 8–12 were used for 24-h evaluations. As to 72-h evaluations, mice with SAH grades 6–9 were used because clots were decreased with time.
Lumen Radius/Wall Thickness (LR/WT) Ratio
Vasospasm was blindly measured using LR/WT ratio in the intracranial ICA as previously described [19]. Briefly, mice were deeply anesthetized and perfused with cold PBS, followed by 4% paraformaldehyde for brain fixation. The brains were removed, embedded in paraffin and coronally cut into 4 μm sections. Sections were deparaffinized, followed by rehydration through decreasing graded ethanol series, and stained with hematoxylin and eosin. Using Image J software (National Institute of Health, Bethesda, MD) [20], the perimeter of intracranial ICA just beneath the bifurcation of ACA and MCA was measured. LR was calculated using the following formula: LR = perimeter /2π. WT was measured at four equally spaced points along the artery circumference, and averaged. Smaller LR/WT ratios meant more severe vasospasm.
Primary Antibodies Used in Western Blotting (WB) and Immunohistochemistry (IHC)
The following primary antibodies were purchased for WB or IHC: anti-beta-tubulin (rabbit polyclonal, 2146, Cell Signaling Technology, Danvers, MA; WB, 1:2000), anti-total (t-) EGFR (rabbit polyclonal, sc-03, Santa Cruz Biotechnology, Santa Cruz, CA; WB, 1:2000; IHC, 1:50), anti-phosphorylated (p-) EGFR (rabbit monoclonal, 4407, Cell Signaling Technology; WB, 1:1000; IHC, 1:50), anti-p-VEGFR2 (rabbit monoclonal, 4991, Cell Signaling Technology; WB, 1:1000; IHC, 1:50), anti-t-extracellular signal-regulated kinase (ERK) 1/2 (rabbit monoclonal, 4695, Cell Signaling Technology; WB, 1:2000), anti-p-ERK1/2 (rabbit polyclonal, sc-16982R, Santa Cruz Biotechnology; WB, 1:1000), anti-p-ERK1/2 (rabbit monoclonal, 4370, Cell Signaling Technology; IHC, 1:400), and anti-VEGF-A (rabbit polyclonal, ab46154, Abcam, Cambridge, MA; WB, 1:2000; IHC, 1:200) antibodies.
WB
WB analysis was performed as previously described [18]. Protein was extracted from all major cerebral arteries, and each protein sample (5 μg) was separated on SDS-PAGE gels and transferred to polyvinylidene fluoride membrane. The membrane was incubated with 5% bovine serum albumin for 30 min, and then with each primary antibody at 4 °C overnight. Reactions with anti-rabbit secondary antibody (PI-1000, Vector, Burlingame, CA) were performed at room temperature for 1 h, visualized with the chemiluminescence method (ECL Prime; Amersham Bioscience, Arlington Heights, IL), and blot bands were photographed. Antibody-specific bands were quantified using Image J software. Beta-tubulin was used as an internal control. The ratios of expression levels of p-EGFR to t-EGFR and p-ERK1/2 to t-ERK1/2 were also calculated.
IHC
Paraffin-embedded brain was coronally cut at 1.0 mm posterior to bregma as previously described [18]. Staining procedures were performed using a commercially available kit (Vectastain Elite Rabbit ABC kit; PK-6101, Vector, Burlingame, CA) except for primary antibodies. After sections were deparaffinized, rehydrated, and incubated with blocking serum at room temperature for 20 min, heat-induced antigen retrieval was performed in 10 mM citrate butter (pH 6.0) for p-ERK 1/2 and VEGF-A, and in 1 mM ethylenediaminetetraacetic acid (pH 8.0) for t-EGFR, p-EGFR, and p-VEGFR2 at 90 °C for 20 min. Then, sections were incubated with each primary antibody at 4 °C overnight, followed by incubation with anti-rabbit secondary antibodies at room temperature for 30 min and with avidin-biotin-complex solution at room temperature for 30 min. Sections were visualized by diaminobenzidine (brown color) and counterstained with hematoxylin (purple) for light microscopic examination. The area of vascular smooth muscle cell layer in each section was determined in a blind manner to the study groups, and the relative quantities of each protein expression were calculated by Image Pro Plus 6.0 software (Media Cybernetics Inc., Rockville, MD) [10].
Statistics
In the statistical analysis, we calculated the power of the tests. The number of animals per group necessary to reach the desired power of 0.800 was in the range of 4 to 6. SAH grades and neurological scores were expressed as median ± 25th–75th percentiles, and were analyzed using Mann-Whitney U-tests or Kruskal-Wallis tests, followed by Steel-Dwass multiple comparisons. Other variables were expressed as mean ± standard deviation (SD), and unpaired t tests or one-way analysis of variance (ANOVA) with Student-Newman-Keuls post hoc tests were used appropriately. Analyses were performed using the statistical package for social science (SPSS). P < 0.05 was considered significant.
Results
AG1478 Prevents Neurological Deterioration and Vasospasm After SAH
There were no differences among groups as to physiological parameters (data not shown). None of 20 sham-operated mice died before euthanasia, and neither neurological deterioration nor vasospasm was observed. Administration of 632.0 ng AG1478 did not influence neurological status in sham-operated animals (Online Resource, Fig. S1). Among 87 SAH mice, 16 died before intracerebroventricular administration of vehicle or drug. By 24 h post-SAH, one SAH+vehicle and one SAH+AG1478 632.0 ng mice died, and 18 mice were excluded due to mild SAH (Online Resource, Table S2). In the remaining SAH mice, SAH grade was similar among groups at 24 h (Fig. 2a).

Therapeutic effects of AG1478 on cerebral vasospasm at 24 h after subarachnoid hemorrhage (SAH). a SAH grade. b Neurological score. c Representative pictures of internal carotid artery with hematoxylin-eosin staining. d Degree of vasospasm assessed by lumen radius/wall thickness ratio (LR/WT). SAH grade and neurological score are expressed as median ± 25th–75th percentiles, and compared with Kruskal-Wallis test. LR/WT is expressed as mean ± standard deviation, and compared with one-way analysis of variance followed by Student-Neuman-Keuls tests. *P < 0.01, **P < 0.05
In the SAH+vehicle group, neurological deterioration and severe vasospasm were observed at 24 h after SAH (Fig. 2b–d). As a higher dose of AG1478 was more effective against post-SAH vasospasm and neurological deterioration, the following experiments were performed using only the SAH+AG1478 632.0 ng group as the treatment group. A higher dosage of AG1478 tended to improve neuroscores at 24 and 48 h, and significantly improved neuroscores at 72 h post-SAH compared with the SAH+vehicle group irrespective of similar SAH grades (Fig. 2b, Fig. 3a, b; Online Resource, Fig. S2). In contrast, post-SAH vasospasm was significantly prevented by higher dosages of AG1478 at 24 h (Fig. 2c). Severe vasospasm was sustained in the SAH+vehicle group at 72 h, and tended to be suppressed by higher dosages of AG1478 (Fig. 3c, d).

Therapeutic effects of AG1478 on cerebral vasospasm at 72 h after subarachnoid hemorrhage (SAH). a SAH grade. b Neurological score. c Representative pictures of internal carotid artery with hematoxylin-eosin staining. d Degree of vasospasm assessed by lumen radius/wall thickness ratio (LR/WT). SAH grade and neurological score are expressed as median ± 25th–75th percentiles, and compared with Mann-Whitney U-test or Kruskal-Wallis test, respectively. LR/WT is expressed as mean ± standard deviation, and compared with one-way analysis of variance followed by Student-Neuman-Keuls tests. *P < 0.01, **P < 0.05
AG1478 Administration Suppresses P-EGFR and P-ERK1/2 Expression After SAH
In Western blotting, t-EGFR expression in cerebral arteries tended to increase after SAH but did not reach statistical significance among the sham, SAH+vehicle and SAH+AG1478 groups, while t-ERK1/2 expression showed no change among the 3 groups. As to p-EGFR and p-ERK1/2, expressions of these molecules were hardly detected in the sham group, but the expressions were upregulated in the SAH+vehicle group. In the SAH+AG1478 group, the levels of these molecules were significantly decreased compared with the SAH+vehicle group (Fig. 4). When expressed as a ratio of t-EGFR or t-ERK1/2 levels, p-EGFR and p-ERK1/2 levels showed similar findings, but the ratio of p-EGFR to t-EGFR levels lost statistically significant differences among the three groups to reflect post-SAH upregulation of t-EGFR (Online Resource, Fig. S3). Immunohistochemistry showed that t-EGFR was expressed in the endothelial cells, smooth muscle cells, and adventitial layers in the cerebral artery in both sham and SAH mice (Fig. 5). Although expression levels of both p-EGFR and p-ERK1/2 were very low in the sham group, the expressions were upregulated in the endothelial and smooth muscle cells in the artery after SAH, and suppressed especially in vascular smooth muscle cells by AG1478 (Fig. 5; Online Resource, Fig. S4).

Representative Western blots and effects of AG1478 on expressions of total (t-) epidermal growth factor receptor (EGFR), phosphorylated (p-) EGFR, and p-extracellular regulated-kinase (ERK) 1/2 in cerebral arteries at 24 h after subarachnoid hemorrhage (SAH). Data are expressed as mean ± standard deviation, and compared with one-way analysis of variance followed by Student-Neuman-Keuls tests. *P < 0.05

Representative immunostaining for total (t-) epidermal growth factor receptor (EGFR), phosphorylated (p-) EGFR, and p-extracellular regulated-kinase (ERK) 1/2 in internal carotid arteries at 24 h after subarachnoid hemorrhage (SAH) showing effects of AG1478. Arrow, immunoreactive endothelial cells; arrow head, immunoreactive smooth muscle cells. Scale bar: 20 μm
SAH Does Not Influence P-VEGFR2 and VEGF-A Expressions
Both Western blotting and immunohistochemistry showed that p-VEGFR2 and VEGF-A expressions were changed by neither SAH nor AG1478 (Online Resource, Figs. S5, S6). VEGFR2 is an endothelial cell-specific receptor [21], and expression of p-VEGFR2 was limited in the endothelial cells in cerebral arteries (Online Resource, Fig. S6). Immunoreactivity of VEGF-A was observed in the smooth muscle cell layer in the artery in addition to endothelial cells, but not significantly changed among groups.
Cetuximab Also Has Therapeutic Effects Against Vasospasm
In experiment 2, cetuximab, an anti-EGFR neutralizing antibody, was administered to confirm the therapeutic effects of the EGFR inhibitor against vasospasm. No significant differences as to physiological parameters were observed among the groups (data not shown). None of 10 sham-operated mice died before euthanasia, and they showed full scores on neurological assessment irrespective of cetuximab administration at 24 h after modeling (Online Resource, Fig. S7). Among 28 SAH mice, six died before an intracerebroventricular injection of vehicle, cetuximab or IgG. After randomization, no mice died, but eight mice were excluded due to mild SAH grade (Online Resource, Table S3). In the remaining SAH animals, SAH grades were similar between the groups (Fig. 6a). The SAH+vehicle group showed neurological deterioration and cerebral vasospasm, but administration of cetuximab significantly prevented neurological deterioration and vasospasm (Fig. 6b–d). Administration of cetuximab showed similar immunohistochemical findings to the administration of AG1478: that is, post-SAH upregulation of p-EGFR and p-ERK1/2 was suppressed especially in the smooth muscle cells in the cerebral artery by cetuximab (Fig. 7; Online Resource, Fig. S8), while expressions of p-VEGFR2 and VEGF-A were unchanged by SAH and cetuximab (Online Resource, Fig. S9). There were no significant differences between the SAH+vehicle and SAH+IgG groups as to neurological scores and vasospasm, excluding the possibility of IgG’s non-specific anti-vasospastic effects (Online Resource, Fig. S10).

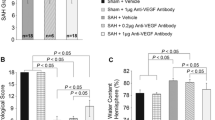
Therapeutic effects of cetuximab on cerebral vasospasm at 24 h after subarachnoid hemorrhage (SAH). a SAH grade. b Neurological score. c Representative pictures of internal carotid artery with hematoxylin-eosin staining. d Degree of vasospasm assessed by lumen radius/wall thickness ratio (LR/WT). SAH grade and neurological score are expressed as median ± 25th–75th percentiles, and compared with Mann-Whitney U-test or Kruskal-Wallis test, respectively. LR/WT is expressed as mean ± standard deviation, and compared with one-way analysis of variance followed by Student-Neuman-Keuls tests. *P < 0.01, **P < 0.05

Representative immunostaining for total (t-) epidermal growth factor receptor (EGFR), phosphorylated (p-) EGFR, and p-extracellular regulated-kinase (ERK) 1/2 in internal carotid arteries at 24 h after subarachnoid hemorrhage (SAH) showing effects of cetuximab. Arrow, immunoreactive endothelial cells; arrow head, immunoreactive smooth muscle cells. Scale bar: 20 μm
Discussion
In this study, we demonstrated that: (1) two kinds of EGFR inhibitors prevented neurological impairments and vasospasm after SAH in mice; (2) EGFR-ERK1/2 pathway might be implicated in the development of cerebral vasospasm; and (3) VEGFR2 might not be involved in cerebral vasospasm in mice.
We intracerebroventricularly administered two kinds of EGFR inhibitors, AG1478 (molecular weight: 315.8) and cetuximab (molecular weight: 145781.6), to assess their therapeutic effects on cerebral vasospasm in this study. AG1478 is a classical EGFR specific tyrosine kinase inhibitor, and its inhibitory effects on EGFR is well examined in experimental animals including mice [8, 9, 16]. Cetuximab-induced EGFR inhibition was also demonstrated in animal models including mice [22, 23]. Mouse cerebral arteries are known to lack vasa vasorum [24, 25], but paravascular space or channel exists just outside of the smooth muscle cells in cerebral arteries, allowing communication with the subarachnoid space, ventricles, and intra-parenchymal para-arteriolar spaces via cerebrospinal fluid [24, 26,27,28]. EGFR inhibitors administered intracerebroventricularly in this study were thought to reach the smooth muscle cells of cerebral arteries via this continuous network and bring anti-vasospastic effects at 24 h after SAH.
In clinical settings, activation of EGFR signaling pathways is suggested to contribute to cardiovascular diseases through blood pressure regulation, endothelial dysfunction, and cardiac remodeling [29]. After ligand binding, EGFR transits from an inactive monomer to an active dimer, and dimerization of EGFR activates its intracellular protein tyrosine kinase, followed by its downstream activation and signaling through intracellular mediators including ERK1/2, phosphoinositide-3 kinase (PI3K) /Akt, phospholipase C-γ, and jun kinase [29, 30]. In addition to the direct activation, EGFR is activated by transactivation through G protein-coupled receptors (GPCRs) [29, 30]. For instance, endothelin-1 (ET-1), thrombin, and angiotensin II activate GPCRs, and then trigger EGFR transactivation through GPCR, leading to several downstream signaling cascades such as ERK1/2 activation [29, 30]. As to vascular contraction, arterial smooth muscle cell contraction is triggered by intracellular signaling pathways such as mitogen-activated protein kinases, protein kinase C, PI3K, and Rho kinase, besides calcium-dependent pathways [31]. Several in vitro studies suggested that EGFR ligands such as heparin-binding (HB)-EGF or EGF caused vasoconstriction through direct activation or transactivation of EGFR as follows [31,32,33,34,35]: (1) HB-EGF released by ET-1 caused EGFR activation and subsequent increases in calcium transients, thereby resulting in constriction of carotid artery smooth muscle cells [32]; (2) administration of HB-EGF brought potassium channel current suppression, leading to cerebral artery smooth muscle cell constriction [33, 34]; and (3) administration of EGF induced PI3K-mediated ERK1/2 activation and resulted in contractile responses to aortic smooth muscle cells of hypertensive rats [31]. Furthermore, EGFR activation induced by cisternal administration of ET-1 [9] or recombinant tenascin-C [8] was demonstrated to increase p-ERK1/2 expression and bring cerebral artery constriction in healthy animals. Based on these previous reports, EGFR-ERK1/2 pathway is convincingly considered to play a role in arterial constriction. This study for the first time demonstrated that EGFR activation possibly in arterial smooth muscle cells is involved in the development of cerebral vasospasm using in vivo SAH models.
Recently, besides cerebral vasospasm, intracranial events arising before the onset of vasospasm which is called as early brain injury (EBI), is suggested to be an important contributor to patients’ poor outcome [10, 35,36,37,38]. Microvasospasm or vasospasm of brain arterioles is regarded as one of constituents of EBI [36]. A recent study revealed that potential spasmogen ET-1 was involved in the development of cerebral vasospasm, but administration of endothelin receptor antagonists did not improve microvasospasm, vasospasm of brain arterioles [37]: according to the authors, the findings could explain why treatment of endothelin receptor antagonist failed to improve SAH patients’ outcomes in clinical trials, although cerebral vasospasm was attenuated [37, 39]. On the other hand, our results showed that improved neurological status persisted at least for 72 h, whereas anti-vasospastic effects of EGFR inhibitors disappeared by 72 h after SAH in mice. Previous studies showed the involvement of EGFR activation in post-SAH persistent constriction of cerebral parenchymal arteriolar myocytes that were harvested from rabbits [35]. Taken together, EGFR may play important roles in both cerebral vasospasm and brain arteriolar vasospasm after SAH, and therefore therapies targeting EGFR could be a candidate for improving final outcomes of SAH patients by suppressing both macro- and micro-vasospasms. More studies are warranted to investigate the mechanisms for EGFR to induce cerebral vasospasm, and possibly microvasospasm-related or -unrelated EBI after SAH including the ligands.
Previous clinical trials targeting EGFR and VEGF in tumors suggested the importance to consider feed-back loops and cross-talk among different signaling pathways to obtain good results [13]. In addition, recent studies suggested that such cross-talk may also exist in non-neoplastic disease models: intraventricular infusion of recombinant human HB-EGF into rats as well as HB-EGF overexpression in mice caused hydrocephalus through activation of VEGF signaling [14, 15]. Thus, it seems important issues to investigate whether such links exist in cerebral vasospasm after SAH, because the combination of EGFR and VEGFR inhibitors could be a strong therapeutic candidate for cerebral vasospasm. In tumor cells, stimulation of EGFR increased expression of VEGF and VEGFR2 via hypoxia-inducible transcription factor-1α, transcription factor second component 1 (Sp1) or ERK1/2 activation [13, 40]. Several studies suggested roles of VEGF(R) in altering vascular contractility under pathological conditions [11, 12]. It has been reported that VEGF increases in brain parenchyma [10, 38] and cerebral arteries [5] after experimental SAH, as well as cerebrospinal fluid after clinical SAH [3, 4]. In addition, an injection of VEGF into cisterna magna caused basilar artery narrowing similar to cisternal blood clot in rabbits [41]. Thus, we hypothesized that VEGF as well as EGFR caused cerebral vasospasm after SAH, but the findings in this study denied the possible involvement of VEGF and VEGFR2 in post-SAH vasospasm.
This study has some limitations. First, an intracerebroventricular injection of inhibitors in this study limits clinical application, and to be more translational, other treatment routes such as intravenous injections should be examined. Second, anti-vasospastic effects of treatment regimen in this study were limited at 72 h, and therefore effects of multiple treatments at different dosages or time courses should be tested. Third, long-term functional outcomes should be investigated in future studies. Lastly, further clarification of the mechanisms between signal activation of EGFR and post-SAH vasospasm or EBI, as well as identification of the ligands is expected.
In conclusion, this study first showed that selective blockage of EGFR prevented post-SAH vasospasm and neurological impairments associated with ERK1/2 inactivation, but without affecting VEGF expression and VEGFR2 activation in mice. The present study suggests EGFR signaling as a new potential molecular target for therapy against post-SAH vasospasm.
References
Suzuki H, Shiba M, Nakatsuka Y, Nakano F, Nishikawa H (2017) Higher cerebrospinal fluid pH may contribute to the development of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Transl Stroke Res 8:165–173
Fujimoto M, Shiba M, Kawakita F, Liu L, Shimojo N, Imanaka-Yoshida K, Yoshida T, Suzuki H (2018) Effects of tenascin-C knockout on cerebral vasospasm after experimental subarachnoid hemorrhage in mice. Mol Neurobiol 55:1951–1958
Borel CO, McKee A, Parra A, Haglund MM, Solan A, Prabhakar V, Sheng H, Warner DS et al (2003) Possible role for vascular cell proliferation in cerebral vasospasm after subarachnoid hemorrhage. Stroke 34:427–433
McGirt MJ, Lynch JR, Blessing R, Warner DS, Friedman AH, Laskowitz DT (2002) Serum von Willebrand factor, matrix metalloproteinase-9, and vascular endothelial growth factor levels predict the onset of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery 51:1128–1135
Yan J, Chen C, Lei J, Yang L, Wang K, Liu J, Zhou C (2006) 2-methoxyestradiol reduces cerebral vasospasm after 48 hours of experimental subarachnoid hemorrhage in rats. Exp Neurol 202:348–356
Midwood KS, Hussenet T, Langlois B, Orend G (2011) Advances in tenascin-C biology. Cell Mol Life Sci 68:3175–3199
Suzuki H, Kinoshita N, Imanaka-Yoshida K, Yoshida T, Taki W (2008) Cerebrospinal fluid tenascin-C increased preceding the development of chronic shunt-dependent hydrocephalus after subarachnoid hemorrhage. Stroke 39:1610–1612
Fujimoto M, Shiba M, Kawakita F, Liu L, Nakasaki A, Shimojo N, Imanaka-Yoshida K, Yoshida T et al (2016) Epidermal growth factor-like repeats of tenascin-C-induced constriction of cerebral arteries via activation of epidermal growth factor receptors in rats. Brain Res 1642:436–444
Kawanabe Y, Masaki T, Hashimoto N (2004) Involvement of epidermal growth factor receptor-protein tyrosine kinase transactivation in endothelin-1-induced vascular contraction. J Neurosurg 100:1066–1071
Liu L, Fujimoto M, Kawakita F, Nakano F, Imanaka-Yoshida K, Yoshida T, Suzuki H (2016) Anti-vascular endothelial growth factor treatment suppressed early brain injury after subarachnoid hemorrhage in mice. Mol Neurobiol 53:4529–4538
Ray L, Mathieu M, Jespers P, Hadad I, Mahmoudabady M, Pensis A, Motte S, Peters IR et al (2008) Early increase in pulmonary vascular reactivity with overexpression of endothelin-1 and vascular endothelial growth factor in canine experimental heart failure. Exp Physiol 93:434–442
Adeoye OO, Butler SM, Hubbell MC, Semotiuk A, Williams JM, Pearce WJ (2013) Contribution of increased VEGF receptors to hypoxic changes in fetal ovine carotid artery contractile proteins. Am J Physiol Cell Physiol 304:C656–C665
Larsen AK, Ouaret D, El Ouadrani K, Petitprez A (2011) Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol Ther 131:80–90
Shim JW, Madsen JR (2018) VEGF signaling in neurological disorders. Int J Mol Sci 19:275
Shim JW, Sandlund J, Hameed MQ, Blazer-Yost B, Zhou FC, Klagsbrun M, Madsen JR (2016) Excess HB-EGF, which promotes VEGF signaling, leads to hydrocephalus. Sci Rep 6:26794
Li XM, Su F, Ji MH, Zhang GF, Qiu LL, Jia M, Gao J, Xie Z et al (2014) Disruption of hippocampal neuregulin 1-ErbB4 signaling contributes to the hippocampus-dependent cognitive impairment induced by isoflurane in aged mice. Anesthesiology 121:79–88
Formisano L, D’Amato V, Servetto A, Brillante S, Raimondo L, Di Mauro C, Marciano R, Orsini RC et al (2015) Src inhibitors act through different mechanisms in non-small cell lung cancer models depending on EGFR and RAS mutational status. Oncotarget 6:26090–26103
Kawakita F, Fujimoto M, Liu L, Nakano F, Nakatsuka Y, Suzuki H (2017) Effects of Toll-like receptor 4 antagonists against cerebral vasospasm after experimental subarachnoid hemorrhage in mice. Mol Neurobiol 54:6624–6633
Sabri M, Ai J, Knight B, Tariq A, Jeon H, Shang X, Marsden PA, Loch Macdonald R (2011) Uncoupling of endothelial nitric oxide synthase after experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab 31:190–199
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to image J: 25 years of image analysis. Nat Methods 9:671–675
Karkkainen MJ, Petrova TV (2000) Vascular endothelial growth factor receptors in the regulation of angiogenesis and lymphangiogenesis. Oncogene 19:5598–5605
Wang WM, Zhao ZL, Ma SR, Yu GT, Liu B, Zhang L, Zhang WF, Kulkarni AB et al (2015) Epidermal growth factor receptor inhibition reduces angiogenesis via hypoxia-inducible factor-1α and Notch1 in head neck squamous cell carcinoma. PLoS One 10:e0119723
Ledón N, Casacó A, Casanova E, Beausoleil I (2011) Comparative analysis of binding affinities to epidermal growth factor receptor of monoclonal antibodies nimotuzumab and cetuximab using different experimental animal models. Placenta 32:531–534
Zervas NT, Liszczak TM, Mayberg MR, Black PM (1982) Cerebrospinal fluid may nourish cerebral vessels through pathways in the adventitia that may be analogous to systemic vasa vasorum. J Neurosurg 56:475–481
Langheinrich AC, Michniewicz A, Bohle RM, Ritman EL (2007) Vasa vasorum neovascularization and lesion distribution among different vascular beds in ApoE-/-/LDL-/- double knockout mice. Atherosclerosis 191:73–81
Bedussi B, van Lier MG, Bartstra JW, de Vos J, Siebes M, VanBavel E, Bakker EN (2015) Clearance from the mouse brain by convection of interstitial fluid towards the ventricular system. Fluids Barriers CNS 12:23
Bedussi B, van der Wel NN, de Vos J, van Veen H, Siebes M, VanBavel E, Bakker EN (2017) Paravascular channels, cisterns, and the subarachnoid space in the rat brain: a single compartment with preferential pathways. J Cereb Blood Flow Metab 37:1374–1385
Abbott NJ (2004) Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int 45:545–552
Makki N, Thiel KW, Miller FJ Jr (2013) The epidermal growth factor receptor and its ligands in cardiovascular disease. Int J Mol Sci 14:20597–20613
Kalmes A, Daum G, Clowes AW (2001) EGFR transactivation in the regulation of SMC function. Ann N Y Acad Sci 947:42–55
Kim J, Lee CK, Park HJ, Kim HJ, So HH, Lee KS, Lee HM, Roh HY et al (2006) Epidermal growth factor induces vasoconstriction through the phosphatidylinositol 3-kinase-mediated mitogen-activated protein kinase pathway in hypertensive rats. J Pharmacol Sci 101:135–143
Chansel D, Ciroldi M, Vandermeersch S, Jackson LF, Gomez AM, Henrion D, Lee DC, Coffman TM et al (2006) Heparin binding EGF is necessary for vasospastic response to endothelin. FASEB J 20:1936–1938
Koide M, Penar PL, Tranmer BI, Wellman GC (2007) Heparin-binding EGF-like growth factor mediates oxyhemoglobin-induced suppression of voltage-dependent potassium channels in rabbit cerebral artery myocytes. Am J Physiol Heart Circ Physiol 293:H1750–H1759
Koide M, Wellman GC (2015) SAH-induced MMP activation and K V current suppression is mediated via both ROS-dependent and ROS-independent mechanisms. Acta Neurochir Suppl 120:89–94
Koide M, Wellman GC (2013) SAH-induced suppression of voltage-gated K(+) (K(V)) channel currents in parenchymal arteriolar myocytes involves activation of the HB-EGF/EGFR pathway. Acta Neurochir Suppl 115:179–184
Friedrich B, Müller F, Feiler S, Schöller K, Plesnila N (2012) Experimental subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: an in-vivo microscopy study. J Cereb Blood Flow Metab 32:447–455
Liu H, Dienel A, Schöller K, Schwarzmaier SM, Nehrkorn K, Plesnila N, Terpolilli NA (2018) Microvasospasms after experimental subarachnoid hemorrhage do not depend on endothelin A receptors. Stroke 49:693–699
Suzuki H, Hasegawa Y, Kanamaru K, Zhang JH (2010) Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke 41:1783–1790
Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I et al (2011) Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid hemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol 10:618–625
Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A (2006) EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res 66:3197–3204
Miller CA, Lombard FW, Wu CT, Hubbard CJ, Silbajoris L, Borel CO, Niklason LE (2006) Role of vascular mitogens in subarachnoid hemorrhage-associated cerebral vasculopathy. Neurocrit Care 5:215–221
Acknowledgments
We thank Ms. Chiduru Yamamoto-Nakamura (Department of Neurosurgery, Mie University Graduate School of Medicine) for her assistance with administrative support.
Funding
This study was funded by a grant-in-aid for Scientific Research from Japan Society for the Promotion of Science to Drs. Suzuki [17K10825] and Shiba [17K16640].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Research Involving Animals
All procedures were approved by the Animal Ethics Review Committee of Mie University and were carried out according to the institution’s Guidelines for Animal Experiments.
Electronic Supplementary Material
ESM 1
(PDF 9712 kb)
Rights and permissions
About this article

Cite this article
Nakano, F., Kawakita, F., Liu, L. et al. Anti-vasospastic Effects of Epidermal Growth Factor Receptor Inhibitors After Subarachnoid Hemorrhage in Mice. Mol Neurobiol 56, 4730–4740 (2019). https://doi.org/10.1007/s12035-018-1400-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1400-6