
Abstract
Pain affects both sensory and emotional aversive responses, often provoking depression and anxiety-related conditions when it becomes chronic. As the opioid receptors in the locus coeruleus (LC) have been implicated in pain, stress responses, and opioid drug effects, we explored the modifications to LC opioid neurotransmission in a chronic constriction injury (CCI) model of short- and long-term neuropathic pain (7 and 30 days after nerve injury). No significant changes were found after short-term CCI, yet after 30 days, CCI provoked an up-regulation of cAMP (cyclic 5′-adenosine monophosphate), pCREB (phosphorylated cAMP response element binding protein), protein kinase A, tyrosine hydroxylase, and electrical activity in the LC, as well as enhanced c-Fos expression. Acute mu opioid receptor desensitization was more intense in these animals, measured as the decline of the peak current caused by [Met5]-enkephalin and the reduction of forskolin-stimulated cAMP produced in response to DAMGO. Sustained morphine treatment did not markedly modify certain LC parameters in CCI-30d animals, such as [Met5]-enkephalin-induced potassium outward currents or burst activity and c-Fos rebound after naloxone precipitation, which may limit the development of some typical opioid drug-related adaptations. However, other phenomena were impaired by long-term CCI, including the reduction in forskolin-stimulated cAMP accumulation by DAMGO after naloxone precipitation in morphine dependent animals. Overall, this study suggests that long-term CCI leads to changes at the LC level that may contribute to the anxiodepressive phenotype that develops in these animals. Furthermore, opioid drugs produce complex adaptations in the LC in this model of chronic neuropathic pain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pain affects both sensory and emotional aversive responses, and when chronic, it often leads to depression and anxiety-related symptoms in humans [1] and rodents [2,3,4]. Hence, it is thought that pain might provoke plastic changes in several brain centers and/or in different neurotransmission systems. The opioid system is one such system and specifically, the mu opioid receptors (MORs), which represent the primary site of action of endogenous opioids, have consistently been implicated in the neurobiology of pain, as well as in stress responses or drug addiction and dependence [5, 6]. Importantly, the pontine locus coeruleus (LC), the principal brain site of noradrenaline synthesis, is targeted by several endogenous opioidergic peptides [7]. Moreover, since MORs are strongly expressed postsynaptically along noradrenergic somatodendritic processes [8], it is likely that chronic pain modifies opioid-related LC activity.
The LC sends numerous projections to the forebrain and spinal cord and thus, it is involved in many physiological functions, including arousal, memory, cognition, behavioral flexibility, stress reactivity, and pain processing [9,10,11,12,13,14,15,16]. Several rodent studies have suggested that the LC system is a possible source of endogenous analgesia in states of acute pain [11, 17, 18]. However, data from rat models of chronic pain (like neuropathic pain) suggest that the role of the LC appears to shift from the inhibition of pain, to the facilitation of pain and related emotional deficits [19,20,21]. In addition, the LC would also seem to be a key nucleus where stress and opioids intersect to mediate vulnerability to opioid drug abuse [11, 17, 18, 22]. Indeed, LC neurons have been widely implicated in physical opioid dependence and withdrawal, and acute exogenous opioid administration dampens the firing rate of rodent LC neurons and their cyclic 5′-adenosine monophosphate (cAMP) signaling. These neurons become tolerant to opioids upon their continued administration, evident through a return to normal neuronal activity and cAMP signaling because of an up-regulation of the cAMP pathway. This plasticity becomes functionally evident on withdrawal of the opiate, which provokes the increase in cAMP activity and firing rates. Significantly, there are several lines of evidence indicating that this sharp activation of the LC contributes to many of the behavioral signs and symptoms of physical opioid withdrawal in rodents [23,24,25].
These findings together suggest that pain could produce neuroplastic changes that affect the opioid system, and that might influence the activity of LC neurons and the effects of opioids in this nucleus. Thus, we set out to interrogate the functionality of the LC in relation to the opioid system in a rat model of neuropathic pain at two different time points: in short term, when animals have already developed the typical neuropathic phenotype (7 days after nerve injury); and in the long-term, when they show an anxiodepressive profile in addition to the neuropathic state (30 days after nerve injury) [2]. Furthermore, we wondered if sustained treatment with the opioid morphine produces the same effects in the LC in conditions of long-term neuropathic pain in animals not affected by chronic pain.
Materials and Methods
Animals
Experiments were carried out on male Sprague-Dawley rats, 250–350 g body weight, under standard laboratory conditions (22 °C, 12 h light/dark cycle, lights on at 08:00 a.m., food and water ad libitum). All procedures and animal handling were carried out in accordance with the guidelines of the European Commission’s directive (2010/63/EC) and Spanish Law regulating animal research (RD 53/2013), and all the experimental protocols were approved by the Committee for Animal Experimentation at the University of Cadiz (Spain).
Drugs
The following drugs were used in this study: ketamine (Merial Laboratorios S.A., Barcelona, Spain); xylazine (Bayer Hispania S.L., Barcelona, Spain); chloral hydrate (Fluka Chemie A.G, Buchs, Switzerland); morphine hydrochloride (MOR agonist: Spanish Agency of Medicines and Medical Devices—AEMPS); naloxone hydrochloride (opioid receptor antagonist: Sigma-Aldrich); [Met5]-enkephalin (ME) and DAMGO (D-ala(2),N-Me-Phe(4),Gly-ol(5))-enkephalin) (MOR agonists: Sigma-Aldrich); and forskolin (F6886: Sigma-Aldrich). All drugs were dissolved in isotonic saline solution (sodium chloride 0.9%).
Neuropathic Pain Model and Experimental Design
As described previously, chronic constriction injury (CCI) of the sciatic nerve was used as a model of neuropathic pain [26, 27]. The rats were anesthetized by ip (intraperitoneal) injection of 100 mg/kg ketamine and 20 mg/kg xylazine, and the left sciatic nerve was exposed at the mid-thigh level, proximal to the sciatic trifurcation. Four chromic gut (4/0) ligatures were tied loosely around the nerve 1.0–1.5 mm apart so as not to compromise the vascular supply. Sham operations were performed in the same manner but with no nerve ligation.
The animals were randomly assigned to the experimental groups, untreated or morphine-treated, and the experiments were carried out at two time points, 7 and 30 days after nerve injury (sham, CCI-7d and CCI-30d). Rats from untreated group were distributed into six independent experimental sets and analyzed by: (i) nociceptive behavioral testing, (ii) depressive-like behavioral testing, (iii) in vivo electrophysiology, (iv) [35S]GTPγS autoradiography, (v) western blotting, and (vi) immunohistochemistry. In the group of rats that received morphine, experiments were carried out 30 days after CCI (sham-saline, sham-morphine, sham-morphine + naloxone, CCI-30d-saline, CCI-30d-morphine, CCI-30d-morphine + naloxone). Each of these groups of animals was distributed into four independent experimental sets that were analyzed by: (i) in vivo electrophysiology, (ii) patch-clamp in brainstem slices, (iii) cyclic AMP assays, and (iv) immunohistochemistry (Fig. 1).

Schematic representation of the experimental design for a untreated or b morphine-treated animals. W, week; CCI-7d, 7 days after chronic constriction injury; CCI-30d, 30 days after chronic constriction injury; behavioral tests, nociceptive tests, and depressive-like behavior; D1-D5, days of morphine treatment; D6, day of naloxone-precipitated withdrawal; NLX, naloxone
Behavioral Tests
Pain-like behavior was evaluated before surgery, and 7 and 30 days after surgery. Mechanical allodynia was measured using the von Frey test (Dynamic Plantar Aesthesiometer, Ugo Basile, Italy), whereby a vertical force was applied to the left hind paw, increasing from 0 to 50 g over a period of 20 s [28]. Mechanical hyperalgesia was measured through the paw pressure test, gradually applying increasing pressure through a motor-driven device (Ugo Basile, Italy) with a 250 g cutoff to avoid tissue damage [29]. To assess thermal allodynia, the rat was placed on a cold metal plate maintained at 4 ± 1 °C (Panlab S.L, Spain), measuring the number of times the animal briskly lifted its left hind paw over a period of 2 min [27]. A 15–20 min interval was maintained between the testing procedures.
Depressive-like behavior was evaluated in the modified forced swimming test (mFST). Animals were placed individually in large plastic cylinders filled to a depth of 30 cm with water at 25 ± 1 °C for a first swimming session (pre-test), and again, 24 h later, for 5 min under the same conditions (test) [30]. Using a time-sampling technique, the predominant behaviors, climbing, swimming, or immobility, in each 5-s period of the 300 s of the swim exposure (test session) were recorded and scored (providing an overall total of 60 scores) using customized software (Red-Mice, Cadiz, Spain). The immobility behavior was determined when no activity was observed other than the movements necessary to keep the animal’s head above water. The climbing behavior was measured when the rats made vigorous upward movements with their forepaws in and out of the water. Swimming was considered the predominant behavior when the rats moved around the cylinder. Depressive-like behavior was defined as an increase in the mean count of the immobility behavior.
Morphine Treatment and Precipitation of the Withdrawal Syndrome
Opioid dependence was induced by repeated morphine injection [31]. The rats were injected with increasing doses of the opioid three times daily (at 8:00, 14:00, and 20:00 h: ip) on five consecutive days: days 1–10, 10 and 10 mg/kg; days 2–10, 10 and 20 mg/kg; days 3–20, 20 and 40 mg/kg; days 4–40, 40 and 80 mg/kg; and days 5–80, 80 and 100 mg/kg. Control animals were injected with an equivalent volume of saline 0.9% over the 5 days. On day 6 after morphine treatment, withdrawal syndrome was precipitated by the administration of naloxone (0.1 mg/kg iv—intravenous) to perform single-unit extracellular recordings of LC neurons (see below). Additionally, naloxone (5 mg/kg ip) was administered to two other sets of animals to precipitate withdrawal syndrome and to then assay cAMP or to perform immunohistochemistry (see below).
Electrophysiology
Recordings in Anesthetized Rats In Vivo
Single-unit extracellular recordings of LC neurons were obtained as described previously [32, 33]. Rats were anesthetized with an intraperitoneal injection of chloral hydrate (400 mg/kg) and anesthesia was maintained using a perfusion pump (chloral hydrate: 60 to 70 mg/kg/h), while the rat’s body temperature was maintained at ~ 37 °C with a heated pad. The rat was then placed in a stereotaxic frame (David Kopf Instrument, USA) with its head oriented at an angle of 15° to the horizontal plane. The recording electrode was lowered into the LC contralateral to the operated hind paw (relative to lambda: anterior-posterior [AP]—3.7 mm, lateral-medial [LM]—1.1 mm, dorsal-ventral [DV]—8.2 mm) and LC neurons were identified on the basis of well-established criteria: a long duration action potential (> 2 ms), regular spontaneous firing, a slow firing rate between 0.5 and 5 Hz, and characteristic spikes with a long-lasting positive-negative waveform [34]. The extracellular signals from the electrode were amplified with a high-input impedance amplifier and monitored. Discriminated spikes were fed into a PC and processed using CED Micro1401 and Spike2 software (Cambridge Electronic Design, UK). The firing patterns were analyzed offline using the Spike2 software and the tonic neuronal activity was assessed as the spontaneous firing rate (Hz). The dose-response curves were constructed for morphine administered acutely (0.15–4.80 mg/kg iv doubling the dose at 2 min intervals) and only one dose-response curve was acquired per rat. In another experimental group, the morphine dose-response curve was obtained after administering electrical pulses in the ipsilateral hind paw. To this end, electrical pulse trains were applied using bipolar needle electrodes (26-gauge, 2 mm separation) inserted subcutaneously into the medial-external surface of the ipsilateral hind paw, corresponding to the zone of innervation of the sciatic nerve. Foot shock stimuli (5.0 ms in duration and 10 mA in intensity) were delivered every 2 s using a stimulator CS-9 and a stimulus isolation unit (Cibertec SA, Spain), with a total of 125 repetitions per train [35]. Thus, eight peripheral foot shock trains were applied to each animal and the morphine dose-response curve (0.15–9.60 mg/kg doubling the dose at 2 min intervals) was obtained 150–250 s after the end of the last stimulus.
In morphine-treated animals, withdrawal syndrome was precipitated by administering naloxone (0.1 mg/kg iv) on day 6 after morphine treatment. The firing rate (Hz) and incidence of burst activity (%) in LC neurons were assessed through single-unit extracellular recordings before and during opioid withdrawal. An LC cell was considered to display burst firing when at least two spikes were evident with an initial interspike interval < 80 ms and subsequent interspike intervals ≥ 160 ms [36].
Whole-Cell Patch-Clamp Recordings
In an independent set of morphine-treated animals, whole-cell patch-clamp recordings of LC neurons were obtained as described elsewhere [37]. Animals were anesthetized with chloral hydrate (400 mg/kg, ip) and decapitated, and their brain was immediately extracted and placed in cooled ACSF saturated with 95% O2 and 5% CO2 [pH 7.3–7.4], and containing (in mM): 125 NaCl, 2.5 KCl, 1.2 MgCl2, 26 NaH2CO3, 1.25 NaH2PO4, 2.4 CaCl2, and 11 D-glucose. Horizontal vibratome sections of the brainstem (220 μm: VT1200S; Leica Microsystems, Germany) that contain the LC were incubated in ACSF for at least 1 h to wash out the drugs applied as chronic treatments that may have persisted in the brain tissue [38]. The slices were mounted in a recording chamber, maintained at 35–37 °C, and bath perfused by gravity with ACSF at a flow rate of 1.5–2 ml/min. LC neurons were visualized on an upright microscope with infrared optics (Eclipse E600FN, Nikon, Spain) as a dense and compact group of cells located at the lateral border of the central gray stratum and the fourth ventricle, just anterior to the genu of the facial nucleus. Whole-cell currents were recorded from rat LC neurons with an Axopatch-200B amplifier (Molecular Devices, USA) in voltage-clamp mode. Glass pipettes (2–4 MΏ) were prepared from borosilicate glass capillaries (World Precision Instruments, USA) on a micropipette puller (PC-10, Narishige CO., LTD, USA), and they were filled with an internal solution containing (in mM): 130 K-Gluconate, 5 NaCl, 1 MgCl2, 1 ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetra acetic acid (EGTA), 10 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 2 adenosine 5′-triphosphate magnesium salt (Mg-ATP), 0.5 guanosine 5′-triphosphate sodium salt hydrate (Na-GTP), and 10 phosphocreatine disodium salt hydrate (pH 7.4, 280 mOsm). Currents were recorded with the membrane potential held at − 50 mV and the LC neurons were identified by the presence of a resting inwardly rectifying potassium (IRK) conductance when stepping the membrane potential from − 40 to − 120 mV in − 10 mV increments. To induce MOR desensitization, a saturating concentration of [Met5] enkephalin (ME, 30 μM) was superfused and the decline in the peak current was measured during a 10-min application. Series resistance was monitored throughout the experiment and the cell was discarded if it exceeded 15 MΩ. Recordings were filtered at 5 kHz and processed with a Digidata 1322A digitizer (Axon Instruments, USA). The data were sampled at 10 kHz and analyzed with Clampex 10.2 software (Molecular Devices, USA), and all the data were extracted using Clampfit 10.3 software.
[35S]GTPγS Autoradiography of MOR Functionality
[35S]GTPγS autoradiography was carried out as described previously [39], with minor modifications. LC coronal sections (20 μm) of untreated animals were preincubated for 30 min at room temperature in a buffer containing: 50 mM Tris-HCl, 0.2 mM EGTA, 3 mM MgCl2, 100 mM NaCl, 1 mM D-dithiothreitol, and 2 mM GDP [pH 7.7]. The slides were subsequently incubated for 2 h in the same buffer containing adenosine deaminase (10 mU/ml) and [35S]GTPγS (0.04 nM). Consecutive sections were incubated in the presence or absence (basal [35S]GTPγS binding) of DAMGO (10 μM: D-ala(2),N-Me-Phe(4),Gly-ol(5)-enkephalin) to study the μ-opioid receptor-mediated stimulation of [35S]GTPγS binding. Specific binding was determined by incubation in the presence of DAMGO (10 μM) and naloxone (10 μM), and non-specific binding was determined in the absence of agonist and in the presence of guanosine-5-O-(3-thio)-triphosphate (10 μM GTPγS). After incubation, the sections were washed twice for 15 min in cold 50 mM Tris–HCl buffer [pH 7.4] at 4 °C, rinsed in distilled cold water and dried under a cold stream of air. The sections were then exposed to autoradiographic film (Biomax MR: GE Healthcare, Spain) at 4 °C for 2 days, together with [14C] microscales (GE Healthcare, Spain). Basal [35S]GTPγS binding is expressed as nCi/g of tissue and DAMGO-stimulated [35S]GTPγS binding is expressed as a percentage of the basal values (100%).
Cyclic AMP Assays
The cAMP assays were performed as described previously [40], with some minor modifications. LC tissue from saline, morphine or morphine + NLX-treated animals were homogenized (1:90 weight/volume dilution) with a Teflon/glass grinder (15 strokes, 800 rpm) in ice-cold homogenization buffer [pH 7.4] (20 mM Tris-HCl, 1 mM EGTA, 5 mM EDTA, 1 mM DTT, 20 μg/ml leupeptin, and 300 mM sucrose). The homogenates were centrifuged at 1500×g (5 min at 4 °C) and the resulting supernatants were centrifuged at 13,000×g (15 min at 4 °C), resuspending the resulting pellets in homogenization buffer. An aliquot of protein (50 mg) was preincubated for 5 min at 37 °C in an assay buffer [pH 7.4] (80 mM Tris-HCl, 0.2 mM EGTA, 1 mM EDTA, 2 mM MgCl2, 100 mM NaCl, 60 mM sucrose, 1 mM DTT, 10 mM GTP, 0.5 mM IBMX, 5 mM phosphocreatine, 50 U/ml creatine phosphokinase, and 5 U/ml myokinase) without (basal adenylyl cyclase—AC activity) or with 10 μM forskolin (FK: FK-stimulated cAMP accumulation). The opioid receptor-mediated inhibition of FK-stimulated cAMP accumulation was determined using the agonist DAMGO (10 μM), and the specificity of the effects were determined by adding the opioid antagonist naloxone (10 μM). In all experimental conditions, Mg-ATP 0.2 mM was added to the membranes and the mixture was incubated for 10 min at 37 °C. The reaction was stopped by boiling for 5 min and the cAMP concentration was determined in a 50-μl sample of the supernatant using the commercial competitive cAMP ELISA kit (Pierce, USA). Assays were performed in triplicate on 4–10 samples per group and each sample was analyzed in two independent experiments, expressing the results as pmol of cAMP/min/mg protein.
Immunohistochemistry
Untreated and morphine + NLX-treated animals were perfused intracardially with 4% paraformaldehyde (PFA) [41]. The brainstem was removed and immersed in fresh PFA for 2–4 h at 4 °C before it was cryoprotected in 30% sucrose (w/v) with 0.1% sodium azide in phosphate buffer 0.1 M for at least 48 h. Freezing microtome sections (30 μm) of the brain region containing the LC were obtained and every fourth free-floating sequential section was incubated with rabbit against phospho-CREB Ser133(pCREB, 1:1000; 06–519, Millipore, Spain) or c-FOS (1:10000; ab99515, Abcam, UK). Subsequently, the sections were incubated with a biotinylated donkey anti-rabbit antibody (1:200, Jackson Immunoresearch, USA) and then an avidin-biotin complex (ABC) conjugated to horseradish peroxidase. Immunostaining was visualized with 3,3-diaminobenzidine tetrahydrochloride (DAB) and the sections were mounted on gelatin-coated slides. The images were acquired on an Olympus BX60 microscope and the number of pCREB or c-FOS inmunoreactive (pCREB-IR or c-FOS-IR) cells of six sections of LC per animal were manually counted. Data was presented as the mean of pCREB-IR or c-FOS-IR cells of six sections per animal.
In order to verify the localization of c-FOS into LC noradrenergic neurons, the detection of c-FOS and dopamine beta hydroxylase (DBH) was performed by immunofluorescence. LC sections were incubated with a mouse anti-DBH (1:1000; MAB308, Millipore, EE.UU) and rabbit c-FOS (1:10000; ab99515, Abcam, UK). Subsequently, the sections were incubated, respectively, with a biotinylated donkey anti-rabbit (1:1000; Invitrogen, Spain) followed by streptavidin 568 and a donkey anti-mouse Alexa Fluor 488 (1:1000; Invitrogen, Spain), before they were washed and cover slipped with fluoro-gel aqueous mounting medium. Merge images were acquired with an inverted fluorescence microscope Axion Observer with Apotome 2.0 (ZEISS) (Carl Zeiss, Germany). All images were processed using the Zen and FI-JI software.
Western Blotting
Untreated animals were killed by decapitation and the LC was removed bilaterally. Equal amounts of protein from tissue homogenates were separated by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and transferred to polyvinylidene difluoride membranes (PVDF). The membranes were probed overnight at 4 °C with primary antibodies against pCREB (1:1000; 06–519, Millipore, Spain), CREB (1:500; #9197, Cell Signaling, USA), tyrosine hydroxylase (TH, 1:5000; ab113 Abcam plc, UK), PKAα cat (1:1000; sc-903, Santa Cruz Biotechnology Inc., USA), or PKAIIα reg (1:1000; sc-909, Santa Cruz Biotechnology Inc., USA). The primary antibodies were detected using the corresponding secondary antibody (1:10,000, Bonsai Advanced Technologies, Spain): IRDye 800CW goat anti-rabbit (green), IRDye 680LT goat anti-mouse (red), or horseradish peroxidase-conjugated secondary antibody (donkey anti-sheep). The protein signals were detected using a LI-COR Odyssey® two-channel quantitative fluorescence imaging system (Bonsai Advanced Technologies, Spain) or a chemiluminescence Versadoc 5000 system (Bio-Rad Laboratories, Spain). Digital images of Western blots were analyzed by densitometry using the ImageJ free access software (National Institutes of Health, USA), and the protein levels were normalized to the level of β-actin or α-tubulin.
Statistical Analysis
All the data are presented as the means ± SEM and the results were all analyzed using STATISTICA 10.0 (StatSoft, USA) or GraphPad Prism 5 software (GraphPad Software, USA) using either a Student’s t test (unpaired or paired, two-tailed), a one-way or two-way analysis of variance (ANOVA) with or without repeated measured followed by the appropriate post hoc tests: Dunnett’s or Newman-Keuls tests. A Fisher’s exact test was used to evaluate the incidence of burst activity. Significance was accepted at p < 0.05 (Table 1).
Results
Neuropathy Induces Biochemical Changes in the Locus Coeruleus
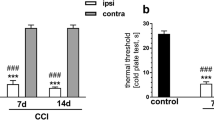
As expected, nerve-injured animals showed pain hypersensitivity to mechanical and thermal stimuli 7 and 30 days post-surgery (Fig. 2a–c). Moreover, CCI-30d animals developed a depressive-like phenotype [2, 3] evident through a significant increase in their immobility (p < 0.001) and a decrease in the climbing behavior (p < 0.001) relative to the sham animals. By contrast, no significant changes were observed in the CCI-7d animals (Fig. 2d).

Sensorial thresholds evaluated before and 7 and 30 days after chronic constriction injury (CCI). a Mechanical allodynia. Withdrawal threshold (g) of the ipsilateral paw in response to von Frey hair stimulation (0 to 50 g, 20 s). b Mechanical hyperalgesia. Withdrawal threshold (g) of the ipsilateral paw in response to paw-pressure stimulation (cutoff 250 g): ***p < 0.001 vs day 0 (before surgery) and sham group, two-way ANOVA with repeated measures followed by Newman-Keuls post-test. c Cold allodynia. Number of lifts of the ipsilateral paw in the cold plate test (2 min, 4 °C), ***p < 0.001 vs day 0 (before surgery), one-way ANOVA with repeated measures followed by Newman-Keuls post-test. Each symbol represents the mean ± SEM of ten animals per group (see Table 1 for detailed statistical analysis). d Depressive-like behavior in the modified forced swimming test. Each bar represents the mean ± SEM of the predominant behavior (immobility, climbing, and swimming) of eight animals/group, ***p < 0.001 vs sham and CCI-7d group assessed by one-way ANOVA followed by Dunnett post-test (see Table 1 for detailed statistical analysis)
In an independent group of animals, we evaluated the possible changes related to the activity of the LC system after nerve injury. Firstly, c-fos was assessed to reflect cell activation in the LC, and there was a significant increase in the number of c-fos labeled neurons in the LC of CCI-30d rats (p < 0.05, Fig. 3a, d), but not in that of CCI-7d animals. Moreover, and in agreement with previous studies [3], TH expression was enhanced in the CCI-30d (p < 0.01, Fig. 3b), but not the CCI-7d animals. As TH is a target gene for CREB in numerous tissues [42], we examined the expression of the active form of this protein, p-CREB. When evaluated by immunohistochemistry, more p-CREB was evident in CCI-30d but not in CCI-7d rats relative to the sham animals (p < 0.05, Fig. 3c–d). Furthermore, this increase in p-CREB expression in the LC of CCI-30d animals was corroborated in western blots (p < 0.001, Fig. 3e), while the total CREB protein remained unaltered in the LC of CCI-30d rats (Fig. 3f). We also evaluated the levels of the cAMP-dependent protein kinase (PKA) as CREB proteins are activated by PKA phosphorylation. An increase in PKAα cat (catalytic subunit) and PKAIIα reg (regulatory subunit) was also evident in CCI-30d rats compared with sham animals (p < 0.01 and p < 0.001 respectively, Fig. 3g, h).

Biochemical changes in the locus coeruleus after chronic constriction injury (CCI). a Quantification of c-FOS positive cells. b Representative blot and quantification of tyrosine hydroxylase (TH). c Quantification of pCREB positive cells. d Photomicrographs showing the expression of c-FOS and pCREB (scale bar 50 μm; 4 V, 4th ventricle). (e–h) Representative blot and quantification of pCREB, CREB, PKAα cat., and PKAIIα reg. Bar graphs represent the mean ± SEM of four animals/group: *p < 0.05, **p < 0.01, and ***p < 0.001 vs sham group assessed by one-way ANOVA followed by Dunnett post-test (see Table 1 for detailed statistical analysis)
In Vitro Effects of Opioids in the Locus Coeruleus Associated with Neuropathy
As CREB is implicated in regulating the mu opioid system in the LC, we studied the action of MOR agonists in vitro through several approaches [43]. Firstly, we evaluated the G protein activation mediated by MORs in [35S]GTPγS binding assays. Both the basal [35S]GTPγS binding and the DAMGO-stimulated [35S]GTPγS binding was similar in all groups of rats (Fig. 4a–b). Secondly, we evaluated acute MORs desensitization using patch-clamp recordings of LC neurons in sham and CCI-30d animals. It is well known that ME activation of MORs in the LC evokes an outward current that rapidly declines in the continued presence of the drug (30 μM during 10 min), which is considered a hallmark of MORs desensitization [38, 44]. No differences were found in the peak currents evoked by ME between the saline groups (Fig. 4c, e), yet stronger ME-induced desensitization was obtained in the CCI-30d rats than in the sham animals (p < 0.05, Fig. 4d–e). We also evaluated MOR desensitization in morphine-dependent rats (after 5 days of morphine treatment) and as reported previously [38], morphine did not significantly alter the peak current evoked by ME in any of the treatment groups (Fig. 4c, e). However, it did increase the amount of MORs desensitization in sham-morphine animals with respect to sham-saline (p < 0.05, Fig. 4d, e) [38]. Strikingly, morphine did not modify the desensitization recorded in CCI-30d animals relative to the CCI-30d-saline (Fig. 4d, e).

In vitro effects of opioids on the locus coeruleus after chronic constriction injury (CCI). a Basal [35S]GTPγS binding in LC sections. b DAMGO-stimulated [35S]GTPγS binding in LC sections (mean ± SEM of stimulation relative to the basal binding). Each column represents the mean ± SEM of 4–5 animals/group: *p < 0.05 vs sham group assessed by one-way ANOVA followed by a Dunnett post-test. c Amplitude of the outward current induced by ME (I-ME; 30 μM) in LC slices from sham or CCI-30d animals after they received saline or morphine for 5 days. d Desensitization. Decline in the peak current induced by ME (30 μM) over 10 min in LC slices from sham or CCI-30d animals after administering saline or morphine for 5 days. Each column represents the mean ± SEM of 8–16 neurons/group: *p < 0.05 vs sham-saline group as assessed by two-way ANOVA followed by a Newman-Keuls post-test. e Current traces showing the outward current induced by ME (30 μM) in slices from saline/morphine-treated sham or CCI-30d animals. f cAMP accumulation (picomoles per minute per milligram of protein) in the LC in the presence or absence (basal) of forskolin (FK), and the inhibition of FK-stimulated cAMP accumulation by DAMGO, a mu opioid receptor agonist (FK + DAMGO), in sham and CCI-30d animals after they had received saline, morphine, or morphine + naloxone. Each column represents the mean ± SEM of 4–5 animals/group: *p < 0.05, **p < 0.01 vs sham-sal-basal, sham-morp + NLX-basal, CCI-30d-sal-basal, CCI-30d-morp + NLX-basal groups; #p < 0.05 vs sham-Sal-FK, sham-morp + NLX-FK groups, assessed by two-way ANOVA with repeated measures followed by a Neuman-Keuls post-test; +p < 0.05, ++p < 0.01 vs sham-sal-basal group, assessed by two-way ANOVA followed by a Dunnett post-test (see Table 1 for detailed statistical analysis). Abbreviations: FK, forskolin; ME, [Met5] enkephalin; Sal, saline; Morp, morphine; NLX, naloxone
Thirdly, we also studied cAMP production in slices of the LC from CCI-30d animals (Fig. 4f), detecting a significant increase in basal cAMP levels in CCI-30d rats relative to the sham animals (sham-saline vs CCI-30d-saline: p < 0.05, Fig. 4f). Forskolin treatment significantly increased the cAMP production in sham (157% of the basal production, p < 0.01) and CCI-30d animals (48% of the basal production, p < 0.05), and although the MOR agonist DAMGO inhibited this forskolin-stimulated cAMP accumulation in sham animals (p < 0.05), it did not reduce cAMP levels in CCI-30d animals. This suggests a functional acute desensitization of MORs in response to DAMGO in CCI-30d animals. We also evaluated the cAMP levels after 5 days of morphine treatment in sham and CCI-30d animals. As expected, morphine significantly increased the basal cAMP levels in the LC of sham animals (sham-saline vs sham-morphine: p < 0.05, Fig. 4f) [45, 46] but it did not further increase the already elevated basal levels of cAMP in CCI-30d rats (CCI-30d-saline vs CCI-30d-morphine). Surprisingly, forskolin and subsequent DAMGO administration did not significantly modify the basal cAMP levels in sham or CCI-30d rats administered with morphine. We also evaluated the cAMP production after a 5-day morphine treatment and subsequent naloxone challenge. As expected, basal cAMP levels remained high in sham animals receiving naloxone (sham-saline vs sham-morphine + NLX: p < 0.05, Fig. 4f) [24] and subsequent administration of forskolin increased the levels of cAMP (56.57%, p < 0.05, Fig. 4f), whereas DAMGO incubation significantly reduced it (p < 0.05). When CCI-30d animals received morphine + naloxone, there was no significant difference in the basal cAMP relative to the CCI-30d-saline rats, but subsequent forskolin administration significantly increased the accumulated levels of cAMP with respect to basal cAMP levels of CCI-30d-morphine + NLX (62.04%, p < 0.01, Fig. 4f). Incubation with DAMGO failed to induce a significant effect on forskolin-stimulated cAMP levels in CCI-30d morphine + NLX animals.
In Vivo Effects of Opioids in the Locus Coeruleus Associated with Neuropathy
Extracellular recordings in anesthetized animals were used to evaluate the function of the LC in vivo. In agreement with previous c-fos data, the spontaneous firing rate in the LC increased significantly in the CCI-30d group (p < 0.01, Fig. 5a), suggesting that long-term nerve injury enhances the electrical activity of LC neurons. Subsequent intravenous morphine administration was used to assess the effect of MORs activation [47]. Morphine dose-response curves did not shift significantly in nerve-injured rats with respect to the sham animals (Fig. 5b–d). However, when similar experiments were performed after electrical stimulation of the ipsilateral hind paw, we found a clear shift to the right in the CCI-30d stimulated group (ED50 = 2.45 ± 0.56 mg/kg) with respect to the CCI-30d non-stimulated (ED50 = 0.59 ± 0.12 mg/kg, p < 0.01) or sham-stimulated animals (ED50 = 0.99 ± 0.17 mg/kg, p < 0.05: Fig. 5b–d). No significant differences were found in either sham or CCI-7d animals.

In vivo effects of opioids on the locus coeruleus after chronic constriction injury (CCI). a Spontaneous firing rate of LC neurons. Each column represents the mean ± SEM of 27–30 neurons/group: **p < 0.01 vs sham group assessed by one-way ANOVA followed by a Dunnett post-test. b Effective dose (ED50) estimated by Parker and Waud’s equation in non-stimulated (NS) and stimulated (S) sham,and CCI-7d and CCI-30d animals. Each column represents the mean ± SEM of 5–7 neurons/group: **p < 0.01 vs CCI-30d-NS; #p < 0.05 vs sham-NS, sham-S, CCI-7d-NS, and CCI-7d-S assessed by two-way ANOVA followed by a Newman-Keuls post-test. c Representative histograms of recordings from LC neurons showing the dose-dependent inhibitory effect of morphine administered intravenously at increasing doses (scale bar, 1 min). d Dose-response curves showing the inhibitory effect of morphine on the rate of LC neuron firing. Each symbol represents the mean ± SEM of the reduction with respect to the basal firing rate of 5–7 neurons/group and the horizontal axis represents the logarithm of the cumulative morphine doses (see Table 1 for detailed statistical analysis)
The firing rate of LC neurons was studied after a 5-day morphine treatment and during opiate withdrawal in anesthetized rats using single-unit extracellular recordings. Similar electrophysiological activity was found in sham and CCI-30d animals treated with morphine, and the subsequent precipitation of opiate withdrawal by naloxone resulted in a rapid and dramatic increase in the LC firing rate that was similar in both groups of animals (p < 0.01 and p < 0.001, Fig. 6a). However, the increase in bursting activity was significantly lower in CCI-30d animals (p < 0.001, Fig. 6b). We also quantified c-fos expression in the LC 2 h after naloxone injection, a reliable measure of neuronal activation [48, 49]. Precipitating opiate withdrawal in sham animals increased c-fos expression in the noradrenergic LC neurons (p < 0.001, Fig. 6c–e), although this increase was significantly weaker in CCI-30d animals (p < 0.05 and p < 0.01, Fig. 6 c–e).

In vivo effects of naloxone-precipitated withdrawal in the locus coeruleus after chronic constriction injury (CCI). Spontaneous firing rate in the a LC and the b burst activity in morphine-treated and morphine + NLX-treated animals (NLX: 0.1 mg/kg iv). Each column represents the mean ± SEM of 4–8 neurons: ***p < 0.001 vs sham-morphine; ##p < 0.01; ###p < 0.001 vs CCI-30d-morphine; +++p < 0.001 vs sham-morphine + NLX as assessed by two-way ANOVA followed by a Newman-Keuls post-test or Fisher’s exact test. c Graph showing c-FOS in the LC, representing the mean ± SEM of five animals/group: *p < 0.05; ***p < 0.001 vs sham group, and ##p < 0.01 vs sham-morphine + NLX as assessed by a Student’s t test. d Photomicrographs of c-FOS expression in the LC (scale bar, 50 μm). e Representative images showing the colocalization of c-FOS into DBH positive neurons in LC (scale bar, 50 μm); magnification indicates an example of colocalization (scale bar, 20 μm). Abbreviations: DBH, dopamine beta hydroxylase; NLX, naloxone. (see Table 1 for detailed statistical analysis)
Discussion
The current study indicates that long-term peripheral nerve injury leads to hyperactivation of the noradrenergic system, engaging the cAMP-CREB pathway, with stronger MOR desensitization in the LC of these animals. Sustained morphine treatment in association with long-term neuropathic pain leads to particular cellular and molecular adaptations, which may contribute to the differential effects of opioid drugs in chronic pain when compared to pain-free conditions.
We show here that the LC is hyperactive 1 month after neuropathy, as reflected by the higher spontaneous firing rate and the up-regulation of c-fos, TH, and CREB phosphorylation. The increase in TH and CREB phosphorylation expression is consistent with previous data [50], and as the CREB-cAMP pathway is thought to contribute to the intrinsic excitability of LC neurons and regulate c-fos expression [51], we also explored the expression of markers of this pathway. As expected, catalytic and regulatory PKA subunits were expressed more strongly in the LC of CCI-30d rats. Indeed, more cAMP was detected under basal conditions in CCI-30d rats compared to sham animals, although administration of the AC activator forskolin elevated the cAMP in this structure in both sham and CCI-30d animals. Interestingly, the cAMP accumulated after forskolin administration was similar in both groups, suggesting a possible ceiling effect in the activation of the cAMP system in long-term pain animals or in the detection limits of the test used. As G protein-coupled receptors also modify cAMP signaling, we explored G protein activation through [35S]GTPγS binding, although no differences were observed. Overall, these data suggest that cAMP signaling is enhanced in CCI-30d animals under basal conditions (in the absence of an exogenous agonist), which could be due to the activation of AC or to changes in G protein coupling to AC. No significant effects in pCREB or PKA subunits expression were found in short-term neuropathic animals (CCI-7d) (Fig. 3), suggesting that the over-activation of the cAMP pathway only occurs after long-term neuropathy.
Considering that CREB phosphorylation is homeostatically regulated by MOR signaling, which inhibits the cAMP pathway [45, 52], we explored the engagement of the opioid system in the LC in association with neuropathy. Firstly, the effect of neuropathy on acute MORs desensitization was studied in vitro, which reflects short-term plasticity of MOR activity. Acute loss of MOR-effector coupling occurs within seconds to minutes of receptor exposure to high concentrations of opioid agonists, such as the endogenous opioid peptides, ME, DAMGO, etorphine, methadone, and fentanyl [43, 53,54,55]. No significant changes were found in MOR-G protein activation by DAMGO. However, the inhibitory effect of DAMGO on forskolin-stimulated AC activity was lost after long-term nerve injury (CCI-30d). Furthermore, MORs desensitization measured as the decline of the peak current induced by ME was greater in the CCI-30d animals compared to that recorded in the sham group. These results suggest that long-term nerve injury leads to stronger MOR desensitization in the LC when activated by maximal concentrations of opioid agonists like DAMGO or ME. Indeed, extracellular recordings in vivo highlighted a reduction in the inhibitory effect of morphine after electrical stimulation of the nerve-injured hind paw in CCI-30d animals. It is likely that the weaker effect of morphine after electrical stimulation in CCI-30d was due to MORs desensitization because endogenous opioids appear to be released in the LC in response to pain stimulation [56,57,58] and acute stress [59]. Accordingly, persistent inflammatory pain decreases the antinociceptive effects of the MOR agonist DAMGO in the LC of rats [60].
The aforementioned data suggests that the LC is more active after long-term pain, as reflected by the overexpression of TH, the CREB-cAMP pathway, and the greater MOR desensitization in response to opioid agonists. These plastic changes might be relevant to stress because the LC system is intensely implicated in the stress response, and its activity is co-regulated by the opposing influences of excitatory and inhibitory neurotransmitters [15]. Thus, when the opioid tone is weaker, the excitatory component would be predominant in enhancing an arousal-like state, which could account for stress vulnerability and that may lead to stress-related disorders [59]. In the case of neuropathic pain, sensorial hypersensitivity appears immediately after nerve injury in rodents, whereas anxiodepressive and cognitive symptoms arise after several weeks (3–8 weeks depending on the species and animal model used) [2,3,4]. Furthermore, an increase in excitatory postsynaptic currents and basal extracellular glutamate concentrations, as well as a down-regulation of GLT-1 (astroglial glutamate transporter-1) in the LC [61, 62] are associated with neuropathic pain, and we now describe the hyperactivation of the LC and stronger desensitization of MOR. These pain-related adaptations might lead to a more enduring activation of the LC system, which would translate into the exaggerated hyperarousal that is a characteristic of chronic pain and that is usually comorbid with stress-related psychiatric disorders (e.g., anxiety and depression) [3, 63].
We also evaluated the effect of a 5-day treatment with morphine in long-term nerve-injured rats. As previously reported, chronic morphine enhanced ME-induced acute MORs desensitization in sham rats [38], yet it did not further augment desensitization in the CCI-30d animals, suggesting that sustained exposure to morphine decreases MOR activity in sham but not in CCI-30d rats. In addition, changes in the cAMP pathway have been widely implicated in the long-term cellular and molecular adaptations to morphine. Thus, and as expected, basal cAMP levels were increased in sham rats that received morphine [45, 46]. However, forskolin did not increase the basal cAMP levels in these animals. Later data are in contradiction with a previous paper, which found an elevation of forskolin-stimulated cAMP levels in LC membranes of morphine-treated rats [45]. Such discrepancy could be due to the different methodological approaches because we have ip-injected increasing doses of the opioid for five consecutive days, and Duman et al., [45] daily implanted subcutaneous morphine pellets (with the same dose) for 5 days. In addition, although our study does not provide information about the mechanism involved in the change of MOR-associated G protein transduction pathway for the AC inhibition, it is interesting to emphasize that opioid receptors are coupled to both Gi and Gs proteins [64,65,66] and thereby, an interaction of stimulatory actions mediated by Gs proteins with forskolin could counteract its effect on AC activity in our experimental conditions. As forskolin was not able to increase cAMP levels, subsequent DAMGO administration did not change them in sham animals treated with morphine. In CCI-30d rats, the already enhanced basal cAMP levels were not significantly modified when these animals received morphine, neither in basal conditions nor in the presence of forskolin nor DAMGO. Overall, these data show that the desensitization of MOR-mediated GIRK currents and the increase of AC activity found in CCI-30d animals are resistant to be further increased by morphine treatment. However, the activator of the AC forskolin behaved similarly in both conditions.
When naloxone-induced morphine withdrawal was explored, the effect of forskolin and forskolin + DAMGO on cAMP levels was recovered in sham rats. Hence, naloxone appears to re-sensitize the MORs in these animals. In CCI-30d rats after naloxone-induced morphine withdrawal, the effect of forskolin on cAMP levels was recovered as happened in sham animals. By contrast, no significant re-sensitization was found when forskolin + DAMGO were administered, which might suggest an impairment in the ability of MORs to recover their function and to re-sensitize cells to opioid agonists in conditions of long-term pain. We also explored the electrophysiological activity of naloxone-induced morphine withdrawal. In vivo, electrophysiological recordings of anesthetized animals highlighted a similar elevation in the spontaneous firing rate after naloxone administration to that in sham and neuropathic animals that received morphine, although the neuropathic rats had a lower incidence of burst activity. Bursts of LC activity more effectively increase terminal noradrenaline release than tonic increases in activity [67]. Thus, less burst activity may dampen noradrenaline release, which may also contribute to a less severe manifestation of withdrawal symptoms. In line with this hypothesis, and using c-fos expression as a hallmark of cell activation, there was a significant increase in c-fos in both sham and CCI-30d rats after naloxone-precipitated morphine withdrawal, although such activation was less intense in the CCI-30d animals.
In summary, we found that chronic neuropathic pain appears to drive the activation of the LC (Fig. 7). It seems that long-term CCI engages specific molecular processes in the LC (TH, cAMP-CREB pathway, potassium channels), and that these may contribute to the anxiodepressive phenotype found in these animals. Alternatively, sustained morphine administration does not substantially modify certain LC specific parameters, which may limit the development of typical opioid withdrawal endpoints. However, other effects seem to be impaired, such as the altered AC activity after naloxone precipitation of withdrawal, which may in turn dampen the recovery of MOR function. Overall, these data suggest that chronic neuropathic pain produces complex plastic changes in LC activity. Importantly, there have recently been a number of papers describing that subpopulations of LC neurons are connected to separate anatomical targets promoting functional diversity [19, 68, 69]. Thus, it is likely that long-term neuropathic pain has modified subsets of LC neurons expressing specific receptors or neurotransmitters (others than noradrenaline), which could alter the properties and conditions of their activation. Thus, further studies using new tools that allow one to know if LC neurons expressing MORs represent a specific cluster of neurons within the LC could help to clarify the LC cellular and molecular adaptations provoked by pain and opioid drugs and their behavioral consequences.

Scheme illustrating the effect of long-term neuropathic pain in the LC. Upward blue arrows summarize the effects of neuropathic pain in the LC. Neuropathic pain augments the cAMP, PKA cat., and PKAII reg. subunits, and phosphoproteins like CREB and TH. These changes may contribute to the increase in the electrical excitability of LC neurons
References
Bair MJ, Robinson RL, Katon W, Kroenke K (2003) Depression and pain comorbidity: a literature review. Arch Intern Med 163(20):2433–2445. https://doi.org/10.1001/archinte.163.20.2433
Alba-Delgado C, Cebada-Aleu A, Mico JA, Berrocoso E (2016) Comorbid anxiety-like behavior and locus coeruleus impairment in diabetic peripheral neuropathy: a comparative study with the chronic constriction injury model. Prog Neuro-Psychopharmacol Biol Psychiatry 71:45–56. https://doi.org/10.1016/j.pnpbp.2016.06.007
Alba-Delgado C, Llorca-Torralba M, Horrillo I, Ortega JE, Mico JA, Sanchez-Blazquez P, Meana JJ, Berrocoso E (2013) Chronic pain leads to concomitant noradrenergic impairment and mood disorders. Biol Psychiatry 73(1):54–62. https://doi.org/10.1016/j.biopsych.2012.06.033
Suzuki T, Amata M, Sakaue G, Nishimura S, Inoue T, Shibata M, Mashimo T (2007) Experimental neuropathy in mice is associated with delayed behavioral changes related to anxiety and depression. Anesth Analg 104(6):1570–1577, table of contents. https://doi.org/10.1213/01.ane.0000261514.19946.66
Drolet G, Dumont EC, Gosselin I, Kinkead R, Laforest S, Trottier JF (2001) Role of endogenous opioid system in the regulation of the stress response. Prog Neuro-Psychopharmacol Biol Psychiatry 25(4):729–741. https://doi.org/10.1016/S0278-5846(01)00161-0
Kanjhan R (1995) Opioids and pain. Clin Exp Pharmacol Physiol 22(6–7):397–403
Christie MJ (1991) Mechanisms of opioid actions on neurons of the locus coeruleus. Prog Brain Res 88:197–205
Van Bockstaele EJ, Colago EE, Cheng P, Moriwaki A, Uhl GR, Pickel VM (1996) Ultrastructural evidence for prominent distribution of the mu-opioid receptor at extrasynaptic sites on noradrenergic dendrites in the rat nucleus locus coeruleus. J Neurosci 16(16):5037–5048
Berridge CW, Waterhouse BD (2003) The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 42(1):33–84. https://doi.org/10.1016/S0165-0173(03)00143-7
Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, Deisseroth K, de Lecea L (2010) Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci 13(12):1526–1533. https://doi.org/10.1038/nn.2682
Llorca-Torralba M, Borges G, Neto F, Mico JA, Berrocoso E (2016) Noradrenergic locus coeruleus pathways in pain modulation. Neuroscience 338:93–113. https://doi.org/10.1016/j.neuroscience.2016.05.057
McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP, Bruchas MR (2015) CRH engagement of the locus coeruleus noradrenergic system mediates stress-induced anxiety. Neuron 87(3):605–620. https://doi.org/10.1016/j.neuron.2015.07.002
Snyder K, Wang WW, Han R, McFadden K, Valentino RJ (2012) Corticotropin-releasing factor in the norepinephrine nucleus, locus coeruleus, facilitates behavioral flexibility. Neuropsychopharmacology 37(2):520–530. https://doi.org/10.1038/npp.2011.218
Tervo DGR, Proskurin M, Manakov M, Kabra M, Vollmer A, Branson K, Karpova AY (2014) Behavioral variability through stochastic choice and its gating by anterior cingulate cortex. Cell 159(1):21–32. https://doi.org/10.1016/j.cell.2014.08.037
Valentino RJ, Van Bockstaele E (2008) Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur J Pharmacol 583(2–3):194–203. https://doi.org/10.1016/j.ejphar.2007.11.062
Vazey EM, Aston-Jones G (2014) Designer receptor manipulations reveal a role of the locus coeruleus noradrenergic system in isoflurane general anesthesia. Proc Natl Acad Sci U S A 111(10):3859–3864. https://doi.org/10.1073/pnas.1310025111
Millan MJ (2002) Descending control of pain. Prog Neurobiol 66(6):355–474
Pertovaara A (2006) Noradrenergic pain modulation. Prog Neurobiol 80(2):53–83. https://doi.org/10.1016/j.pneurobio.2006.08.001
Hirschberg S, Li Y, Randall A, Kremer EJ, Pickering AE (2017) Functional dichotomy in spinal- vs prefrontal-projecting locus coeruleus modules splits descending noradrenergic analgesia from ascending aversion and anxiety in rats. eLife 6:e29808. https://doi.org/10.7554/eLife.29808
Hughes SW, Hickey L, Hulse RP, Lumb BM, Pickering AE (2013) Endogenous analgesic action of the pontospinal noradrenergic system spatially restricts and temporally delays the progression of neuropathic pain following tibial nerve injury. Pain 154(9):1680–1690. https://doi.org/10.1016/j.pain.2013.05.010
Llorca-Torralba M, Mico JA, Berrocoso E (2018) Behavioral effects of combined morphine and MK-801 administration to the locus coeruleus of a rat neuropathic pain model. Prog Neuropsychopharmacol Biol Psychiatry 84 (Pt A) 84:257–266. https://doi.org/10.1016/j.pnpbp.2018.03.007
Van Bockstaele EJ, Reyes BA, Valentino RJ (2010) The locus coeruleus: a key nucleus where stress and opioids intersect to mediate vulnerability to opiate abuse. Brain Res 1314:162–174. https://doi.org/10.1016/j.brainres.2009.09.036
Mazei-Robison MS, Nestler EJ (2012) Opiate-induced molecular and cellular plasticity of ventral tegmental area and locus coeruleus catecholamine neurons. Cold Spring Harb Perspect Med 2(7):a012070. https://doi.org/10.1101/cshperspect.a012070
Rasmussen K, Beitner-Johnson DB, Krystal JH, Aghajanian GK, Nestler EJ (1990) Opiate withdrawal and the rat locus coeruleus: behavioral, electrophysiological, and biochemical correlates. J Neurosci 10(7):2308–2317
Shaw-Lutchman TZ, Barrot M, Wallace T, Gilden L, Zachariou V, Impey S, Duman RS, Storm D et al (2002) Regional and cellular mapping of cAMP response element-mediated transcription during naltrexone-precipitated morphine withdrawal. J Neurosci 22(9):3663–3672 doi:20026223
Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33(1):87–107. https://doi.org/10.1016/0304-3959(88)90209-6
Berrocoso E, De Benito MD, Mico JA (2007) Role of serotonin 5-HT1A and opioid receptors in the antiallodynic effect of tramadol in the chronic constriction injury model of neuropathic pain in rats. Psychopharmacology 193(1):97–105. https://doi.org/10.1007/s00213-007-0761-8
Berrocoso E, Mico JA, Vitton O, Ladure P, Newman-Tancredi A, Depoortere R, Bardin L (2011) Evaluation of milnacipran, in comparison with amitriptyline, on cold and mechanical allodynia in a rat model of neuropathic pain. Eur J Pharmacol 655(1–3):46–51. https://doi.org/10.1016/j.ejphar.2011.01.022
Randall LO, Selitto JJ (1957) A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther 111(4):409–419
Detke MJ, Rickels M, Lucki I (1995) Active behaviors in the rat forced swimming test differentially produced by serotonergic and noradrenergic antidepressants. Psychopharmacology 121(1):66–72
Pineda J, Torrecilla M, Martin-Ruiz R, Ugedo L (1998) Attenuation of withdrawal-induced hyperactivity of locus coeruleus neurones by inhibitors of nitric oxide synthase in morphine-dependent rats. Neuropharmacology 37(6):759–767
Alba-Delgado C, Borges G, Sanchez-Blazquez P, Ortega JE, Horrillo I, Mico JA, Meana JJ, Neto F et al (2012) The function of alpha-2-adrenoceptors in the rat locus coeruleus is preserved in the chronic constriction injury model of neuropathic pain. Psychopharmacology 221(1):53–65. https://doi.org/10.1007/s00213-011-2542-7
Berrocoso E, Mico JA, Ugedo L (2006) In vivo effect of tramadol on locus coeruleus neurons is mediated by alpha2-adrenoceptors and modulated by serotonin. Neuropharmacology 51(1):146–153. https://doi.org/10.1016/j.neuropharm.2006.03.013
Cedarbaum JM, Aghajanian GK (1976) Noradrenergic neurons of the locus coeruleus: inhibition by epinephrine and activation by the alpha-antagonist piperoxane. Brain Res 112(2):413–419
Hirata H, Aston-Jones G (1994) A novel long-latency response of locus coeruleus neurons to noxious stimuli: mediation by peripheral C-fibers. J Neurophysiol 71(5):1752–1761. https://doi.org/10.1152/jn.1994.71.5.1752
Grace AA, Bunney BS (1984) The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci 4(11):2877–2890
Bruzos-Cidon C, Llamosas N, Ugedo L, Torrecilla M (2015) Dysfunctional inhibitory mechanisms in locus coeruleus neurons of the Wistar Kyoto rat. Int J Neuropsychopharmacol 18(7):pyu122. https://doi.org/10.1093/ijnp/pyu122
Quillinan N, Lau EK, Virk M, von Zastrow M, Williams JT (2011) Recovery from mu-opioid receptor desensitization after chronic treatment with morphine and methadone. J Neurosci 31(12):4434–4443. https://doi.org/10.1523/JNEUROSCI.4874-10.2011
Sim LJ, Selley DE, Childers SR (1995) In vitro autoradiography of receptor-activated G proteins in rat brain by agonist-stimulated guanylyl 5′-[gamma-[35S]thio]-triphosphate binding. Proc Natl Acad Sci U S A 92(16):7242–7246
Valdizan EM, Diaz A, Pilar-Cuellar F, Lantero A, Mostany R, Villar AV, Laorden ML, Hurle MA (2012) Chronic treatment with the opioid antagonist naltrexone favours the coupling of spinal cord mu-opioid receptors to Galphaz protein subunits. Neuropharmacology 62(2):757–764. https://doi.org/10.1016/j.neuropharm.2011.08.029
Bravo L, Mico JA, Rey-Brea R, Perez-Nievas B, Leza JC, Berrocoso E (2012) Depressive-like states heighten the aversion to painful stimuli in a rat model of comorbid chronic pain and depression. Anesthesiology 117(3):613–625. https://doi.org/10.1097/ALN.0b013e3182657b3e
Cambi F, Fung B, Chikaraishi D (1989) 5′ flanking DNA sequences direct cell-specific expression of rat tyrosine hydroxylase. J Neurochem 53(5):1656–1659. https://doi.org/10.1111/j.1471-4159.1989.tb08567.x
Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ et al (2013) Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol Rev 65(1):223–254. https://doi.org/10.1124/pr.112.005942
Fiorillo CD, Williams JT (1996) Opioid desensitization: Interactions with G-protein-coupled receptors in the locus coeruleus. J Neurosci 16(4):1479–1485
Duman RS, Tallman JF, Nestler EJ (1988) Acute and chronic opiate-regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J Pharmacol Exp Ther 246(3):1033–1039
Mostany R, Diaz A, Valdizan EM, Rodriguez-Munoz M, Garzon J, Hurle MA (2008) Supersensitivity to mu-opioid receptor-mediated inhibition of the adenylyl cyclase pathway involves pertussis toxin-resistant Galpha protein subunits. Neuropharmacology 54(6):989–997. https://doi.org/10.1016/j.neuropharm.2008.02.004
Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M (1999) Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron 23(4):737–746
Beckmann AM, Matsumoto I, Wilce PA (1995) Immediate early gene expression during morphine withdrawal. Neuropharmacology 34(9):1183–1189. https://doi.org/10.1016/0028-3908(95)00089-O
Georges F, Stinus L, Le Moine C (2000) Mapping of c-fos gene expression in the brain during morphine dependence and precipitated withdrawal, and phenotypic identification of the striatal neurons involved. Eur J Neurosci 12(12):4475–4486
Martin TJ, Kim SA, Buechler NL, Porreca F, Eisenach JC (2007) Opioid self-administration in the nerve-injured rat: relevance of antiallodynic effects to drug consumption and effects of intrathecal analgesics. Anesthesiology 106(2):312–322
Ginty DD, Bonni A, Greenberg ME (1994) Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell 77(5):713–725
Guitart X, Thompson MA, Mirante CK, Greenberg ME, Nestler EJ (1992) Regulation of cyclic AMP response element-binding protein (CREB) phosphorylation by acute and chronic morphine in the rat locus coeruleus. J Neurochem 58(3):1168–1171
Arttamangkul S, Quillinan N, Low MJ, von Zastrow M, Pintar J, Williams JT (2008) Differential activation and trafficking of micro-opioid receptors in brain slices. Mol Pharmacol 74(4):972–979. https://doi.org/10.1124/mol.108.048512
Bailey CP, Couch D, Johnson E, Griffiths K, Kelly E, Henderson G (2003) Mu-opioid receptor desensitization in mature rat neurons: lack of interaction between DAMGO and morphine. J Neurosci 23(33):10515–10520. https://doi.org/10.1523/JNEUROSCI.23-33-10515.2003
Virk MS, Williams JT (2008) Agonist-specific regulation of mu-opioid receptor desensitization and recovery from desensitization. Mol Pharmacol 73(4):1301–1308. https://doi.org/10.1124/mol.107.042952
Bruehl S, Chung OY, Burns JW, Diedrich L (2007) Trait anger expressiveness and pain-induced beta-endorphin release: support for the opioid dysfunction hypothesis. Pain 130(3):208–215. https://doi.org/10.1016/j.pain.2006.11.013
Zangen A, Herzberg U, Vogel Z, Yadid G (1998) Nociceptive stimulus induces release of endogenous beta-endorphin in the rat brain. Neuroscience 85(3):659–662
Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, Meyer CR, Koeppe RA et al (2001) Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science 293(5528):311–315. https://doi.org/10.1126/science.1060952
Valentino RJ, Van Bockstaele E (2015) Endogenous opioids: the downside of opposing stress. Neurobiol Stress 1:23–32. https://doi.org/10.1016/j.ynstr.2014.09.006
Jongeling AC, Johns ME, Murphy AZ, Hammond DL (2009) Persistent inflammatory pain decreases the antinociceptive effects of the mu opioid receptor agonist DAMGO in the locus coeruleus of male rats. Neuropharmacology 56(6–7):1017–1026. https://doi.org/10.1016/j.neuropharm.2009.02.005
Kimura M, Suto T, Morado-Urbina CE, Peters CM, Eisenach JC, Hayashida K (2015) Impaired pain-evoked analgesia after nerve injury in rats reflects altered glutamate regulation in the locus coeruleus. Anesthesiology 123(4):899–908. https://doi.org/10.1097/ALN.0000000000000796
Rohampour K, Azizi H, Fathollahi Y, Semnanian S (2017) Peripheral nerve injury potentiates excitatory synaptic transmission in locus coeruleus neurons. Brain Res Bull 130:112–117. https://doi.org/10.1016/j.brainresbull.2017.01.012
Emery PC, Wilson KG, Kowal J (2014) Major depressive disorder and sleep disturbance in patients with chronic pain. Pain Res Manag 19(1):35–41
Crain SM, Shen KF (1996) Modulatory effects of Gs-coupled excitatory opioid receptor functions on opioid analgesia, tolerance, and dependence. Neurochem Res 21(11):1347–1351
Cruciani RA, Dvorkin B, Morris SA, Crain SM, Makman MH (1993) Direct coupling of opioid receptors to both stimulatory and inhibitory guanine nucleotide-binding proteins in F-11 neuroblastoma-sensory neuron hybrid cells. Proc Natl Acad Sci U S A 90(7):3019–3023
Liu JG, Anand KJ (2001) Protein kinases modulate the cellular adaptations associated with opioid tolerance and dependence. Brain Res Brain Res Rev 38(1–2):1–19
Florin-Lechner SM, Druhan JP, Aston-Jones G, Valentino RJ (1996) Enhanced norepinephrine release in prefrontal cortex with burst stimulation of the locus coeruleus. Brain Res 742(1–2):89–97
Schwarz LA, Luo L (2015) Organization of the locus coeruleus-norepinephrine system. Curr Biol 25(21):R1051–R1056. https://doi.org/10.1016/j.cub.2015.09.039
Uematsu A, Tan BZ, Ycu EA, Cuevas JS, Koivumaa J, Junyent F, Kremer EJ, Witten IB et al (2017) Modular organization of the brainstem noradrenaline system coordinates opposing learning states. Nat Neurosci 20(11):1602–1611. https://doi.org/10.1038/nn.4642
Acknowledgments
We are very grateful to Ms. Raquel Rey-Brea, Mr. José Antonio García Partida, Mr. Santiago Muñoz, and Ms. Paula Reyes Perez for their excellent technical assistance. This work is dedicated in memory of Elsa Valdizán, PhD.
Funding and Disclosure
This work was supported by Spain’s Ministerio de Economía y Competitividad, co-financed by “Fondo Europeo de Desarrollo Regional” FEDER “A way to build Europe” (SAF2015-68647-R, SAF2011-25020); the “Centro de Investigación Biomédica en Red de Salud Mental-CIBERSAM” (Spain, G18); the “Consejería de Economía, Innovación, Ciencia y Empleo de la Junta de Andalucía” (CTS-510, CTS-7748); the Basque Government (IT 747-13); “Fundación Progreso y Salud de la Junta de Andalucía (PI-0080-2017); Fundación Española del Dolor (PI2015-FED-007); and a 2015 NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation (NARSAD 23982).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Animal handling and all the procedures were carried out in accordance with the guidelines of the European Commission’s directive (2010/63/EC) and Spanish Law (RD 53/2013) regulating animal research. Furthermore, all the experimental protocols were approved by the Committee for Animal Experimentation at the University of Cadiz (Spain).
Conflict of Interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Llorca-Torralba, M., Pilar-Cuéllar, F., Bravo, L. et al. Opioid Activity in the Locus Coeruleus Is Modulated by Chronic Neuropathic Pain. Mol Neurobiol 56, 4135–4150 (2019). https://doi.org/10.1007/s12035-018-1361-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1361-9