
Abstract
The term proteostasis reflects the fine-tuned balance of cellular protein levels, mediated through a vast network of biochemical pathways. This requires the regulated control of protein folding, post-translational modification, and protein degradation. Due to the complex interactions and intersection of proteostasis pathways, exposure to stress conditions may lead to a disruption of the entire network. Incorrect protein folding and/or modifications during protein synthesis results in inactive or toxic proteins, which may overload degradation mechanisms. Further, a disruption of autophagy and the endoplasmic reticulum degradation pathway may result in additional cellular stress which could ultimately lead to cell death. Neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and Amyotrophic Lateral Sclerosis all share common risk factors such as oxidative stress, aging, environmental stress, and protein dysfunction; all of which alter cellular proteostasis. The differing pathologies observed in neurodegenerative diseases are determined by factors such as location-specific neuronal death, source of protein dysfunction, and the cell’s ability to counter proteotoxicity. In this review, we discuss how the disruption in cellular proteostasis contributes to the onset and progression of neurodegenerative diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative disordes, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), Huntington’s disease (HD), Amyotrophic Lateral Sclerosis (ALS), and prion diseases (pD), are caused by neuronal dysfunction and death. Although neurodegenerative diseases share common risk factors, such as oxidative stress, environmental factors, aging, and protein dysfunction, these manifest distinctly in each disease and produce unique pathologies [1].
Protein misfolding and aggregation is a common characteristic in many neurodegenerative diseases, therefore maintaining intracellular protein homeostasis (proteostasis), by balancing protein folding and misfolding, is paramount in protecting the functionality of the proteome [2]. Although misfolded proteins are generally inactive, the accumulation of these misfolded proteins can cause stress responses in cells and organelles. Indeed, the endoplasmic reticulum (ER) is a key organelle in maintaining proteostasis and the unfolded protein response (UPR) is triggered in response to protein accumulation and ER stress. The UPR promotes protein folding and reduces ER protein levels by proteosomal degradation, translation mitigation, and autophagy [3]. Autophagy also plays an essential role in proteostasis because of its ability to degrade protein aggregates that cannot be processed by the proteasome [4]. For example, the autophagosome can degrade and recycle entire organelles in an effort to promote cell survival. Protein degradation by autophagy and the proteasome utilize ubiquitination to recruit target proteins [5]. Ubiquitination along with other covalent attachments, such as phosphorylation, SUMOylation, and oxidation, regulate normal proteome function. The Post-Translational Modifications (PTMs) of proteins have also been shown to influence protein aggregation in many neurodegenerative diseases [6]. The accumulation of disease-associated proteins, resulting in protein aggregation and/or inclusions, can lead to proteotoxicity which is especially problematic in post-mitotic neurons highlighting the clear relationship between neurodegeneration and age [7].
Mitochondrial proteostasis is also critical for cell survival and its dysfunction can lead to the accumulation of reactive oxygen species (ROS), which in turn can be disruptive to overall cellular proteostasis. Indeed, a disruption of mitochondrial proteostasis may lead to the irreversible induction of apoptosis, a highly regulated set of pathways leading to cell death. Although cell-specific, stress-induced apoptosis is designed to avoid the damaging of adjacent cells, it is detrimental to the nervous system as differentiated neurons cannot be reproduced. Although neuronal loss cannot be fully attributed to apoptosis, it is a common culprit in many neurodegenerative disorders.
In this review, we summarize important factors that affects proteostasis and further describe how proteostasis dysfunction ultimately affects neurodegeneration.
Post-Translational Modifications
PTMs play a pivotal role in protein structure and function where they increase the diversity and complexity of protein function by altering protein tertiary structures. PTMs include, but are not limited to, phosphorylation, acetylation, glycosylation, ubiquitination, NEDDylation, and SUMOylation. They play a crucial role in many cellular functions and for this reason need to be tightly regulated. As a result, disruption in PTMs can lead to a cascade of pathological events that disrupt cellular proteostasis [8]. In neurodegenerative diseases, PTM pathways are often compromised which can have toxic effects on cells, and therefore cause neuronal cell death [9]. In the following sections we will discuss the link between aberrant PTMs of proteins, and their downstream effects with respect to specific neurodegenerative diseases.
Parkinson’s Disease
The formation of Lewy bodies (LBs) within dopaminergic neurons represent one of the hallmarks of PD. LBs are large protein deposits enriched in α-synuclein, a natively unfolded protein, which is found in aggregated states in PD neurons. α-Synuclein is a soluble 14-kDa protein and its primary structure contains five phosphorylation sites (Ser87, Ser129, Tyr125, Tyr133, and Tyr 136) [10]. Under normal physiological conditions, phosphorylation at Ser129 occurs at a frequency of approximately 4%; however, studies have shown that approximately 90% of α-synuclein is phosphorylated at Ser129 in LBs [11]. This finding has propelled the hypothesis that the hyperphosphorylation of Ser129 has a strong connection with the formation of aggregated α-synuclein. Phosphorylation of Ser129 has also been shown to play a cytotoxic role by facilitating trafficking between the cytosol and nucleus [12]. In the nucleus, the phospho-Ser129 α-synuclein variant can interact with histones masking them from histone deacetylases thereby inducing neurotoxicity. Although the phosphorylation status of α-synuclein on Ser87, Tyr125, Tyr133, and Tyr 136 is also altered in PD patients, these PTMs do not seem to contribute to proteins aggregation [13].
Histone acetylation is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs add an acetyl group to histones and promote transcription. By contrast, HDACs remove the acetyl group. The link between acetylation and the pathogenesis of PD has been shown with respect to increased levels of histone acetylation. A study was conducted to investigate environmental effects on PD pathogenesis by treating SHSY neuroblastoma cells with 1-methy-4-phenylpyridinium (MPP+), and it was found that MPP+-treated cells exhibited increased histone acetylation [14]. This increase in histone acetylation also showed a decrease in the levels of HDACs alluding to the neuroprotective effects of HDACs. However, there have been conflicting studies that indicate that HDAC inhibitors attribute to neuroprotective effects in cellular models and this has been explored in clinical trials with the aim of mitigating α-synuclein accumulation [15].
The degradation of proteins is essential for maintaining cellular proteostasis. Ubiquitination is an enzymatically catalized PTM that targets and marks proteins for degradation by the attachment of ubiquitin (Ub) [16]. Ubiquitination consists of three main enzyme classes including, Ub activating enzyme (E1), Ub conjugating enzyme (E2), and Ub ligase (E3) [17]. Proteins tagged for degradation can be mono-ubiquitinated or poly-ubiqutinated where the latter targets proteins to the 26S proteasome for degradation. Parkin, a protein associated with PD, is an E3 ubiquitin ligase that will tag misfolded proteins for ubiquitination. Further, PINK1 (PTEN-induced putative kinase 1) is a mitochondrial Ser/Thr kinase which activates and recruits Parkin by phosphorylation. Studies have shown that PINK1 phosphorylates ubiquitin at serine 65, which binds to Parkin and recruits Parkin to the mitochondria so that it can ubiquitinate misfolded proteins, which ultimately leads to proteasomal degradation of the proteins [18, 19]. Parkin and PINK1 mutations and dysfunction are linked to early-onset familial PD, as well as sporadic PD. This dysfunction has been shown to cause excessive ROS production in mitochondria leading to inappropriate cellular proteostasis. Studies have also shown that the protein kinase c-Abl can downregulate Parkin activity by phosphorylating Tyr143, thus affecting its E3 ubiquitin ligase activity. This inactivation of Parkin by c-Abl has also been reported in post mortem PD brains [20]. Parkin has also been shown to interact with and ubiquitinate p62, a major shuttle protein and regulator of protein quality control [21]. p62 plays an important role in transporting polyubiquitinated proteins for proteasomal degradation [4] and in response to mitochondrial stress Parkin tags p62 for degradation. When this balance is interrupted, p62 can cause accumulation of aggregated proteins and disturb mitochondrial dynamics and cellular homeostasis.
Phosphorylation, nitration, and SUMOylation of α-synuclein are altered in PD and may serve as potential biomarkers for the disease [22]. Indeed, Tyr 125 phosphorylation and Y39 nitration are significantly increased in PD patients as compared to healthy individuals, whereas SUMO-1 is significantly reduced in PD patients [22]. Interestingly, SUMOylation reduces α-synuclein aggregation in vitro and in vivo, which may explain the decreased SUMO-1 levels in PD patients [23]. Although more analysis is required, these findings suggest that alterations in PTMs may represent promising diagnostic and prognostic biomarkers for PD.
Alzheimer’s Disease
AD is characterized by the presence of neurofibrillary tangles (NFTs), which are aggregates of abnormally phosphorylated tau protein as well as amyloid β-peptide plaques. Tau is a microtubule-associated protein mainly expressed in neurons and its main function is to stabilize the neuronal cytoskeleton [24]. Tau exists as six isoforms, each with a different number of N-terminal inserts and C-terminal repeats. Phosphorylation is the most common PTM of the tau protein, which contains 85 potential phosphorylation sites. Of these, 45 are serine (53%), 35 are threonine (41%), and 5 are tyrosine (6%). Hyperphosphorylation of tau occurs at several sites throughout the protein, and this precedes the formation of NFTs. This hyperphosphorylation disrupts tau’s ability to efficiently bind to microtubules, which can result in neurotoxicity [25]. AD-associated kinase glycogen synthase kinase-3β (GSK3β) phosphorylates tau at T231. Reduction of tau phosphorylation by GSK3β inhibition in mice has been shown to block the formation of NFTs [26]. The activity of phosphatases, specifically protein phosphatase 2A (PP2A), is decreased in AD patients due to the upregulation of PP2A, I1 and I2 inhibitors [27]. PP2A is a major phosphatase and accounts for more than 70% of cellular phosphatase activity [28]. This cascade of events in the dysregulation of phosphorylation contributes greatly to the pathogenesis of AD.
Aggregation of tau and amyloid beta (Aβ) is mediated by ubiquitination of these proteins as well as other proteins. The formation of Aβ plaques occurs by the cleavage of the amyloid precursor protein (APP) in various compartments throughout the cell. APP is produced in the ER and has been shown to interact with HRD1, an E3 ligase which clears newly synthesized proteins in the ER. In patients with AD, a decrease in HRD1 expression, which induces ER stress, leads to accumulation of APP and Aβ and subsequent apoptosis [29]. APP is then internalized into the trans-golgi network where it is cleaved, producing Aβ plaques [30]. These Aβ plaques have been suggested to stimulate GSK3β which will subsequently result in the hyperphosphorylation of tau [31].
SUMOylation is another PTM that has been linked to AD pathogenesis. The small ubiquitin-like modifier is similar to ubiquitin, but binds covalently to lysine residues on target proteins and does not overlap with the ubiquitin pathway. In comparison to ubiquitination, SUMOylation requires the action of E1, E2, and E3 to attach SUMO to the target proteins, and these target proteins can be altered by the SUMOylation pathway. SUMOylation has been recognized as playing an important role in oxidative stress [32], and its dysregulation is associated with neurodegenerative diseases. Reports have shown that both SUMO-1 and SUMO-2 covalently attach to lysines 587 and 595 of APP respectively, and this regulates levels of Aβ aggregates [25].
APP is also modulated by NEDDylation, another ubiquitin-like pathway, where lysine residues of APP are modified [33]. These sites are important because they are within the Binding protein-1 (BP-1) sites of APP, and BP-1 has been shown to be a member of the NEDDylation-conjugation pathway [34]. The downregulation of BP-1 inhibits NEDDylation, which subsequently results in accumulation of APP and APP β-secretase fragment; this suggests that NEDDylation of APP can also signal for the degradation of APP [35, 36].
Recent data also suggests an important role of ER calcium homeostasis in AD [37], specifically, dysfunction in the ryanodine receptors (RyR) in the ER causing leakage of Ca2+ which has been reported in AD. Hyperphosphorylation and hypernitrosylation of the RyR complex results in calstabin depletion and leakage of Ca2+ from the RyR channel [38, 39]. It is plausible to suggest that targeting these “leaky” RyR channels may represent a possible future therapeutic approach in the treatment of AD.
Amyotrophic Lateral Sclerosis
ALS, also known as Lou Gehrig’s disease, is a fatal neurodegenerative disease that affects motor neurons. ALS pathogenesis is characterized by the loss of motor neurons in the brain and spinal cord, which can lead to progressive paralysis and ultimately death [40]. Key factors that contribute to the pathogenesis of ALS are mutations in superoxide dismutase (SOD1) and the dysregulation of the astroglial glutamate transporter, EAAT2 [41] [42]. Ubiquitination, SUMOylation, and acetylation of SOD1 and EAAT2 have been shown to contribute to late onset ALS [43]. Most ALS cases are sporadic, while approximately 10% are familial. The combination of rapid deterioration of motor neurons accompanied by short life-span of patients makes it difficult to perform longitudinal studies with the ultimate aim of developing therapeutic strategies.
In familial cases of ALS, one of the genes that is mutated is UBQLN2, which is located on the X chromosome and functions in proteostasis [44]. Indeed, it appears that UBQLN2 mutations interfere with proteasomal degradation [44]. Mutation in the UBQLN2 gene, encoding ubiquilin-2 protein, results in less efficient binding of ubiquitinated proteins to the proteasome [45], leading to protein accumulation. SOD1 plays an important role in eliminating free superoxide species and SOD1 has been linked to both familial ALS where the SOD1 gene is mutated, and sporadic ALS where PTMs seem to play a role in the late onset of the disease [46]. The ubiquitination of SOD1 occurs via the E3 ligase E6-AP and mouse studies have shown decreased levels of E6-AP and increased SOD1 before the onset of ALS [47]. Recent studies have also suggested that the acetylation of SOD1 at lysine 123 (K123) [43], a region for copper binding and protein folding [48], may play a role in ALS; however, the relationship between SOD1 acetylation dysregulation and ALS is not fully understood.
SUMOylation of SOD1 at lysine 75 occurs via SUMO-1, SUMO-2, and SUMO-3 [49]. EAAT2, which is cleaved by caspase 3 and accumulates in spinal cord astrocytes [42], is also SUMOylated. Studies show that SUMO-3 modification of SOD1 mutants contributes to the aggregate formation of SOD1, which leads to ALS pathogenesis. The resulting inability of SOD1 to remove ROS causes oxidative stress to cells and affects cellular proteostasis, and elevated oxidative damage is observed in ALS patients [50]. Fei and colleagues have further shown that SUMO-1 can SUMOylate SOD1 at Lys9, and that this modification increases the susceptibility of SOD1 aggregation.
EAAT2 is a glutamate transporter that is expressed in the spinal cord and its expression is decreased in ALS patients [51]. The SUMOylation of EAAT2 occurs by SUMO-1 on its C-terminal intracellular domain. The accumulation of these SUMOylated domains has been shown in astrocytes at the end-stage of ALS and a generic dysfunction of astrocytic physiology, leading to motor neuronal loss, has been suggested [52].
Fused in sarcoma (FUS), a protein primarily involved in RNA metabolism, has recently gained attention because of its connection with ALS. Mislocalization and aggregation of pathological FUS occurs in ALS patients [53], and has been found in aggregates along with SOD1 [54]. Arising data suggests that PTMs may play a role in the localization of FUS, primarily serine/threonine phosphorylation [55]. It is not fully understood how the PTMs affect localization and aggregation of FUS due to the extensive and complex modification sites.
PTMs such as phosphorylation, acetylation, and ubiquitination are important biological processes involved in protein-protein interactions, protein degradation, and protein localization, respectively. Therefore, it is not surprising that dysfunction in these processes play a role in neurodegenerative diseases. Many of the neurodegenerative diseases share commonalities in terms of PTMs. For example, phosphorylation of α-synuclein at ser129 has been linked to its neurotoxicity in PD while the phosphorylation of tau increases AD pathogenesis. Ubiquitination is also a major contributor in both familial and sporadic cases of PD and in ALS pathogenesis mutations in UBQLN2 cause less efficient binding of ubiquitin to its substrates. While commonalities exist, much of the dysfunction caused by PTMs manifests distinctly in different neurodegenerative diseases.
Aggregation and Proteotoxicity
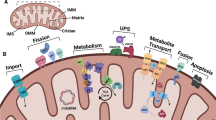
The aggregation of disease-specific proteins is a hallmark for several neurodegenerative disorders. The presence of pathological aggregates, often called “bodies” or inclusions, classify PD, AD, HD, ALS, and pD as proteopathies (Fig. 1 ). Pathological aggregates result from the accumulation of misfolded proteins where the protein and type of neuron affected determine the disease.

Altered proteostatic levels and protein conformations lead to hallmark pathological neurodegenerative pathways. Damaged and excessive protein directly causes ER stress and aggregation as well as mitochondrial dysfunction and oxidative stress indirectly. Additionally, feedback loops (I, II) highlight the destructive nature of damaged proteins propagating ER stress and mitochondrial dysfunction leading to loss of functionality further altering proteostasis. The cellular defense response to these pathways includes proteosome degradation, autophagy/mitophagy, and apoptosis
Although protein aggregates are a pathological feature of proteopathies, it is largely unknown whether they are toxic or neuroprotective. It has been postulated that the collection of misfolded protein into aggregates represents a cellular defense mechanism to combat proteotoxicity and maintain proteostasis [56]. These inclusions, however, have been deemed proteotoxic either through loss-of-function, where aggregated proteins are unable to carry out their normal physiological function, and/or by gain-of-function, acquiring new functions that contributes to cell death [57].
Protein aggregates in PD, AD, HD, and pD are formed as beta sheet-rich insoluble structures called amyloid fibrils. Although it is known that aggregation is a result of protein misfolding, the initiation and formation of amyloid fibrils are not well understood. Studying the formation of amyloid fibrils, their gain-of-function, loss-of-function, and neuroprotectivity are critical to the advancement of understanding neurodegenerative diseases and developing future therapeutics.
Parkinson’s Disease
In PD, LBs are enriched in α-synuclein and aggregation has been correlated with inappropriate PTMs, protein accumulation, and misfolding. In addition to α-synuclein, LBs have been shown to harbor a plethora of other proteins including α-synuclein binding proteins, the PD-associated proteins DJ-1, LRRK2, parkin, PINK-1, GBA, and synphilin-1, in addition to components of the ubiquitin-proteosome pathway. The complex composition of LBs implies that they acquire cytotoxicity by attracting proteins and thereby disrupting proteostasis. Interestingly, PD progression is influenced by neuron-to-neuron transmission of α-synuclein fibrils or non-fibrillar multimers through a prion-like mechanism, which can presumptively skip or accelerate the early stages of LB formation [58, 59].
α-Synuclein proteotoxicity has traditionally been attributed to LB formation in neurons. However, more recently studies have emerged suggesting that smaller, non-fibrillar, multimeric species of α-synuclein are more significant to PD pathogenesis [60, 61]. For example, there is evidence suggesting that α-synuclein multimers interact and damage cell membranes and further that the toxicity of α-synuclein multimers is associated with the inhibition of the axonal transport machinery [62]. If the aggregation of α-synuclein multimers reduces their toxicity, LB formation could be viewed as a neuroprotective mechanism rather neurotoxic. However, as both α-synuclein aggregation and LB formation represent temporal processes, it is likely that the consequences of protein aggregation in PD is multidimensional.
Although the aggregation of α-synuclein is generally viewed as a toxic mechanism, its loss-of-function during aggregation may have detrimental effects on neurons. While not fully understood, the function of α-synuclein has been shown to include suppression of apoptosis, regulation of glucose, modulation of calmodulin activity, promotion of SNARE assembly, chaperone activity, polyunsaturated fatty acid control, antioxidation, neuronal differentiation, and regulation of dopamine synthesis, all of which may be compromised in response to aggregation [63].
Alzheimer’s Disease
AD is associated with two forms of aggregation: extracellular Aβ plaques and intracellular neurofibrillary tangles composed of the tau protein. AD is considered both an amyloidopathy and a tauopathy, and although early research implicated Aβ aggregation as the primary cause of AD, it has been shown that the formation of neurofibrillary tangles is highly correlated with the progression of the disease.
Plaque formation has been attributed to mutations in amyloid precursor metabolic protein encoding genes, the ratio of two forms of Aβ, and Aβ accumulation. However, it is unclear whether AD pathogenesis is driven by non-fibrillar Aβ multimers or amyloid plaques [64, 65]. In AD patients, the proteostatic equilibrium of Aβ42 and Aβ40 is disrupted in the mesenchyme. As the Aβ42/Aβ40 ratio increases, proteotoxic gain-of-function of Aβ inhibits synaptic function and eventually becomes neurotoxic. The increased ratio also results in increased aggregation; however, non-toxic aggregates have been shown independent of AD, leading to the conclusion that fibril toxicity is caused by conformational changes. In fact, higher levels of Aβ40 aggregates have been shown to be neuroprotective. A plausible mechanism for this protection is the hoarding and inactivation of toxic Aβ42 into aggregates rich in Aβ40. The function of Aβ is not well understood, therefore, any loss-of-function due to aggregation has not been described.
Aβ has shown cytotoxicity with tau. Not only can tau dysfunction cause neurodegeneration independent of Aβ, supporting the hypothesis that it is downstream of plaque formation in AD pathogenesis, but it has been shown that soluble extracellular Aβ and soluble intracellular tau codependently cause synaptic dysfunction and neuronal death. Tau can also facilitate Aβ toxicity through a feedback loop [66].
Tau aggregation into NFTs or paired helical filaments (PHFs) might be neuroprotective, due to the toxic nature of tau being neutralized by the PHF structure [67]. PHFs gain-of-function inhibition of fast axonal transport indicates pathogenic proteotoxicity linked to AD [68]. Conversely, loss-of-function toxicity by PHFs, due to microtubule instability and cellular transport dysfunction, does not appear to be significant [69].
Huntington’s Disease
HD is associated with the aggregation of the huntingtin (HTT) protein to form inclusion bodies. HTT aggregation is due to a 5′ CAG repeat in the IT15 gene encoding huntingtin. Normal HTT contains 6–34 CAG (glutamine) repeats; however, HD is developed in individuals with at least 40 glutamine repeats (polyQ) [70]. The rate of development of HD and aggregation of HTT correlates with the amount of polyQ repeats. Inclusion bodies are independent of neurotoxicity and the accumulation of toxic misfolded HTT is found to be neuroprotective [71]. The physiological roles of HTT include, but are not limited to, vesicle trafficking, ciliogenesis, endocytosis, autophagy, transcription, and cell survival [72]. HD-like pathology can be reversed in mice by reducing HTT levels, implying a toxic gain-of-function form of the protein [73]; however, the various functions of HTT, especially in central nervous system development [74], suggest that a loss-of-function might contribute to HD.
Prion Diseases
Prion diseases are caused by extracellular toxic protein accumulation due to genetic mutations or environmental factors. α-Synuclein, tau, beta-amyloid, HTT, TAR DNA-binding protein, and SOD1 are considered prions due to their transmissible ability. The widely accepted pD model involves PrPSC, an infectious, abnormal, toxic protein. Physiologically normal human PrPC is indeed a substrate, which is converted into PrPSC upon infection. PrPSC is prone to β-sheet formation rather than the α-helix-rich PrPC, leading to the development of amyloid fibrils. It is debated whether the proteotoxicity is due to PrPC loss-of-function, resulting in anti-apoptotic activity, or amyloid fibril gain-of-function by blocking axonal transport, impairing synaptic function or apoptotic activity [75]. While not often suggested, aggregation of PrPSC might halt or delay the spread of infectious protein and conversion of PrPC, resulting in a neuroprotective effect.
Amyotrophic Lateral Sclerosis
ALS is associated with TAR DNA-binding protein (TDP-43) or SOD1 abnormalities and insoluble aggregation in neurons and glial cells. In ALS, TDP-43 most often forms disordered aggregates; however, skein-like inclusions, which share amyloid fibril characteristics, have been shown in a subset of ALS patients [76]. ALS pathology can be mimicked by both overexpression and partial knockout of TDP-43, suggesting both loss-of and gain-of-function toxicity [77]. Interestingly, the mechanics of TDP-43 loss-of-function (insufficient RNA splicing and transport) are similar to its gain-of-function toxicity (recruitment of nuclear TDP-43 to the cytoplasm resulting in improper localization). Mutations in SOD1 are linked to familial ALS, and WT and mutant SOD1 form amyloid fibrils of different conformations, which might vary in neurotoxicity [78]. SOD1 mutants exhibit a gain-of-toxic function when localized near mitochondria and loss-of-function neurotoxicity by insufficient defense against oxidative stress [79, 80]. These gain-of and loss-of-function characteristics underline the common challenge to maintain protein equilibrium and homeostasis in neurodegenerative diseases.
The aggregation phenomena in all five neurodegerative diseases described here result in beta sheet-rich amyloid fibrils. The formation of amyloid fibrils is an event which requires the formation of a nucleus, which seeds subsequent aggregation [81]. Recent research has identified low complexity regions (LCRs) of proteins as prion-like and aggregate-prone. LCRs are intrinsically disordered regions which, through weak intermolecular forces, are independently sufficient to produce phase separation into liquid droplets [82,83,84]. These droplets have been shown to act as a site of nucleation for fibril formation and conversion into solid-state aggregates [82]. The main protein used in these early experiments has been the ALS-associated protein FUS; however, it is expected that α-synuclein, mutant HTT, PrPSC, TDP-43, and Aβ will be included in future studies of LCR phase separation. This new field is generating much excitement as LCRs may represent a potential common, primary event in neurodegenerative disease pathology.
Endoplasmic Reticulum Stress
The ER is the center for protein quality control and proteostasis. Functions of ER include correct folding of newly synthesized peptides, degradation or chaperone-mediated refolding of misfolded proteins, and control of cellular calcium homeostasis. ER stress is caused by the accumulation of misfolded protein and in response the UPR pathway promotes correct folding through chaperones, proteosomal degradation by the ERAD pathway, antioxidation, and ER genesis. In cases where the stress is too extreme for adaptation, apoptosis is initiated (Fig. 2 ). ER stress and the UPR are major topics of study in relation to neurodegenerative diseases due to the common pathology of protein misfolding and its downstream effects. As misfolded proteins accumulate during neurodegeneration, the ER undergoes stress and initiates UPR, which eventually proves insufficient leading to proteotoxicity and aggregation. At baseline, the ER is capable of maintaining proteostasis in healthy cells with incidental misfolding events; however, the chronic protein misfolding in neurodegenerative diseases not only overruns the capability of the ER but promotes pathways which contribute to cytotoxicity [85].

Pathway of protein throughout the ER. Protein is synthesized in the rough ER by ribosomes. Native protein is predominantly exported, while some can aggregate. Misfolded protein aggregates are sent to proteasome for degradation. Misfolded protein and aggregates promote the UPR,which includes UPR associated gene regulation, ERAD pathway, chaperone response, and apoptosis all in an effort to mitigate the effects of toxic aggregates
Parkinson’s Disease
PD pathology induces ER stress and inhibits its response mechanism, exacerbating the effect. Accumulation of misfolded α-synuclein occurs in various cell compartments including the ER lumen [86]. α-Synuclein has been shown to inhibit UPR and ERAD resulting in apoptosis (Fig. 2 ) [87]. ERAD functions by exporting target proteins to the ubiquitin-proteosome system (UPS) in the cytosol and the ERAD E3-ligase, HRD1, is elevated in PD brain tissue [88]. α-Synuclein also inhibits the ERAD pathway, promoting pathogenesis, in a similar, parallel manner as the PD protein, parkin, inhibits the UPS. Rotenone, which induces PD conditions in vitro and in vivo, increases the phosphorylation of ER stress markers as well as the upregulation of UPR components and ER chaperones [89].
Alzheimer’s Disease
AD and ER stress have been linked by the presence of UPR components in patient-derived brain tissue [90]. Aβ42 activates AMPK, which inhibits the mTOR pathway, shown to promote AD [91, 92]. Interestingly, some UPR components regulate AD pathways directly. For example, the UPR transcription factor, XBP1, regulates gamma secretase complex components, cyclin-dependent kinase 5 (CDK5) as well as other APP processing factors [93]. Furthermore, the PERK-eIF2-alpha pathway directly regulates beta-secretase 1 (BACE1), resulting in increased processing of APP and the production of beta-amyloid [94]. Interestingly, deletion of eIF2 activators, PERK, GCN2, and PKR, in AD models reduce symptoms [95]. Indeed, a “vicious cycle” has been described in which ER stress is induced by Aβ42 which in turn promotes Aβ42 production by the UPR component, JNK3 [92].
Huntington’s Disease
HD pathology correlates with the upregulation of the UPR components BiP, CHOP, and XBP1 [96]; this occurs in response to impairment of ERAD by mHTT inactivating the ERAD proteins Npl4, Ufd1, p97, and gp78 [97, 98]. mHTT proteotoxicity overloads the UPS and results in the accumulation of misfolded proteins [99,100,101]. The connection between ER stress, XBP1, and autophagy has been investigated within the context of HD [102].
Prion Diseases
Prion diseases, as with other neurodegenerative diseases, is correlated with ER stress and upregulation of the UPR; however, the specific modes of action remain to be elucidated [103,104,105]. Inhibition of the PERK-eIF2-alpha UPR pathway by the eIF2-alpha phosphatase GADD34 and by PERK inhibitors results in neuroprotection and slowed disease progression while treatment with Salubrinal, which promotes the PERK-eIF2-alpha stress response, enhances pD progression [106, 107].
Interestingly, there are commonalities in how neurodegenerative diseases stress the ER and inhibit proteostasis. For instance, AD and HD are both associated with the upregulation of XBP1 while HD and pD are both associated with the upregulation of PERK. As these neurodegenerative diseases are clearly distinct, it is interesting to contemplate other molecular commonalities that may exist in neurodegeneration.
Autophagy and Mitophagy
Autophagy is a cellular process that involves the efficient and selective degradation of misfolded/aggregated proteins [108]. Autophagy is a lysosomal-mediated degradation pathway that is activated under stress conditions to degrade and eliminate damaged organelles and proteins (Fig. 3 ) [109]. This process begins with the formation of the autophagosome, which is a double-membrane vesicle that engulfs and degrades damaged cellular components [110]. The process of autophagy is driven by a group of autophagy-related proteins (ATGs) that are responsible for autophagosome formation, vesicle expansion, infusion with the lysosome, and cargo recruitment [111]. A complex of protein kinases initiates the signaling mechanism that controls the activation of autophagy, one of which is the mammalian target of rapamycin (mTOR). Rapamycin, a widely used molecule, is an inhibitor of mTOR and has been tested in terms of enhancing autophagy activity [112]. Defects in autophagy are associated with the pathogenesis of many neurodegenerative diseases and controlled modifications of autophagy may prove very useful in the development of possible therapeutic intervention strategies [113]. Quality control of the autophagic pathway is of great importance to the maintenance of cellular proteostasis due to compounding evidence that the alteration of various regulatory steps in autophagy may lead to global dysfunction in cells [114]. In neurodegenerative disorders, dysfunction in the autophagy pathway leads to accumulation of pathogenic proteins and damaged organelles (Fig. 3 ).

Neurodegenerative disorders alter specific steps in the autophagic pathway, which ultimately leads to neuronal cell death. The altered steps include reduced induction levels due to protein aggregation and defective mTOR inhibition; Defects in cargo recognition resulting in accumulation of toxic proteins; Mutation in VCP leads to inhibited transport of autophagic vesicle in ALS; Defects in lysosome/autophagasome fusion and acidification. All defective steps, leading to impairment of protein degradation by lysosomal degradation, ultimately leads to the release of cathepsin’s and apoptosis. Huntington’s disease (HD); Alzheimer’s disease (AD); Parkinson’s disease (PD); spino-cerebelar ataxia (SCA); spino-muscular atrophy (SMA); dementia with Lewy bodies (DLB); Amyotrophic Lateral Sclerosis (ALS); Valsin-containing protein (VCP)
In AD, the accumulation of damaged mitochondria in neurons occurs due to the translocation of misfolded proteins into the mitochondria [115]. This leads to disruption in the oxidative phosphorylation cycle triggering an autophagic response by the cell. This response, mitophagy, is a type of macroautophagy where damaged mitochondria are engulfed by the autophagosome and degraded. If this response is hindered, it can lead to accumulation of protein aggregates, specifically Aβ oligomers. Mitophagy can indeed protect cells from apoptosis by degrading damaged mitochondria [116].
Studies suggest that specific proteins involved in the production of the autophagosome are disrupted in early stages of sporadic AD [117]. Microtubule-associated light chain 3-ll (LC3-ll), along with multiple proteins, is involved in the early formation of the autophagosome. Upregulation of LC3-ll is seen in early stages of AD, which suggests that autophagy induction is an early response to AD progression; dysfunction in the induction could influence the pace of neuronal cell death [117]. The reduction of lysosomal activity, which can impair autophagy, is also observed in the pathogenesis of AD. Evidence suggests that decreased lysosomal acidification can be directly linked to mutations in vacuolar ATPase (v-ATPase), a proton pump responsible for lowering the pH in lysosomes, in patients with early-onset AD (Fig. 3 ) [118, 119]. The defective clearance of many AD related proteins is greatly emphasized by mutations of Presenilin 1 (PS1), a common cause of early-onset AD (Fig. 3 ) [120]. PS1 is a transmembrane protein and one of the four proteins in the gamma secretase complex. Mutations in PS1 result in the increase of Aβ deposits by cleavage of APP. Previous studies show that PS1 is required for lysosomal turnover and that deletion of PS1 causes a complete loss of autophagy [120].
Autophagy can also be inhibited. For example, the pathogenic A53T and A30P mutants of α-synuclein block the uptake of damaged proteins or substances by the lysosome [121]. This inhibition is one of the key aspects in the pathogenesis and progression of PD. Another example is illustrated by mitochondrial dysfunction where enlarged mitochondria do not get removed by mitophagy [122]. Many PD related proteins such as Parkin, PINK1, and Htra2 (Park13) have been shown to be associated with mitochondrial dysfunction. Mutations in PINK1 cause a cascade of events leading to mitochondrial dysfunction and ultimately mitophagy [18]. The loss of function of mitophagy is caused by elevated ROS levels and decreased lysosomal-autophagic degradation resulting from mutations in DJ-1, a protein involved in cellular protection against oxidative stress [123]. Recent therapeutic strategies has been focused on key autophagy-lysosomal pathways (ALP) instead of autophagy within a boad context. Focusing on specific targets, such as chaperone-mediated autophagy and lysosomes, may enhance drug specificity and decrease adverse side effects. In conclusion, defects in autophagy or mitophagy can be sufficient to cause PD [124].
In HD, mitochondrial defects have been hypothesized to have a pivotal role in the cause of the disease [125]. While mitophagy and autophagy induction combats damaged mitochondria and organelles, which can lead to neuronal cell death, studies hypothesize that these autophagosome cannot trap the aggregated HTT protein [126]; this results in an influx of the autophagosome as well as aggregates in the cytoplasm acting as possible sources of neuronal cell damage (Fig. 3 ). Studies have also shown that overexpression of lysosomal cathepsins helps clear HTT protein aggregates from HEK cells which is consistent with in vivo studies that show the favorable effects of autophagy [127, 128]. Despite these findings, the role of autophagy in HD remains unclear.
Factors contributing to ALS onset and pathogenesis include oxidative stress, ER stress, mitochondrial dysfunction, and genetic factors [129]. The induction of autophagy is due to impairment of the UPS found in animal and patient samples [130]. Aggregated proteins can cause the impairment of the UPS as well as mitochondrial dysfunction. It has been shown that mutations in Valsin-containing protein (VCP), a chaperon associated with autophagosome maturation, may be linked to patients with autosomal dominant ALS [131]. Consequently, this could hinder autophagy and mitochondrial quality control. Autophagy induction is clearly beneficial to slowing the progression of ALS [132] and autophagy inducers, such as trehalose and resveratrol [133, 134], have been shown to decrease protein aggregation and promote cell survival.
Combined, these results point to the importance of autophagy, a regulated and orderly recycling of unnecessary or dysfunctional components of the cell, in terms of neurodegenerative diseases. This process needs to be tightly regulated by chaperons and proteins to maintain cellular proteostasis.
Mitochondrial Dysfunction and Oxidative Stress
Mitochondrial proteostasis is critical to the proper functioning of this organelle. In highly metabolic post-mitotic neurons, mitochondrial function is key due to ATP demand and the organelle’s role in regulating apoptosis. The major source of mitochondrial dysfunction in cells is oxidative stress, which originates in mitochondria, but are tightly regulated through antioxidant pathways. Dysfunctioning mitochondria either produce more ROS or are unable to sustain proper regulation. ROS, extremely reactive molecules containing one or more free or unpaired electrons, can damage lipids, DNA, and proteins. ROS toxicity provides a link between mitochondrial proteostasis, which regulates oxidative stress, to cellular proteostasis, which can be damaged and disrupted by ROS.
The consequences of oxidative stress are overarching and devastating to the cell; due to its deleterious effects, oxidative stress is a key element in elucidating the link between neurodegenerative diseases and age [135]. In relation to neurodegenerative diseases and proteostasis, oxidative stress can alter transcription and translation of disease proteins by reacting with transcription factors or altering enzymes, respectively. ROS are also known to affect components of autophagy and proteosome degradation. A feedback loop is therefore obvious where these effects propagate the aggregation of disease proteins, which in turn have been shown to cause mitochondrial dysfunction, leading to oxidative stress and disease progression (Fig. 1 ). In some cases, such as in ALS, SOD1 holds a key function in mitochondrial proteostasis and its loss of function may represent a key step of neuropathology [136].
Parkinson’s Disease
The PD-associated proteins PINK1, PARK13, Parkin, DJ-1, and α-synuclein have been shown to regulate mitochondrial function. PINK1 along with its targets, Parkin and Miro, regulate mitophagy in response to mitochondrial dysfunction. PARK13, also activated by PINK1, is a protease, which removes damaged mitochondrial proteins. PARK13 mutations have been associated with oxidative stress and PD phenotypes, while overexpression and knockout mouse models have shown both neuroprotection and PD phenotypes [137, 138]. DJ-1, a predominantly cytosolic antioxidant protein, can be recruited to mitochondria, including damaged mitochondria where it induces mitophagy [139]. The accumulation of α-synuclein on the outer and inner membranes of mitochondria in dopaminergic neurons decreased mitochondrial Complex I activity and induced ROS levels [136]. Interestingly, under oxidative stress, α-synuclein upregulates PGC-1-alpha, the “master” mitochondrial biogenesis regulator which may represent a therapeutic target for neurodegenerative diseases [140].
Alzheimer’s Disease
In AD the earliest pathological events have not been identified; however, mitochondrial dysfunction has been suggested as a primary event leading to its pathology [141]. Indeed, reduced energy levels and decreased activity of mitochondrial complexes I, III, and IV have been reported in AD patient brains [142, 143]. Aβ overproduction leads to dysfunctional mitochondrial dynamics in human neuroblastoma cells [144] and its localization to mitochondria in AD patients and in AD models impairs complexes I, III, and IV [145]. Furthermore, antioxidants have been shown to decrease the accumulation of Aβ and restore mitochondrial function [146].
Aβ also facilitates the reduction of Cu2+ and Fe2+, which in turn produces H2O2 [147]. A feedback effect has been suggested where ROS induces the accumulation of Aβ, which as previously stated, causes mitochondrial dysfunction and oxidative stress [148]. The same effect might be involved in the observed aggregation and oxidative stress in other proteopathies such as HD, ALS, and pD.
Concluding Remarks
It is evident from the literature that impaired proteostasis in neuronal cells is a key contributor to the pathogenesis of neurodegenerative diseases. Alterations in cellular proteostasis, due to the accumulation of misfolded proteins, ER stress, autophagy, and mitochondrial dysfunction, are a few of the main contributors of neuronal cell death. Therefore, maintaining cellular proteostasis is key in ensuring cell survival and longevity. In recent years, the increased understanding of intracellular pathways and how they contribute to protein quality control to offset the progression of age-related diseases has been the focus of many recent research programs. This collective strategy seems logical, as the traditional neurodegenerative disease proteins have proven to be challenging as targets for therapeutic intervention strategies. By broadening the scope of research to include the manipulation of proteostatic pathways may lead to promising ways to reduce or reverse the pathology of neurodegenerative diseases.
References
Coppede F, Migliore L (2015) DNA damage in neurodegenerative diseases. Mutat Res 776:84–97
Hipp MS, Park SH, Hartl FU (2014) Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 24(9):506–514
Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13:89–102
Komatsu M, Ueno T, Waguri S, Uchiyama Y, Kominami E, Tanaka K (2007) Constitutive autophagy: vital role in clearance of unfavorable proteins in neurons. Cell Death Differ 14:887
Tanaka K, Matsuda N (2014) Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochim Biophys Acta 1843(1):197–204
Drazic A, Myklebust LM, Ree R, Arnesen T (2016) The world of protein acetylation. Biochim Biophys Acta 1864(10):1372–1401
Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95(1):55–66
Didonna A, Benetti F (2015) Post-translational modifications in neurodegeneration. AIMS Biophysics 3(1):27–49
Pennuto M, Palazzolo I, Poletti A (2009) Post-translational modifications of expanded polyglutamine proteins: impact on neurotoxicity. Hum Mol Genet 18(R1):R40–R47
Waxman EA, Giasson BI (2011) Characterization of kinases involved in the phosphorylation of aggregated alpha-synuclein. J Neurosci Res 89(2):231–247
Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ, Barbour R, Huang J et al (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281(40):29739–29752
Goncalves S, Outeiro TF (2013) Assessing the subcellular dynamics of alpha-synuclein using photoactivation microscopy. Mol Neurobiol 47(3):1081–1092
Cavallarin N, Vicario M, Negro A (2010) The role of phosphorylation in synucleinopathies: focus on Parkinson’s disease. CNS Neurol Disord Drug Targets 9(4):471–481
Park G, Tan J, Garcia G, Kang Y, Salvesen G, Zhang Z (2016) Regulation of histone acetylation by autophagy in Parkinson disease. J Biol Chem 291(7):3531–3540
Harrison IF, Dexter DT (2013) Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson’s disease? Pharmacol Ther 140(1):34–52
Wilkinson KD (1987) Protein ubiquitination: a regulatory post-translational modification. Anticancer Drug Des 2(2):211–229
Pickart CM (1997) Targeting of substrates to the 26S proteasome. FASEB J 11(13):1055–1066
Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205(2):143–153
Kazlauskaite A, Martinez-Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, Johnson C, Zhang J et al (2015) Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep 16(8):939–954
Ko HS, Lee Y, Shin JH, Karuppagounder SS, Gadad BS, Koleske AJ, Pletnikova O, Troncoso JC et al (2010) Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc Natl Acad Sci U S A 107(38):16691–16696
Song P, Li S, Wu H, Gao R, Rao G, Wang D, Chen Z, Ma B et al (2016) Parkin promotes proteasomal degradation of p62: implication of selective vulnerability of neuronal cells in the pathogenesis of Parkinson’s disease. Protein Cell 7(2):114–129
Vicente Miranda H, Cássio R, Correia-Guedes L, Gomes MA, Chegão A, Miranda E, Soares T, Coelho M et al (2017) Posttranslational modifications of blood-derived alpha-synuclein as biochemical markers for Parkinson’s disease. Sci Rep 7(1):13713
Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao HH, Bossis G, Urlaub H, Zweckstetter M et al (2011) Sumoylation inhibits alpha-synuclein aggregation and toxicity. J Cell Biol 194(1):49–60
Almos PZ, Horváth S, Czibula A, Raskó I, Sipos B, Bihari P, Béres J, Juhász A et al (2008) H1 tau haplotype-related genomic variation at 17q21.3 as an Asian heritage of the European Gypsy population. Heredity (Edinb) 101(5):416–419
Zhang Y, Tian Q, Zhang Q, Zhou X, Liu S, Wang JZ (2009) Hyperphosphorylation of microtubule-associated tau protein plays dual role in neurodegeneration and neuroprotection. Pathophysiology 16(4):311–316
Serenó L, Coma M, Rodríguez M, Sánchez-Ferrer P, Sánchez MB, Gich I, Agulló JM, Pérez M et al (2009) A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol Dis 35(3):359–367
Tanimukai H, Grundke-Iqbal I, Iqbal K (2005) Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am J Pathol 166(6):1761–1771
Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K (2000) Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem 275(8):5535–5544
Kaneko M, Koike H, Saito R, Kitamura Y, Okuma Y, Nomura Y (2010) Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J Neurosci 30(11):3924–3932
Thinakaran G, Koo EH (2008) Amyloid precursor protein trafficking, processing, and function. J Biol Chem 283(44):29615–29619
Llorens-Martín M, Jurado J, Hernández F, Avila J (2014) GSK-3beta, a pivotal kinase in Alzheimer disease. Front Mol Neurosci 7:46
Feligioni M, Brambilla E, Camassa A, Sclip A, Arnaboldi A, Morelli F, Antoniou X, Borsello T (2011) Crosstalk between JNK and SUMO signaling pathways: deSUMOylation is protective against H2O2-induced cell injury. PLoS One 6(12):e28185
Lee MR, Lee D, Shin SK, Kim YH, Choi CY (2008) Inhibition of APP intracellular domain (AICD) transcriptional activity via covalent conjugation with Nedd8. Biochem Biophys Res Commun 366(4):976–981
Gong L, Yeh ET (1999) Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J Biol Chem 274(17):12036–12042
Chen Y, Bodles AM, McPhie DL, Neve RL, Mrak RE, Griffin WST (2007) APP-BP1 inhibits Abeta42 levels by interacting with Presenilin-1. Mol Neurodegener 2:3
Laifenfeld D, Patzek LJ, McPhie DL, Chen Y, Levites Y, Cataldo AM, Neve RL (2007) Rab5 mediates an amyloid precursor protein signaling pathway that leads to apoptosis. J Neurosci 27(27):7141–7153
Del Prete D, Checler F, Chami M (2014) Ryanodine receptors: physiological function and deregulation in Alzheimer disease. Mol Neurodegener 9:21
Liu X, Betzenhauser MJ, Reiken S, Meli AC, Xie W, Chen BX, Arancio O, Marks AR (2012) Role of leaky neuronal ryanodine receptors in stress-induced cognitive dysfunction. Cell 150(5):1055–1067
Bussiere R, Lacampagne A, Reiken S, Liu X, Scheuerman V, Zalk R, Martin C, Checler F et al (2017) Amyloid beta production is regulated by beta2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J Biol Chem 292(24):10153–10168
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362(6415):59–62
Fei E, Jia N, Yan M, Ying Z, Sun Q, Wang H, Zhang T, Ma X et al (2006) SUMO-1 modification increases human SOD1 stability and aggregation. Biochem Biophys Res Commun 347(2):406–412
Gibb SL, Boston-Howes W, Lavina ZS, Gustincich S, Brown RH Jr, Pasinelli P, Trotti D (2007) A caspase-3-cleaved fragment of the glial glutamate transporter EAAT2 is sumoylated and targeted to promyelocytic leukemia nuclear bodies in mutant SOD1-linked amyotrophic lateral sclerosis. J Biol Chem 282(44):32480–32490
Kaliszewski M, Kennedy AK, Blaes SL, Shaffer RS, Knott AB, Song W, Hauser HA, Bossy B et al (2016) SOD1 lysine 123 acetylation in the adult central nervous system. Front Cell Neurosci 10:287
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F et al (2011) Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 477(7363):211–215
Chang L, Monteiro MJ (2015) Defective proteasome delivery of polyubiquitinated proteins by ubiquilin-2 proteins containing ALS mutations. PLoS One 10(6):e0130162
Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP, Cashman NR, Kondejewski LH et al (2002) Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem 277(49):47551–47556
Mishra A, Maheshwari M, Chhangani D, Fujimori-Tonou N, Endo F, Joshi AP, Jana NR, Yamanaka K (2013) E6-AP association promotes SOD1 aggresomes degradation and suppresses toxicity. Neurobiol Aging 34(4):1310 e11–1310 e23
Pardo CA, Xu Z, Borchelt DR, Price DL, Sisodia SS, Cleveland DW (1995) Superoxide dismutase is an abundant component in cell bodies, dendrites, and axons of motor neurons and in a subset of other neurons. Proc Natl Acad Sci U S A 92(4):954–958
Niikura T, Kita Y, Abe Y (2014) SUMO3 modification accelerates the aggregation of ALS-linked SOD1 mutants. PLoS One 9(6):e101080
Nakamura T, Cho DH, Lipton SA (2012) Redox regulation of protein misfolding, mitochondrial dysfunction, synaptic damage, and cell death in neurodegenerative diseases. Exp Neurol 238(1):12–21
Bristol LA, Rothstein JD (1996) Glutamate transporter gene expression in amyotrophic lateral sclerosis motor cortex. Ann Neurol 39(5):676–679
Foran E, Rosenblum L, Bogush A, Pasinelli P, Trotti D (2014) Sumoylation of the astroglial glutamate transporter EAAT2 governs its intracellular compartmentalization. Glia 62(8):1241–1253
Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC et al (2016) ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7:10465
Renton AE, Chio A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17(1):17–23
Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS (2013) The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem 288(34):24731–24741
Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S et al (2007) Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem 282(33):23818–23828
Winklhofer KF, Tatzelt J, Haass C (2008) The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J 27(2):336–349
Helwig M, Klinkenberg M, Rusconi R, Musgrove RE, Majbour NK, el-Agnaf OMA, Ulusoy A, di Monte DA (2016) Brain propagation of transduced alpha-synuclein involves non-fibrillar protein species and is enhanced in alpha-synuclein null mice. Brain 139(Pt 3):856–870
Luk KC, Song C, O'Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VMY (2009) Exogenous α-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci 106(47):20051–20056
Abdullah R, Patil KS, Rosen B, Pal R, Prabhudesai S, Lee S, Basak I, Hoedt E et al (2017) Subcellular Parkinson’s disease-specific alpha-synuclein species show altered behavior in neurodegeneration. Mol Neurobiol 54(10):7639–7655
Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T et al (2011) In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A 108(10):4194–4199
Prots I, Veber V, Brey S, Campioni S, Buder K, Riek R, Böhm KJ, Winner B (2013) alpha-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J Biol Chem 288(30):21742–21754
Emamzadeh FN (2016) Alpha-synuclein structure, functions, and interactions. J Res Med Sci 21:29
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I et al (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci 95(11):6448–6453
Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH (2006) A specific amyloid-[beta] protein assembly in the brain impairs memory. Nature 440(7082):352–357
Leroy K, Ando K, Laporte V, Dedecker R, Suain V, Authelet M, Héraud C, Pierrot N et al (2012) Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am J Pathol 181(6):1928–1940
Kopeikina KJ, Hyman BT, Spires-Jones TL (2012) Soluble forms of tau are toxic in Alzheimer’s disease. Transl Neurosci 3(3):223–233
Kanaan NM, Pigino GF, Brady ST, Lazarov O, Binder LI, Morfini GA (2013) Axonal degeneration in Alzheimer’s disease: when signaling abnormalities meet the axonal transport system. Exp Neurol 246:44–53
Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA (2008) Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J Neurosci 28(7):1682–7
Arrasate M, Finkbeiner S (2012) Protein aggregates in Huntington’s disease. Exp Neurol 238(1):1–11
Miller J, Arrasate M, Shaby BA, Mitra S, Masliah E, Finkbeiner S (2010) Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into Huntington’s disease molecular pathogenesis. J Neurosci 30(31):10541–10550
Saudou F, Humbert S (2016) The biology of huntingtin. Neuron 89(5):910–926
Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, Yang L, Kotin RM et al (2005) RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A 102(16):5820–5825
O'Kusky JR, Nasir J, Cicchetti F, Parent A, Hayden MR (1999) Neuronal degeneration in the basal ganglia and loss of pallido-subthalamic synapses in mice with targeted disruption of the Huntington’s disease gene. Brain Res 818(2):468–479
Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, Pasinelli P, Trotti D (2016) Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep 17(3):645–652
Robinson JL, Geser F, Stieber A, Umoh M, Kwong LK, van Deerlin VM, Lee VMY, Trojanowski JQ (2013) TDP-43 skeins show properties of amyloid in a subset of ALS cases. Acta Neuropathol 125(1):121–131
Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Velde CV, Bouchard JP, Lacomblez L et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Chan PK, Chattopadhyay M, Sharma S, Souda P, Gralla EB, Borchelt DR, Whitelegge JP, Valentine JS (2013) Structural similarity of wild-type and ALS-mutant superoxide dismutase-1 fibrils using limited proteolysis and atomic force microscopy. Proc Natl Acad Sci 110(27):10934–10939
Pedrini S, Sau D, Guareschi S, Bogush M, Brown RH Jr, Naniche N, Kia A, Trotti D et al (2010) ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet 19(15):2974–2986
Sau D, de Biasi S, Vitellaro-Zuccarello L, Riso P, Guarnieri S, Porrini M, Simeoni S, Crippa V et al (2007) Mutation of SOD1 in ALS: a gain of a loss of function. Hum Mol Genet 16(13):1604–1618
Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL (2000) Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289(5483):1317–1321
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J et al (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162(5):1066–1077
Brangwynne CP, Tompa P, Pappu RV (2015) Polymer physics of intracellular phase transitions. Nat Phys 11:899–904
Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP (2015) Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163(1):123–133
Iurlaro R, Munoz-Pinedo C (2016) Cell death induced by endoplasmic reticulum stress. FEBS J 283(14):2640–2652
Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK (2012) Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J Neurosci 32(10):3301–3305
Credle JJ, Forcelli PA, Delannoy M, Oaks AW, Permaul E, Berry DL, Duka V, Wills J et al (2015) alpha-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol Dis 76:112–125
Omura T, Kaneko M, Okuma Y, Matsubara K, Nomura Y (2013) Endoplasmic reticulum stress and Parkinson’s disease: the role of HRD1 in averting apoptosis in neurodegenerative disease. Oxidative Med Cell Longev 2013:239854
Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA (2002) Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci 22(24):10690–10698
Hetz C, Chevet E, Harding HP (2013) Targeting the unfolded protein response in disease. Nat Rev Drug Discov 12:703–719
Chano T, Okabe H, Hulette CM (2007) RB1CC1 insufficiency causes neuronal atrophy through mTOR signaling alteration and involved in the pathology of Alzheimer’s diseases. Brain Res 1168:97–105
Yoon SO, Park DJ, Ryu JC, Ozer HG, Tep C, Shin YJ, Lim TH, Pastorino L et al (2012) JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron 75(5):824–837
Acosta-Alvear D, Zhou Y, Blais A, Tsikitis M, Lents NH, Arias C, Lennon CJ, Kluger Y et al (2007) XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol Cell 27(1):53–66
O'Connor T, Sadleir KR, Maus E, Velliquette RA, Zhao J, Cole SL, Eimer WA, Hitt B et al (2008) Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 60(6):988–1009
Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, Cavener DR, Klann E (2013) Suppression of eIF2alpha kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat Neurosci 16(9):1299–1305
Carnemolla A, Fossale E, Agostoni E, Michelazzi S, Calligaris R, de Maso L, del Sal G, MacDonald ME et al (2009) Rrs1 is involved in endoplasmic reticulum stress response in Huntington disease. J Biol Chem 284(27):18167–18173
Duennwald ML, Lindquist S (2008) Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev 22(23):3308–3319
Yang D, Wang CE, Zhao B, Li W, Ouyang Z, Liu Z, Yang H, Fan P et al (2010) Expression of Huntington’s disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum Mol Genet 19(20):3983–3994
Lajoie P, Snapp EL (2011) Changes in BiP availability reveal hypersensitivity to acute endoplasmic reticulum stress in cells expressing mutant huntingtin. J Cell Sci 124(Pt 19):3332–3343
Wang CE, Tydlacka S, Orr AL, Yang SH, Graham RK, Hayden MR, Li S, Chan AWS et al (2008) Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum Mol Genet 17(17):2738–2751
Wang J, Wang CE, Orr A, Tydlacka S, Li SH, Li XJ (2008) Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J Cell Biol 180(6):1177–1189
Jiang Y, Chadwick SR, Lajoie P (2016) Endoplasmic reticulum stress: the cause and solution to Huntington's disease? Brain Res 1648:650–657
Hetz C, Mollereau B (2014) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15(4):233–249
Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C (2003) Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J 22(20):5435–5445
Xu K, Zhu XP (2012) Endoplasmic reticulum stress and prion diseases. Rev Neurosci 23(1):79–84
Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE et al (2013) Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med 5(206):206ra138
Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J et al (2012) Sustained translational repression by eIF2α–P mediates prion neurodegeneration. Nature 485(7399):507–511
Menzies FM, Moreau K, Rubinsztein DC (2011) Protein misfolding disorders and macroautophagy. Curr Opin Cell Biol 23(2):190–197
Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self-digestion. Nature 451(7182):1069–1075
Lum JJ, DeBerardinis RJ, Thompson CB (2005) Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol 6(6):439–448
Wong E, Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13(7):805–811
Sarkar S (2013) Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem Soc Trans 41(5):1103–1130
Vidal RL, Matus S, Bargsted L, Hetz C (2014) Targeting autophagy in neurodegenerative diseases. Trends Pharmacol Sci 35(11):583–591
Marino G, Madeo F, Kroemer G (2011) Autophagy for tissue homeostasis and neuroprotection. Curr Opin Cell Biol 23(2):198–206
Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H et al (2009) Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci U S A 106(47):20057–20062
Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D et al (2005) Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25(3):1025–1040
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M et al (2005) Macroautophagy—a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 171(1):87–98
Coffey EE, Beckel JM, Laties AM, Mitchell CH (2014) Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 263:111–124
Mindell JA (2012) Lysosomal acidification mechanisms. Annu Rev Physiol 74:69–86
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M et al (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141(7):1146–1158
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D (2004) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305(5688):1292–1295
Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ (2008) The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A 105(5):1638–1643
Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J, Wolburg H, Gizatullina Z et al (2010) Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One 5(2):e9367
Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5):795–803
Pickrell AM, Fukui H, Wang X, Pinto M, Moraes CT (2011) The striatum is highly susceptible to mitochondrial oxidative phosphorylation dysfunctions. J Neurosci 31(27):9895–9904
Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E et al (2010) Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 13(5):567–576
Liang Q, Ouyang X, Schneider L, Zhang J (2011) Reduction of mutant huntingtin accumulation and toxicity by lysosomal cathepsins D and B in neurons. Mol Neurodegener 6:37
Wang P, Li B, Zhou L, Fei E, Wang G (2011) The KDEL receptor induces autophagy to promote the clearance of neurodegenerative disease-related proteins. Neuroscience 190:43–55
Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7(9):710–723
Sasaki S (2011) Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 70(5):349–359
Johnson JO, Mandrioli J, Benatar M, Abramzon Y, van Deerlin VM, Trojanowski JQ, Gibbs JR, Brunetti M et al (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68(5):857–864
Crippa V, Sau D, Rusmini P, Boncoraglio A, Onesto E, Bolzoni E, Galbiati M, Fontana E et al (2010) The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum Mol Genet 19(17):3440–3456
Gomes C, Escrevente C, Costa J (2010) Mutant superoxide dismutase 1 overexpression in NSC-34 cells: effect of trehalose on aggregation, TDP-43 localization and levels of co-expressed glycoproteins. Neurosci Lett 475(3):145–149
Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, Delalle I, Baur JA et al (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J 26(13):3169–3179
Abdullah R, Basak I, Patil KS, Alves G, Larsen JP, Møller SG (2014) Parkinson's disease and age: the obvious but largely unexplored link. Exp Gerontol 68:33–38
Johri A, Beal MF (2012) Mitochondrial dysfunction in neurodegenerative diseases. J Pharmacol Exp Ther:619–630
Kawamoto Y, Kobayashi Y, Suzuki Y, Inoue H, Tomimoto H, Akiguchi I, Budka H, Martins LM et al (2008) Accumulation of HtrA2/Omi in neuronal and glial inclusions in brains with alpha-synucleinopathies. J Neuropathol Exp Neurol 67(10):984–993
Moisoi N, Klupsch K, Fedele V, East P, Sharma S, Renton A, Plun-Favreau H, Edwards RE et al (2009) Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ 16(3):449–464
Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, Miller D, Maric D et al (2011) DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet 20(1):40–50
Siddiqui A, Chinta SJ, Mallajosyula JK, Rajagopolan S, Hanson I, Rane A, Melov S, Andersen JK (2012) Selective binding of nuclear alpha-synuclein to the PGC1alpha promoter under conditions of oxidative stress may contribute to losses in mitochondrial function: implications for Parkinson’s disease. Free Radic Biol Med 53(4):993–1003
Swerdlow RH, Burns JM, Khan SM (2010) The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimers Dis 20(Suppl 2):S265–S279
Beal MF (2005) Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58(4):495–505
Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA, Murri L, Rapoport SI et al (2002) Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiol Aging 23(3):371–376
Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X (2008) Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A:19318–19323
Munguia ME, Govezensky T, Martinez R, Manoutcharian K, Gevorkian G (2006) Identification of amyloid-beta 1–42 binding protein fragments by screening of a human brain cDNA library. Neurosci Lett 397(1):79–82
Ono K, Hamaguchi T, Naiki H, Yamada M (2006) Anti-amyloidogenic effects of antioxidants: implications for the prevention and therapeutics of Alzheimer’s disease. Biochim Biophys Acta (BBA) - Mol Basis Dis 1762(6):575–586
Jiang D, Li X, Williams R, Patel S, Men L, Wang Y, Zhou F (2009) Ternary complexes of iron, amyloid-beta, and nitrilotriacetic acid: binding affinities, redox properties, and relevance to iron-induced oxidative stress in Alzheimer’s disease. Biochemistry 48(33):7939–7947
Misonou H, Morishima-Kawashima M, Ihara Y (2000) Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 39(23):6951–6959
Funding
Research in our laboratory is funded by The Norwegian Center for Movement Disorders, The Norwegian Parkinson’s Foundation, and St. John’s University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kurtishi, A., Rosen, B., Patil, K.S. et al. Cellular Proteostasis in Neurodegeneration. Mol Neurobiol 56, 3676–3689 (2019). https://doi.org/10.1007/s12035-018-1334-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1334-z