
Abstract
Obesity is associated with an increase in the brain levels of saturated free fatty acids, such as palmitic acid (PA). Previous studies have shown that PA exerts proinflammatory actions and reduces cell viability in astrocyte cultures. In this study, we have assessed whether an alteration in autophagy is involved in the effects of PA on astrocytes. Primary astrocytes were obtained from the cerebral cortex of male and female CD1 mouse pups and were incubated for 4.5 or 24 h with 250–500 μM PA. PA increased the levels of LC3-II, an autophagosome marker, and reduced LC3-II flux in astrocytes, suggesting a blockade of autophagy. This effect was observed both after 4.5 and 24 h of treatment with PA. PA had additional effects after treatment for 24 h, increasing the expression of proinflammatory cytokines, decreasing cell viability, and increasing the levels of an endoplasmic reticulum stress marker. In addition, PA decreased the expression of estrogen receptors, but only in female astrocytes. However, the treatment with estradiol, estrogen receptor agonists, or inhibitor of estradiol synthesis did not counteract the action of PA on cell viability. Rapamycin, an autophagy inducer, was unable to prevent the effect of PA on cell viability. In addition, hydroxychloroquine, an autophagy blocker, did not cause per se astrocyte death. These findings suggest that the effect of PA on autophagy is not sufficient to induce astrocyte loss, which is only observed when prolonged PA treatment causes other alterations in astrocytes, such as increased inflammation and endoplasmic reticulum stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the last decades, obesity has become a global epidemic. Between 1975 and 2016, the worldwide prevalence of obesity nearly tripled. The World Health Organization (WHO) estimates that 13% of the world’s adult population (11% of men and 15% of women) was obese in 2016. Obesity is defined as an abnormal or excessive accumulation of fat that can negatively affect health. Indeed, it is associated with increased risk of cardiovascular diseases, diabetes, musculoskeletal disorders, and certain types of cancer [1]. In addition, several studies suggest a link between obesity and cognitive decline and dementia. For instance, high values of body mass index (BMI), the clinical parameter used to determine the presence of overweight and obesity, are associated with a diminished brain volume in middle-aged adults [2] and with a reduction in gray matter volume in elderly subjects [3]. In relation to brain function, overweight and obesity correlate with poorer executive function and reduced activity of memory areas [4, 5]. Therefore, obesity is considered as a risk factor for Alzheimer’s disease [6].
The specific pathophysiological and molecular mechanisms involved in the effect of obesity on brain function have not been fully elucidated yet, although diverse animal models have been used. Diet-induced obesity (DIO) models are characterized by the presence of a variety of metabolic disturbances such as an increased body weight, accumulation of adipose tissue, and insulin resistance [7]. Besides these peripheral effects, some alterations have been detected in the central nervous system (CNS) of these animals. For instance, the integrity of the blood–brain barrier (BBB) is compromised [8] and there is a reduction in the levels of brain-derived neurotrophic factor [9] and in the number of dendritic spines, as well as an impairment of spatial learning and memory [10].
There is evidence that inflammation plays a crucial role in the pathophysiology of obesity, as it does in many disorders of the CNS. Obesity was firstly associated with peripheral low-grade inflammation due to an increased expression of tumor necrosis factor (TNF) α in adipose tissue of obese humans and mice [11]. Afterwards, it was discovered that high-fat diet (HFD) induces a proinflammatory response in the hypothalamus of obese rodents and humans [12]. The hypothalamus is a key structure for energy balance and the control of body weight, and HFD-induced hypothalamic inflammation is involved in impaired metabolic control [13]. Moreover, other brain regions, such as the hippocampus and the amygdala, also present an increased expression of proinflammatory markers after HFD consumption [14].
Furthermore, obesity is characterized by the presence of an abnormal lipid profile in blood, consistent of increased levels of triglycerides and free fatty acids (FFAs) [15]. Plasmatic FFAs can also cross the BBB and reach brain parenchyma [16], where they can exert pathological effects. Levels of saturated FFAs, such as the proinflammatory palmitic acid (PA), increase in the brain of animal models of obesity [17] and in obese patients [18] and contribute to the development of HFD-associated neuroinflammation. Treatment of primary astrocytes and microglia with PA has been used as an in vitro model to study the impact of the increase in saturated fatty acids in the brain during obesity and the importance of glial cells in this metabolic process [17, 19,20,21,22,23]. Astrocytes are the most abundant cells in the CNS and they were initially considered as mere supporters for neuronal function. However, they participate in essential processes such as the transport of substances across the BBB, the storage of energy, and the regulation of neurotransmission. Astrocytes are able to maintain brain homeostasis in response to metabolic alterations through the sensing of nutrients, hormones, and other metabolites [24]. Specifically, lipids are sensed by astrocytes, which have a higher capacity to uptake and metabolize them than neurons [25]. Previous studies have shown that PA downregulates glucose uptake and lactate release by astrocytes [20]; induces the generation of reactive oxygen species [22]; increases the production of proinflammatory cytokines, such as interleukin (IL)-1β, IL-6, and TNF-α [17, 23]; and decreases cell viability in astrocyte cultures by apoptosis [21, 22].
In this study, we have assessed whether autophagy is involved in the effects of PA on astrocytes. Autophagy is a cellular process for recycling large macromolecules and damaged cellular components, as well as for obtaining energy under metabolically challenging conditions [26]. The more prevalent and best characterized autophagy variant, called macroautophagy, involves the formation of autophagosomes, special transient double-membrane structures that engulf cellular debris and macromolecules and that fuse with lysosomes to deliver their content for degradation [27]. Autophagy is an essential mechanism to maintain cellular homeostasis. Thus, the pharmacological or genetic inhibition of autophagy results in apoptotic cellular death [28] and autophagy impairment has been associated with neurodegenerative and metabolic diseases, among other pathological conditions [29].
Although previous studies have shown that PA impairs autophagy in different cell types [30,31,32,33], causing the generation of reactive oxygen species [34], enhanced inflammatory response [35], endoplasmic reticulum stress [31, 32], and finally cell death [28, 31, 32], the possible effect of PA on autophagy in astrocytes has not been explored. Our aim, therefore, was to determine whether PA impairs autophagy in astrocytes. Thus, in the present study, we have explored autophagy in PA-treated primary astrocyte cultures. As sex differences in the metabolic alterations induced by HFD have been described [36, 37], we have explored the effect of PA separately in male and female astrocytes.
Materials and Methods
Animals
Postnatal day 0 (PND0)–PND2 male and female CD1 mouse pups were raised in our in-house colony at the Cajal Institute. Male pups were distinguished from female pups by a larger genital papilla and longer anogenital distance. All the procedures applied to the animals used in this study were in accordance with the European Commission guidelines (2010/63/UE) and the Spanish regulation (R. D. 53/2013) on the protection of animals for experimental use. These procedures were approved by our institutional animal care and use committee (Comité de Ética de Experimentación Animal del Instituto Cajal) and the Consejeria del Medio Ambiente y Territorio (Comunidad de Madrid, PROEX 200/14).
Cortical Astrocyte Cultures
Astrocytes were cultured from male and female PND0–PND2 pups, separately. The brain was extracted, meninges were removed, and the cerebral cortex was isolated under a dissecting microscope and then mechanically dissociated and washed twice in Hank’s balanced salt solution (Sigma-Aldrich, Tres Cantos, Madrid). After complete dissociation in Dulbecco’s modified Eagle’s medium/Nutrient mixture F-12 (DMEM/F-12) culture medium with phenol red (Sigma-Aldrich) containing 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA) and 1% antibiotic–antimycotic (Invitrogen), the cells were filtered through a 40-μm nylon cell strainer (Corning Inc., Corning, NY). The cells were centrifuged, resuspended in the same medium, and plated onto poly-L-lysine-coated 75-cm2 flasks at 37 °C and 5% CO2. The medium was replaced after the first day in vitro and two times per week until the cells reached confluence (~ 7 days). Then, the cell cultures were shaken overnight at 37 °C and 280 rpm on a tabletop shaker (Infors HT, Bottmingen, Switzerland) in order to minimize oligodendrocyte and microglia contamination. The astrocytes were incubated with 0.5% trypsin (Sigma-Aldrich), centrifuged, resuspended in phenol red-free DMEM/F-12 with 10% FBS and 1% antibiotic–antimycotic, and seeded in poly-L-lysine-coated 75-cm2 flasks at 37 °C and 5% CO2. When the cells reached confluence for the second time (~ after 5 days), the subculture process was repeated but the astrocytes were plated onto poly-L-lysine-coated plates (6, 48, or 96 wells) at a density of 25,000 cells/cm2 using phenol red-free DMEM/F-12 with 10% FBS and 1% antibiotic–antimycotic. Twenty-four hours after plating, the astrocytes were treated as indicated below.
Cell Treatments
PA (Sigma-Aldrich) was dissolved in methanol in a concentration of 0.5 M by heating and mixing. Then, PA was diluted in saline solution containing 5% fatty acid-free bovine serum albumin (BSA, Sigma-Aldrich) to obtain a 5-mM stock solution. PA stock solution was agitated at 37 °C overnight, filtered thorough 22-μm syringe filter (Pall Corporation, Alcobendas, Madrid), aliquoted, and maintained at − 20 °C. Vehicle stock solution was prepared using the same protocol without PA. Male and female astrocytes were treated with 250 and 500 μM PA in phenol red-free DMEM/F-12 medium without FBS and antibiotic–antimycotic. Control astrocytes were incubated with vehicle solution. The duration of vehicle/PA treatment was 4.5 or 24 h depending on the experimental approach.
In some experiments, male and female astrocytes were treated with 17β-estradiol (10−10, 10−9, and 10−8 M; Sigma-Aldrich), the ERα agonist PPT (10−9 and 10−8 M; Tocris Bioscience, Bristol), the ERβ agonist DPN (10−9 and 10−8 M, Tocris Bioscience), the GPER agonist G1 (10−7 and 10−6 M; Tocris Bioscience), or the aromatase inhibitor letrozole (10−7 M; Sigma-Aldrich). The astrocytes were treated with these molecules for 24 h. Then, they were exposed to PA or its vehicle and these compounds for additional 24 h. The doses of estrogenic compounds are based on previous studies [38, 39]. These compounds were dissolved in dimethyl sulfoxide (Sigma-Aldrich) and then diluted to the final concentration in phenol red-free DMEM/F-12 without FBS and antibiotic–antimycotic.
Some cultures were treated with 30 μg/mL hydroxychloroquine (Laboratorios Rubió, Castellbisbal, Barcelona), an inhibitor of autophagosome–lysosome fusion, for 4.5 h when cells were treated with PA for this time period or for the last 4 h in the experiments in which cells were treated with PA for 24 h. To increase autophagic activity, astrocytes were pretreated with 0.2 μg/mL rapamycin (Sigma-Aldrich) for 2 h and then treated with a combination of rapamycin and 250 μM PA for 24 h. Furthermore, some astrocytes were treated with 1 μg/mL tunicamycin (Sigma-Aldrich) for 6 h as a positive control of endoplasmic reticulum stress.
Immunocytochemistry
Cells were fixed for 20 min at room temperature in 4% paraformaldehyde, washed with 0.12% gelatin in phosphate-buffered saline (PBS), and permeabilized for 4 min with 0.12% Triton-X plus 0.12% gelatin in PBS. The cells were then washed with PBS/gelatin and incubated at room temperature for 1.5 h with anti-GFAP mouse monoclonal antibody (dilution 1:500 in PBS/gelatin; #G3893, Sigma-Aldrich) and with anti-p62 guinea pig polyclonal antibody (dilution 1:150 in PBS/gelatin; #GP62-C, Progen, Heidelberg). After washing in the same buffer, the astrocytes were incubated for 45 min at room temperature with goat anti-mouse Alexa 488 conjugated antibody (dilution 1:500; Jackson Immuno-Research Europe Ltd., Ely, Cambridgeshire) for the detection of GFAP and with goat anti-guinea pig Cy3 conjugated antibody (dilution 1:250; Jackson Immuno-Research Europe Ltd) for detection of p62. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired with ×63/1.40 oil immersion Leica DM6000B objective in a Leica SP-5 confocal microscope.
Cell Viability Assays
To assess cell survival, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, Sigma-Aldrich) assay was used. Male and female astrocytes were plated in 96-well microplates. At the end of the experimental treatments, the culture medium was removed and the cells were incubated with MTT dissolved in DMEM/F-12 (0.5 mg/mL) without phenol red or additives. After 3 h of incubation at 37 °C and 5% CO2, cell culture media was removed and 100 μL of dimethyl sulfoxide was added to each well. Absorbance was measured at 595 nm wavelength in a multiwell plate reader (Thermo Fisher, Madrid).
To confirm cell viability changes, fluorescein diacetate (FDA, Sigma-Aldrich) assay was performed. Astrocytes from males and females were plated in 48-well microplates. At the end of the experimental treatments, the cells were incubated for 50 min at 37 °C and 5% CO2 with FDA (100 μM in DMEM/F-12 without phenol red or additives). Then, the cells were washed twice with culture medium and fluorescence emission was measured in a microplate reader (Thermo Fisher, Madrid).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
After the experimental treatments, the cells were harvested from six-well plates and total RNA was extracted using an Illustra RNAspin Mini kit (GE Healthcare, Madrid). First-strand cDNA was prepared from 2 μg RNA using M-MLV reverse transcriptase (Promega, Alcobendas, Madrid) according to the manufacturer’s protocol. After reverse transcription, cDNAs were amplified by real-time PCR in 15 μL reaction volume using SYBR Green Master Mix (Applied Biosystems, Foster City, CA) and the ABI Prism 7500 Sequence Detection System (Applied Biosystems) with conventional Applied Biosystems cycling parameters (40 cycles of changing temperatures, first at 95 °C for 15 s and then 60 °C for a minute). All the primer sequences were designed using Primer Express software (Applied Biosystems) and are shown in Table 1. For each primer pair, an appropriate dilution of cDNA was chosen in order to achieve the same amplification efficiency as that of the housekeeping genes (18S rRNA, β-actin, and Rpl13A). Changes in mRNA expression were calculated following the DDCt method [40], using the Best Keeper index [41] as total mRNA load reference for each sample.
Western Blotting
After the experimental treatments, cells were lysed and solubilized in 200 μL of 50 mM Tris-HCl pH 6.8, containing 2% SDS, 10% glycerol, 100 mM dithiothreitol, protease/phosphatase inhibitor cocktail, and bromophenol blue. Samples were boiled and sonicated for 5 min. Solubilized proteins (20 μL) were resolved by 6, 12, or 15% SDS–PAGE and then electrophoretically transferred to 0.2 μm Trans-Blot Turbo PVDF or nitrocellulose membranes (BioRad, Alcobendas, Madrid). The membranes were blocked at room temperature for 90 min in Tris-buffered saline containing 0.1% Tween 20 and 5% BSA. Then, the membranes were incubated overnight at 4 °C with primary antibodies diluted in the same blocking solution. After that, the membranes were incubated with horseradish peroxidase-conjugated antibodies (diluted 1:10,000; Jackson Immunoresearch, West Grove, PA) or infrared dye-conjugated antibodies (diluted 1:10,000; LI-COR Biosciences, Lincoln, NE). Specific proteins were visualized by enhanced chemiluminescence detection reagent (GE Healthcare) or by Odyssey Infrared Imaging System (LI-COR Biosciences). The following primary antibodies were used: anti-LC3B (diluted 1:3500; #L7543, Sigma-Aldrich), anti-p62 (diluted 1:1000; #5114, Cell Signaling Technology, Leiden), anti-phospho-mTOR (diluted 1:1000; #2971, Cell Signaling), anti-mTOR (diluted 1:1000; #2983; Cell Signaling), anti-CHOP (diluted 1:1000; #2895, Cell Signaling), anti-β-actin (diluted 1:4000; #A5316, Sigma-Aldrich), and anti-α-tubulin (diluted 1:5000; #T5168, Sigma-Aldrich). Densitometric analyses were performed with ImageJ software (freely available at https://imagej.nih.gov/ij/). Data were normalized to the mean of β-actin and α-tubulin expression.
Statistical Analysis
Data shown in the figures are presented as the mean ± standard error of the mean (SEM). The size of the experimental groups corresponds to the number of independent culture preparations, and it is indicated in each figure legend. Statistical analyses were carried out using SPSS Statistics 23 software (IBM, Armonk, NY). Normal distribution and homoscedasticity were assessed using Shapiro-Wilk test and Levene’s test, respectively. Statistical significance was evaluated by two-way analysis of variance (ANOVA) followed by Bonferroni or Games-Howell post hoc tests (depending on whether variances were homogeneous or not, respectively) for multiple comparisons. When an interaction between two factors was not detected, data were split and each factor was analyzed by one-way ANOVA (treatment effect) or Student’s t test (treatment effect or sex effect). The statistical significance level was set at p < 0.05.
Results
Autophagy Is Modulated by PA in Astrocytes
As it has been established that PA can modulate autophagy in neurons [42], we were interested in determining if PA could also modify this process on astrocytes. For this purpose, cultured cortical astrocytes were treated with different doses (250 and 500 μM) of PA for 24 h. The doses of PA used are within the range of PA levels in mouse serum under physiological conditions and after the administration of a HFD [43, 44]. We first evaluated autophagosome levels by assessing the amount of LC3-II by Western blot [45]. Male astrocytes treated with both doses of PA had higher levels of LC3-II than the control group. In the case of female astrocytes, only 500 μM PA induced a significant increase of LC3-II levels (Fig. 1a).

Autophagic activity was impaired by PA treatment in cultured astrocytes. Male and female astrocytes were treated with vehicle (C), HCQ, 250 μM PA (PA 250), or 500 μM PA (PA 500) for 24 h. a LC3-II protein levels. b p62 protein levels. c MAP1LC3B mRNA levels. d SQSTM1 mRNA levels. e Representative image of LC3-II detection by Western blot used to quantify LC3-II flux. f LC3-II flux measured as the ratio of LC3-II levels in cells treated with HCQ to LC3-II levels in cells untreated with HCQ of the same experimental group. For LC3-II protein levels, two-way ANOVA revealed a significant treatment effect on LC3-II levels [F(2,52) = 14.366; p = 0.000]. As there was no interaction between treatment and sex, the effect of treatment was analyzed for each sex by one-way ANOVA. Treatment had a significant effect in astrocytes from males [F(2,26) = 9.303; p = 0.001] and females [F(2,26) = 5.374; p = 0.011]. For p62 protein levels, two-way ANOVA revealed a significant treatment effect [F(2,28) = 63.450; p = 0.000] without sex effect or interaction between treatment and sex. Treatment effect was analyzed by one-way ANOVA in each sex, showing a significant effect on both males [F(2,16) = 21.307; p = 0.000] and females [F(2,12) = 80.860; p = 0.000]. For MAP1LC3B mRNA levels, two-way ANOVA revealed a significant treatment effect [F(2,24) = 9.948; p = 0.001], with no effect of sex or interaction between the two factors. One-way ANOVA of data split by sex showed a significant treatment effect on MAP1LC3B expression in astrocytes from males [F(2,12) = 3.958; p = 0.048] and females [F(2,12) = 7.427; p = 0.008]. For SQSTM1 mRNA levels, two-way ANOVA showed a significant effect of treatment [F(2,24) = 13.417; p = 0.000], with no effect of sex or interaction between treatment and sex. To study the effect of treatment, one-way ANOVA was carried out for each sex and it revealed a significant effect in male [F(2,12) = 4.514; p = 0.035] and female astrocytes [F(2,12) = 10.216; p = 0.003]. For LC3-II flux, two-way ANOVA revealed an effect of the treatment [F(2,24) = 16.773; p = 0.000]. Due to the lack of interaction between sex and treatment, one-way ANOVA was performed to study the effect of the treatment in each sex. There was a significant treatment effect on LC3-II flux in male [F(2,12) = 5.951; p = 0.016] and female [F(2,12) = 11.103; p = 0.002] astrocytes. Data are represented as mean ± SEM. Sample size N ≥ 3. Significant differences with the post hoc test: one asterisk, two asterisks, and three asterisks, significant differences (p < 0.05, p < 0.01, and p < 0.001) versus control male astrocytes. One number sign, two number signs, and three number signs, significant differences (p < 0.05, p < 0.01, and p < 0.001) versus control female astrocytes. Three at signs, significant differences (p < 0.001) versus female astrocytes treated with PA 250
As increased levels of LC3-II can indicate both increased formation and decreased degradation of autophagosomes, we next assessed p62, an autophagy adaptor that is degraded by autophagy [46]. Male astrocytes treated with either 250 or 500 μM PA had higher levels of p62 compared to control values. Both doses of PA also increased p62 levels in female astrocytes (Fig. 1b). The increase in p62 levels was further corroborated by fluorescence microscopy. While in control GFAP immunostained astrocytes p62 immunoreactivity showed a speckled appearance (consisting of numerous small puncta distributed along the whole cytoplasm), PA-treated astrocytes contained larger p62 immunoreactive bodies clustered in restricted locations in the cytoplasm (Fig. 2). These findings suggest that a treatment for 24 h with PA induces the accumulation of autophagosomes in astrocytes.

PA induced the formation of large p62 immunoreactive bodies in the cytoplasm of astrocytes. Astrocytes were incubated for 4.5 h with PA or vehicle and immunostained for p62 (red), a marker of autophagy, and for the astrocytic marker GFAP (green). Cell nuclei were stained with DAPI (blue). In control male astrocytes (C), numerous small p62 immunoreactive bodies were observed along the whole cytoplasm. The incubation of astrocytes with 250 μM PA (PA 250) or 500 μM PA (PA 500) caused the clustering of p62 immunoreactive bodies in some cytoplasmic regions. In addition, p62 immunoreactive bodies showed a much larger size in PA-treated astrocytes compared to control astrocytes. Representative images of male astrocytes. Scale bar 20 μm
The mRNA expression of LC3 and p62 (encoded by MAP1LC3B and SQSTM1, respectively) was also assessed. Treatment with 500 μM PA decreased MAP1LC3B expression in female astrocytes. In male astrocytes, the effect of 500 μM PA did not reach statistical significance (p = 0.054) (Fig. 1c). Treatment with 500 μM PA increased the levels of SQSTM1 mRNA in astrocytes from males. In astrocytes from females, both doses of PA increased the expression of SQSTM1 (Fig. 1d). These findings indicate that a treatment for 24 h with PA affects not only protein levels and distribution but also LC3 and p62 mRNA expression in astrocytes.
Autophagy is a very dynamic process, so the detection of LC3-II levels is not sufficient to assess autophagy activity in cells. To figure out whether the increase in LC3-II protein levels was due to an increased activity in autophagy or a lysosomal blockade, we evaluated the autophagic flux using hydroxychloroquine (HCQ) as an inhibitor of autophagosome–lysosome fusion. Astrocytes were treated with vehicle, 250 μM PA, or 500 μM PA for 24 h, and HCQ was added to the culture medium for the last 4 h of PA treatment. LC3-II levels were analyzed by Western blot to measure LC3-II flux (corresponding to the ratio between the densitometric value of LC3-II in cells treated with HCQ to the densitometric value of LC3-II in cells untreated with HCQ of the same experimental group). Both doses of PA decreased LC3-II flux in male astrocytes in comparison to control astrocytes. However, the effect of PA did not reach statistical significance in female astrocytes (p = 0.054 for 500 μM PA) (Fig. 1e, f). These results show that the accumulation of LC3-II caused by HCQ in control astrocytes is reduced by PA. This, together with the accumulation of p62, indicates that PA induces a blockade of autophagy.
PA Induces Inflammation in Cultured Cortical Astrocytes
Since autophagy regulates the inflammatory response, we explored the mRNA levels of selected inflammatory molecules in astrocytes exposed to 250 and 500 μM PA for 24 h. Treatment with 500 μM PA significantly increased TNF-α mRNA expression in male astrocytes versus control values. In addition, control female astrocytes showed lower expression of TNF-α mRNA compared to control male astrocytes (Fig. 3a).

PA induced an inflammatory response in cultured cortical astrocytes after 24 h of treatment. Male and female astrocytes were treated with vehicle (C), 250 μM PA (PA 250), or 500 μM PA (PA 500). a TNF-α mRNA levels. b IL-1β mRNA levels. c IL-6 mRNA levels. Regarding TNF-α, two-way ANOVA showed a significant effect of both treatment [F(2,24) = 5.710; p = 0.009] and sex [F(1,24) = 4.819; p = 0.038]. As there was no interaction between these two factors, the effect of each one was analyzed individually. One-way ANOVA split by sex revealed a significant treatment effect in male astrocytes [F(2,12) = 3.933; p = 0.049], but not in female cells. For IL-1β, two-way ANOVA revealed significant effects of treatment [F(2,21) = 9.991; p = 0.001] and sex [F(1,21) = 9.829; p = 0.005], without interaction between these two factors. One-way ANOVA split by sex showed a significant treatment effect in male astrocytes [F(2,11) = 7.498; p = 0.009], but not in female astrocytes. For IL-6 mRNA levels, two-way ANOVA revealed a general treatment effect [F(2,23) = 24.962; p = 0.000] and sex effect [F(1,23) = 5.270; p = 0.031], with no interaction between the two factors. One-way ANOVA showed a significant effect of treatment in male [F(2,12) = 12.787; p = 0.001] and female [F(2,11) = 30.913; p = 0.000] astrocytes. Data are represented as mean ± SEM. Sample size N ≥ 4. One asterisk and two asterisks, significant differences (p < 0.05 and p < 0.01) versus control male astrocytes. Significant differences with the post hoc test: one number sign and three number signs, significant differences (p < 0.05 and p < 0.001) versus control female astrocytes. Dollar sign, significant differences (p < 0.05) versus male astrocytes treated with PA 250. Two at signs, significant differences (p < 0.01) versus female astrocytes treated with PA 250. One accent symbol, significant differences (p < 0.05) versus male astrocytes treated with PA 500
Treatment with 500 μM PA increased IL-1β mRNA expression in male astrocytes to a value that was not significantly different to control levels (p = 0.050). The mRNA expression levels of IL-1β were significantly higher in male astrocytes treated with either vehicle or 500 μM PA in comparison to female astrocytes (Fig. 3b).
IL-6 mRNA expression was also modified by PA. IL-6 mRNA expression was increased by 500 μM PA in male astrocytes and by 250 and 500 μM PA in female astrocytes. Moreover, significant differences in the expression of IL-6 between the two doses of PA were found in male and female astrocytes (Fig. 3c). These findings indicate that PA treatment for 24 h induces an inflammatory response in cultured cortical astrocytes. PA modulates the expression of proinflammatory cytokines, such as TNF-α, IL-6, and IL-1β, with different effects in male and female astrocytes.
PA Induces Cell Death in Cultured Cortical Astrocytes
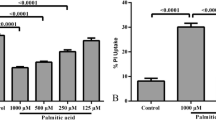
To determine if PA affected cell viability, MTT and FDA assays were performed. Treatment with 250 or 500 μM PA significantly decreased MTT values in male and female astrocytes compared to control values (Fig. 4a). Similar results were obtained with the FDA assay (Fig. 4b). Therefore, the results of both MTT and FDA assays indicate that PA treatment for 24 h decreases astrocyte viability.

Astrocyte viability was reduced after 24 h of treatment with PA. Male and female astrocytes were treated with vehicle (C), 250 μM PA (PA 250), or 500 μM PA (PA 500). a Cell viability measured by MTT assay. b Cell viability measured by FDA staining. For MTT data, two-way ANOVA revealed a significant effect of both treatment [F(2,24) = 60.553; p = 0.000] and sex [F(1,24) = 23.126; p = 0.000], with a significant interaction between the two factors [F(2,24) = 5.944; p = 0.008]. For FDA staining, two-way ANOVA showed a significant effect of both treatment [F(2,24) = 34.809; p = 0.000] and sex [F(1,24) = 5.138; p = 0.033], without a significant interaction between the two factors. To analyze the effect of treatment, data were split by sex and one-way ANOVA was carried out. There was a significant treatment effect in both male [F(2,12) = 17.806; p = 0.000] and female [F(2,12) = 17.348; p = 0.000] astrocytes. Data are represented as mean ± SEM. Sample size N = 5. Significant differences with the post hoc test: two asterisks and three asterisks, significant differences (p < 0.01 and p < 0.001) versus control male astrocytes. Two number signs and three number signs, significant differences (p < 0.01 and p < 0.001) versus control female astrocytes. Three dollar signs, significant differences (p < 0.001) versus male astrocytes treated with PA 250
PA Decreases the Expression of Estrogen Receptors in Female Astrocytes
Due to the close relationship between estradiol and brain metabolism and to the existence of sex differences in the effect of PA on the expression of inflammatory genes, we decided to study the expression of estrogen receptors (ERs) after PA treatment for 24 h. Compared to control values, the treatment with either 250 or 500 μM PA decreased the expression of ERα, ERβ, and GPER, but exclusively in female astrocytes (Fig. 5).

PA decreased the expression of estrogen receptors in female astrocytes. Male and female astrocytes were treated with vehicle (C), 250 μM PA (PA 250), or 500 μM PA (PA 500) for 24 h. a ERα mRNA levels. b ERβ mRNA levels. c GPER mRNA levels. For ERα mRNA levels, two-way ANOVA revealed a significant treatment effect [F(2,23) = 4.946; p = 0.016] without sex effect or interaction between treatment and sex. One-way ANOVA showed a significant effect of treatment, but only in female astrocytes [F(2,11) = 21.163; p = 0.000]. In the case of ERβ mRNA levels, treatment was the only factor that had a significant effect in two-way ANOVA [F(2,22) = 6.116; p = 0.008]. Due to the lack of interaction between sex and treatment, one-way ANOVA was performed on data split by sex. A significant effect of treatment was found in female astrocytes [F(2,11) = 32.022; p = 0.000]. For GPER mRNA levels, two-way ANOVA revealed significant effects of both treatment [F(2,23) = 5.398; p = 0.012] and sex [F(1,23) = 6.757; p = 0.016], with no interaction between the two factors. The effect of treatment was analyzed for each sex by one-way ANOVA, showing a significant treatment effect, but only in female astrocytes [F(2,12) = 11.961; p = 0.001]. Data are represented as mean ± SEM. Sample size N ≥ 4. Significant differences with the post hoc test: One number sign, two number signs, three number signs, significant differences (p < 0.05, p < 0.01, and p < 0.001) versus control female astrocytes. Accent symbol, significant differences (p < 0.05) versus male astrocytes treated with PA 500
Estrogenic Compounds Are Unable to Prevent PA-Induced Cell Death
Since estrogenic compounds exert anti-inflammatory effects in astrocytes and since PA modified the expression of ERs in female astrocytes, we assessed whether the activation of ERs was able to prevent PA-induced cell death. Male and female astrocytes were pretreated with different concentrations of estradiol (10−10, 10−9, and 10−8 M) for 24 h and then exposed to 250 μM PA and estradiol for an additional 24 h. As shown previously, PA induced a significant cell death assessed by MTT assay in both male and female astrocytes. Estradiol per se did not affect MTT levels compared to control values. Astrocytes treated with both estradiol and PA showed similar MTT levels as astrocytes treated with PA alone (Fig. S1).
Since ERα, ERβ, and GPER may have different transcriptional effects and even opposite actions, we assessed the effect of selective ER agonists. Male and female astrocytes were pretreated with the ER agonists (ERα agonist PPT, ERβ agonist DPN, and GPER agonist G1) for 24 h and then exposed to 250 μM PA in the presence of the ER agonists for an additional 24 h. Neither PPT, DPN, nor G1 affected MTT levels in vehicle-treated or PA-treated astrocytes (Fig. S2).
Since astrocytes in vitro express the enzyme aromatase and are able to produce estradiol, we evaluated the possibility for a protective effect of endogenous estradiol synthesis. Thus, we treated male and female astrocytes with 10−7 M letrozole, an aromatase inhibitor, hypothesizing that the inhibition of the synthesis of estradiol will increase cell death induced by PA. However, letrozole did not significantly affect the levels of MTT induced by PA (Fig. S3). These results suggest that the functional impairment induced by PA in primary astrocytes is so strong as to preclude the protective action of estrogenic compounds, which are known to reduce damage in these cells when exposed to other insults.
Rapamycin Does Not Prevent the Effect of PA on Astrocyte Viability
Since our results suggested that PA was blocking autophagy, we tested whether rapamycin, an autophagy inducer [47], could prevent the effect of PA on cell viability. First, we assessed the levels of phospho-mTOR after 26 h of treatment with rapamycin, to verify that rapamycin was inhibiting its target, mTOR (Fig. 6a). Furthermore, we also analyzed LC3-II levels to confirm that rapamycin was increasing autophagy (Fig. 6b). Then, astrocytes were pretreated with rapamycin for 2 h and then in combination with 250 μM PA for 24 h and cell viability was analyzed by MTT and FDA assays. Rapamycin per se did not affect MTT and FDA values compared to control cells and when combined with PA did not alter the decrease in MTT and FDA values induced by PA alone (Fig. 6c, d). Therefore, rapamycin was unable to prevent PA-induced cell death.

Rapamycin did not protect astrocytes from PA-induced cell death. Male and female astrocytes were treated with vehicle (C), rapamycin (R), 250 μM PA (PA), or rapamycin combined with PA (PA + R) for 24 h. a Representative image of phospho-mTOR (p-mTOR) detection by Western blot to verify mTOR inhibition by rapamycin. b Representative image of LC3 detection by Western blot to verify that rapamycin was inducing autophagy. c Cell viability measured by MTT assay. d Cell viability measured by FDA staining. Two-way ANOVA of MTT results showed a significant effect of treatment [F(3,24) = 32.266; p = 0.000] and sex [F(1,24) = 9.462; p = 0.005]. As there was no interaction between treatment and sex, the effect of treatment was analyzed by one-way ANOVA in data split by sex. This revealed a significant effect of treatment in males [F(3,8) = 19.789; p = 0.000] and females [F(3,16) = 17.202; p = 0.000]. Two-way ANOVA of FDA data showed a significant effect of treatment [F(3,24) = 26.307; p = 0.000] without sex effect or interaction between sex and treatment. One-way ANOVA was carried out, and treatment effect was detected separately in males [F(3,8) = 10.559; p = 0.004] and females [F(3,16) = 16.893; p = 0.000]. Data are represented as mean ± SEM. Sample size N ≥ 3. Significant differences with the post hoc test: one asterisk and two asterisks, significant differences (p < 0.05 and p < 0.01) versus control male astrocytes. Two number signs and three number signs, significant differences (p < 0.01 and p < 0.001) versus control female astrocytes. One percent symbol and two percent symbols, significant differences (p < 0.05 and p < 0.01) versus male astrocytes treated with rapamycin. Two ampersands and three ampersands, significant differences (p < 0.01 and p < 0.001) versus female astrocytes treated with rapamycin
PA Blocks Autophagic Activity Before the Induction of Inflammation and Astrocytic Cell Death
Due to the interconnection between autophagy and cell death, especially with the mechanism of apoptosis [48], changes in cell viability could be considered as a confounding factor in our experimental model. To avoid this potential problem, we studied the effect of PA on autophagy using a treatment of only 4.5 h, since at this time point PA did not reduce cell viability in astrocytes (Fig. S4) and did not significantly affect the expression of TNF-α, IL-1β, IL-6, or IL-10 (Fig. S5).
The treatment with 500 μM PA for 4.5 h induced an increase in LC3-II protein levels in female astrocytes (Fig. 7a). The treatment with 250 or 500 μM PA for 4.5 h induced an increase in p62 expression in astrocytes from males. Moreover, only 500 μM PA induced a significant increase in the levels of p62 protein in astrocytes from females (Fig. 7b). The mRNA expression of MAP1LC3B and SQSTM1 was also assessed, but no significant effect of the treatment was detected (Fig. 7c, d).

PA promoted a blockade in autophagic activity after a treatment for 4.5 h. Male and female astrocytes were treated with vehicle (C), HCQ, 250 μM PA (PA 250), or 500 μM PA (PA 500). a LC3-II protein levels. Representative images for LC3-II blot were run in the same membrane for each sex. b p62 protein levels. Representative images for p62 blot were run in the same membrane for each sex. c MAP1LC3B mRNA levels. d SQSTM1 mRNA levels. e Representative image of LC3-II detection by Western blot used to quantify LC3-II flux. f LC3-II flux measured as the ratio of LC3-II levels in cells treated with HCQ to LC3-II levels in cells untreated with HCQ of the same experimental group. For LC3-II protein levels, two-way ANOVA revealed a significant treatment effect [F(2,18) = 8.284; p = 0.003]. One-way ANOVA of data split by sex showed a significant treatment effect only in astrocytes from females [F(2,9) = 7.242; p = 0.013]. For p62 protein expression, two-way ANOVA showed a significant treatment effect [F(2,18) = 17.086; p = 0.000]. After splitting data by sex, one-way ANOVA revealed a significant treatment effect in both male [F(2,9) = 11.841; p = 0.003] and female [F(2,9) = 5.485; p = 0.028] astrocytes. For the mRNA levels of MAP1LC3B and SQSTM1, two-way ANOVA revealed no significant effects of treatment or sex. For LC3-II flux, two-way ANOVA revealed a significant effect of the treatment [F(2,18) = 40.403; p = 0.000]. Treatment effect was studied by one-way ANOVA for each sex, which showed a significant effect in male [F(2,9) = 43.339; p = 0.000] and female [F(2,9) = 10.110; p = 0.005] astrocytes. Data are represented as mean ± SEM. Sample size N ≥ 3. Significant differences with the post hoc test: one asterisk, two asterisks, and three asterisks, significant differences (p < 0.05, p < 0.01, and p < 0.001) versus control male astrocytes. One number sign and two number signs, significant differences (p < 0.05 and p < 0.01) versus control female astrocytes
We were also interested in determining the flux of autophagy after a short treatment with PA, as we did previously with a treatment for 24 h. Astrocytes were co-treated with PA and HCQ for 4.5 h for this purpose, and protein expression of LC3-II was analyzed to determine LC3-II flux. Both doses of PA decreased LC3-II flux in male and female astrocytes (Fig. 7e, f). These findings suggest that PA causes a blockade in the autophagic flux before the induction of cell death and inflammation.
Hydroxychloroquine Does Not Affect Cell Viability but Increases the Toxic Effect of PA
To determine whether the blockade of autophagic flux per se causes decreased astrocyte viability, the cultures were treated for 4.5 or 24 h with HCQ to inhibit the fusion of autophagosomes with lysosomes. First, we verified that HCQ treatment for 4.5 h was able to block autophagy and generate LC3-II accumulation: HCQ induced an increase in LC3-II levels in male and female astrocytes (Fig. 8a). Then, cell viability was analyzed by MTT after a treatment with HCQ and PA for 4.5 or 24 h. No significant effect of the treatments was detected at 4.5 h (Fig. 8b). However, HCQ decreased cell viability in male and female astrocytes treated for 24 h with 250 or 500 μM PA compared to the effect of PA alone (Fig. 8c). Therefore, these findings indicate that HCQ per se does not affect cell viability but increases the toxic effect of PA at 24 h.

HCQ increased the toxic effect of PA after a treatment for 24 h. Male and female astrocytes were treated with vehicle (C), HCQ, 250 μM PA (PA 250), or 500 μM PA (PA 500) for 4.5 or 24 h. a LC3-II protein levels after HCQ treatment for 4.5 h. b Cell viability measured by MTT assay after a treatment for 4.5 h. c Cell viability measured by MTT assay after a treatment for 24 h. For LC3-II protein levels, two-way ANOVA revealed a significant effect of treatment [F(1,12) = 82.074; p = 0.000] without sex effect or interaction between treatment and sex. For MTT data after the treatment with PA and HCQ for 4.5 h, two-way ANOVA showed a significant effect of sex [F(1,48) = 17.107; p = 0.000] but no effect of treatment. In contrast for MTT data after the treatment with PA and HCQ for 24 h, two-way ANOVA showed a significant effect of the treatment [F(5,48) = 163.123; p = 0.000] without interaction between sex and treatment. To study the effect of treatment, data were split by sex and a significant effect of treatment was found in male [F(5,24) = 110.512; p = 0.000] and female [F(5,24) = 64.338; p = 0.000] astrocytes. Data are represented as mean ± SEM. Sample size N ≥ 4. Significant differences with the post hoc test: two asterisks and three asterisks, significant differences (p < 0.01 and p < 0.001) versus control male astrocytes. One number sign, two number signs, and three number signs, significant differences (p < 0.05, p < 0.01, and p < 0.001) versus control female astrocytes. Three dollar signs, significant differences (p < 0.001) versus male astrocytes treated with PA 250. Three at signs, significant differences (p < 0.001) versus female astrocytes treated with PA 250. Three accent symbols, significant differences (p < 0.001) versus male astrocytes treated with PA 500. Three plus signs, significant differences (p < 0.001) versus female astrocytes treated with PA 500. Letter C with cedilla (Ç), significant differences (p < 0.05) versus male astrocytes treated with HCQ
Prolonged Exposure to PA Increases the Expression of a Marker of Endoplasmic Reticulum Stress
Since autophagy, endoplasmic reticulum stress, and cell viability are interrelated events, we assessed the effect of PA on the induction of C/EBP-homologous protein (CHOP), a transcription factor associated to apoptosis when endoplasmic reticulum is seriously impaired [49]. CHOP expression was not induced in astrocytes after the treatment with PA for 4.5 h (Fig. 9). In contrast, CHOP expression was increased by PA after 24 h of treatment and by tunicamycin, an inducer of endoplasmic reticulum stress, used as a positive control. Cells were also treated with HCQ to determine if the blockade of autophagy per se induced CHOP expression in astrocytes. HCQ treatment did not induce CHOP and did not modify the effect of PA on CHOP levels (Fig. 9). The same results were obtained in male and female astrocytes (data not shown). These findings suggest that CHOP is induced in astrocytes after a long exposure to PA, but the cause of this induction is not the blockade of autophagy per se.

PA induced CHOP expression in astrocytes after 24 h of treatment. Astrocytes were treated with tunicamycin (Tun), vehicle (C), HCQ, 250 μM PA (PA), and PA combined with HCQ (PA + HCQ). Representative image of CHOP detection by Western blot. The experiment was repeated four times
Discussion
The results of this study indicate that PA blocks autophagy in primary cortical astrocytes from male and female mice pups. The effect on autophagy was observed after 4.5 or 24 h of treatment with concentrations of 250 or 500 μM PA. In addition, PA increased the expression of proinflammatory cytokines, decreased cell viability, and increased the levels of the endoplasmic reticulum stress marker CHOP, but only after 24 h of treatment. The effect of PA on cell viability was probably not a direct consequence of the autophagy impairment, since rapamycin, an autophagy inducer, was unable to prevent the effect of PA acid on cell viability and HCQ, an autophagy blocker, did not cause per se astrocyte cell death.
In agreement with previous studies [21,22,23, 50], we have observed that the treatment for 24 h with PA induced cell death in astrocytes. Our results also extend previous findings obtained in brain rat astrocytes, in which 100–400 μM PA increased the release of IL-6 and TNF-α [23]. PA has been also reported to increase the expression of IL-1β and TNF-α in rat cortical astrocytes [51]; of IL-6, TNF-α, and IL-1β in mouse cortical astrocytes [21, 52]; of IL-6 in rat hippocampal astrocytes [53]; and of IL-6 and IL-1β in mouse hypothalamic astrocytes [17].
In our study, we have examined the inflammatory response of astrocytes, analyzing separately the results in male and female cells. Sex differences in the inflammatory response of astrocytes have been previously described in response to PA [17], lipopolysaccharide (LPS) [54, 55], and the insecticide dimethoate (DMT) [56]. In these studies, the inflammatory response was in general higher in male than in female astrocytes. Thus, in hypothalamic mouse astrocytes, PA increases the expression of IL-6 and IL-1β in male and female cells, but the effect was statistically higher in male cells [17]. In primary cortical mouse astrocytes, LPS induced an increased expression of IL-6, TNF-α, and IL-1β in male cells compared to female cells [54]. A similar effect was observed with DMT in primary mouse cortical astrocytes; DMT increased the expression of IL-6, TNF-α, and IL-1β in male but not in female astrocytes [56]. In rat cortical astrocyte cultures, LPS increased the expression of IL-1β in both male and female cells, but the response was higher in male astrocytes [55]. Our present results, showing that the treatment with PA (500 μM) for 24 h resulted in a significant increase in the expression of TNF-α and IL-1β, but only in male astrocytes, extend these previous findings and confirm that PA also elicits a sex-dimorphic inflammatory response in cortical astrocytes, as shown previously for hypothalamic astrocytes [17].
Morselli et al. [17] have shown that PA induces a decrease in the expression of ERα in male hypothalamic astrocytes but not in female astrocytes. They postulated that the decreased ERα expression in male astrocytes contributes to their increased inflammatory response compared to female cells. Estradiol is known to reduce the inflammatory response of astrocytes [57]. Furthermore, primary astrocytes express the enzyme aromatase and are able to produce estradiol [58]. Therefore, it is possible that estradiol synthesized by astrocytes may contribute to reduce inflammation when exposed to PA. In the present study, we have assessed the expression of estrogen receptors (ERα, ERβ, and GPER) in astrocytes exposed to vehicle or PA. In agreement with the results obtained in hypothalamic astrocytes [17], treatment with PA caused a decreased expression of ERα in cortical astrocytes. However, an important difference is that in cortical astrocytes PA decreased ERα expression in female cells, while in hypothalamic astrocytes, PA decreased ERα expression in male cells. The decreased expression of ERα in female astrocytes exposed to PA was accompanied by a decreased expression of ERβ and GPER in these cells. In contrast, PA did not affect the expression of ERs in male cortical astrocytes. Our findings, together with those of Morselli et al. [17], indicate that hypothalamic and cortical astrocytes respond to PA with sex differences in the expression of ERs. However, this sex-specific response is different in hypothalamic and cortical astrocytes. The functional implications of this difference remain to be established. In the hypothalamus, astrocytes may contribute to the generation of sex differences in the inflammatory response caused by high-fat diet [17]. On the other hand, PA levels increase in the cerebral cortex after brain injury [59] and in the parietal cortex of Alzheimer’s disease patients [60]. Since astrocytes mediate the effect of PA on the development of Alzheimer’s disease-like alterations in cortical neurons [20, 51], the sex differences observed in the response of cortical astrocytes to PA may be also relevant for the generation of sex differences in neurodegenerative diseases.
Given the existence of sex differences in the expression of ERs in cortical astrocytes after PA treatment and since estradiol is known to decrease the inflammatory response of astrocytes [17, 57], we assessed the potential protective effects of estradiol, selective ER agonists, and local estradiol synthesis by astrocytes against the effects of PA on cell viability. Our findings, showing that the toxic effect of PA is not prevented by estrogenic molecules that are known to decrease inflammation in astrocytes, suggest that in addition to inflammation other mechanisms operate to cause PA-induced cell death.
To explore the mechanisms that mediate PA-induced astrocyte cell death, we assessed the effect of PA on autophagy. Under physiological conditions, autophagy is a mechanism to mobilize lipids in cells by digesting lipid droplets, where FFAs are stored [61, 62]. However, an excess of FFAs, in particular high levels of PA, are known to suppress autophagy in different cell types, such as pancreatic β-cells [30, 32, 33] and hepatocytes [31, 63]. PA impairs lysosomal acidification and therefore the fusion of autophagosomes with lysosomes [30, 33, 64], and this seems to be the cause of the inhibition of the autophagic flux [33].
In H9C2 cardiomyoblasts, PA impairs fatty acid oxidation and induces the accumulation of lipids in the endoplasmic reticulum, causing endoplasmic reticulum stress and cell death [65]. Since autophagy inhibits endoplasmic reticulum stress [66], the suppression of autophagy by long treatments with PA results in increased endoplasmic reticulum stress in hepatocytes and pancreatic β-cells, which finally causes cell death [31, 32]. Therefore, our hypothesis was that an impairment of autophagy is involved in the toxic effect of PA on astrocytes.
We have found that PA increases the protein levels of LC3-II and p62 in astrocytes. LC3-II, a phosphatidylethanolamine conjugate of LC3-I, is recruited to autophagosomal membranes where it binds to p62 [67]. Therefore, the increased protein levels of LC3-II suggest an increased formation or an increased accumulation of autophagosomes. To discriminate between these two possibilities, we determined the effect of PA on autophagic flux. For this purpose, we treated astrocytes with HCQ, which blocks the fusion of autophagosomes with lysosomes [28, 67]. PA significantly reduced the ratio between LC3-II protein levels in presence of HCQ and LC3-II protein levels in absence of HCQ. Since this ratio is an indicator of autophagic flux [28], our results suggest a suppression of autophagy by PA in both male and female astrocytes. This is also supported by the increased accumulation of large p62 immunoreactive bodies in the cytoplasm of PA-treated astrocytes, since p62 accumulation is observed in the cytoplasm of cells with impaired autophagy [46, 68, 69].
In addition, PA decreased the mRNA levels of MAP1LC3B (the gene encoding for LC3) in female astrocytes and increased the mRNA levels of SQSTM1 (the gene encoding for p62) in astrocytes of both sexes. These mRNA changes were observed after the treatment for 24 h with PA but not after the treatment with PA for 4.5 h, suggesting that changes in mRNA levels at 24 h may be a transcriptional response elicited by a long period of autophagy inhibition as an attempt to increase autophagy by the upregulation of p62 [70, 71]. Interestingly, by 4.5 h of treatment with PA, cell viability was not affected and the expression of inflammatory markers in astrocytes was not increased yet. This indicates that the impairment in autophagy precedes the effects of PA on inflammation and cell viability.
The observation that the effect of PA in reducing cell viability at 24 h was enhanced by the blockage of autophagic flux with HCQ suggests that autophagy impairment is involved in the effect of PA on astrocyte cell death. However, the treatment of astrocytes with rapamycin, an autophagy inductor [47], did not decrease the effect of PA on cell viability. Furthermore, the inhibition of autophagy by HCQ did not decrease per se cell viability. These observations, together with the finding that the inhibition of autophagic flux by PA at 4.5 h did not result in increased cell death, suggest that impairment of autophagy in astrocytes is not sufficient per se to decrease cell viability. Indeed, the effect of PA on cell viability was only observed after a treatment for 24 h, when PA also induced an increase in inflammation and in the expression of CHOP. These observations are in agreement with the results obtained in other cell types in which prolonged treatment with PA induced cell death in parallel to the suppression of autophagic flux and the stimulation of endoplasmic reticulum stress [31, 32]. Therefore, our findings suggest that the PA induces cell death when, in addition to blocking autophagy, it causes other alterations in astrocytes, such as increased inflammation and endoplasmic reticulum stress. These additional alterations may be, at least in part, a consequence of the permanent autophagic impairment. For instance, the progressive accumulation of lipids in the endoplasmic reticulum of cells treated by PA [65], together with the lack of control of endoplasmic reticulum stress by the impaired autophagy [66], may overcome the endogenous cellular homeostatic mechanisms to promote cell survival, causing cell death.
References
World Health Organization (2017) Obesity and overweight. WHO Media Center. http://www.who.int/mediacentre/factsheets/fs311/en. Accessed 19 March 2018
Ward MA, Carlsson CM, Trivedi MA, Sager MA, Johnson SC (2005) The effect of body mass index on global brain volume in middle-aged adults: a cross sectional study. BMC Neurol 5:23. https://doi.org/10.1186/1471-2377-5-23
Raji CA, Ho AJ, Parikshak NN, Becker JT, Lopez OL, Kuller LH, Hua X, Leow AD et al (2010) Brain structure and obesity. Hum Brain Mapp 31:353–364. https://doi.org/10.1002/hbm.20870
Gunstad J, Paul R, Cohen R, Tate D, Spitznagel M, Gordon E (2007) Elevated body mass index is associated with executive dysfunction in otherwise healthy adults. Compr Psychiatry 48:57–61. https://doi.org/10.1016/j.comppsych.2006.05.001
Cheke LG, Bonnici HM, Clayton NS, Simons JS (2017) Obesity and insulin resistance are associated with reduced activity in core memory regions of the brain. Neuropsychologia 96:137–149. https://doi.org/10.1016/j.neuropsychologia.2017.01.013
Profenno LA, Porsteinsson AP, Faraone SV (2010) Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiatry 67:505–512. https://doi.org/10.1016/j.biopsych.2009.02.013
Montgomery MK, Hallahan NL, Brown SH, Liu M, Mitchell TW, Cooney GJ, Turner N (2013) Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia 56:1129–1139. https://doi.org/10.1007/s00125-013-2846-8
Stranahan AM, Hao S, Dey A, Yu X, Baban B (2016) Blood–brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J Cereb Blood Flow Metab 36:2108–2121. https://doi.org/10.1177/0271678X16642233
Molteni R, Barnard RJ, Ying Z, Roberts CK, Gómez-Pinilla F (2002) A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience 112:803–814. https://doi.org/10.1016/S0306-4522(02)00123-9
Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP (2008) Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 18:1085–1088. https://doi.org/10.1002/hipo.20470
Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95:2409–2415. https://doi.org/10.1172/JCI117936
Thaler J, Yi C, Schur E, Guyenet S, Hwang B, Dietrich M, Zhao X, Sarruf D et al (2012) Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122:153–162. https://doi.org/10.1172/JCI59660
Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D (2008) Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135:61–73. https://doi.org/10.1016/j.cell.2008.07.043
Almeida-Suhett CP, Graham A, Chen Y, Deuster P (2017) Behavioral changes in male mice fed a high-fat diet are associated with IL-1β expression in specific brain regions. Physiol Behav 169:130–140. https://doi.org/10.1016/j.physbeh.2016.11.016
Franssen R, Monajemi H, Stroes ES, Kastelein JJ (2011) Obesity and dyslipidemia. Med Clin North Am 95:893–902. https://doi.org/10.1016/j.mcna.2011.06.003
Smith QR, Nagura H (2001) Fatty acid uptake and incorporation in brain: studies with the perfusion model. J Mol Neurosci 16:167–172; discussion 215–21. https://doi.org/10.1385/JMN:16:2-3:167
Morselli E, Fuente-Martin E, Finan B, Kim M, Frank A, Garcia-Caceres C, Navas CR, Gordillo R et al (2014) Hypothalamic PGC-1α protects against high-fat diet exposure by regulating ERα. Cell Rep 9:633–645. https://doi.org/10.1016/j.celrep.2014.09.025
Karmi A, Iozzo P, Viljanen A, Hirvonen J, Fielding BA, Virtanen K, Oikonen V, Kemppainen J et al (2010) Increased brain fatty acid uptake in metabolic syndrome. Diabetes 59:2171–2177. https://doi.org/10.2337/db09-0138
Yanguas-Casás N, Crespo-Castrillo A, de Ceballos ML, Chowen JA, Azcoitia I, Arevalo MA, Garcia-Segura LM (2018) Sex differences in the phagocytic and migratory activity of microglia and their impairment by palmitic acid. Glia 66:522–537. https://doi.org/10.1002/glia.23263
Patil S, Melrose J, Chan C (2007) Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur J Neurosci 26:2131–2141. https://doi.org/10.1111/j.1460-9568.2007.05797.x
Wang Z, Liu D, Wang J, Liu S, Gao M, Ling EA, Hao A (2012) Cytoprotective effects of melatonin on astroglial cells subjected to palmitic acid treatment in vitro. J Pineal Res 52:253–264. https://doi.org/10.1111/j.1600-079X.2011.00952.x
Wong KL, Wu YR, Cheng KS, Chan P, Cheung CW, Lu DY, Su TH, Liu ZM et al (2014) Palmitic acid-induced lipotoxicity and protection by (+)-catechin in rat cortical astrocytes. Pharmacol Reports 66:1106–1113. https://doi.org/10.1016/j.pharep.2014.07.009
Gupta S, Knight AG, Gupta S, Keller JN, Bruce-Keller AJ (2012) Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J Neurochem 120:1060–1071. https://doi.org/10.1111/j.1471-4159.2012.07660.x
Freire-Regatillo A, Argente-Arizón P, Argente J, García-Segura LM, Chowen JA (2017) Non-neuronal cells in the hypothalamic adaptation to metabolic signals. Front Endocrinol (Lausanne) 8:51. https://doi.org/10.3389/fendo.2017.00051
Blázquez C, Galve-Roperh I, Guzmán M (2000) De novo-synthesized ceramide signals apoptosis in astrocytes via extracellular signal-regulated kinase. FASEB J 14:2315–2322. https://doi.org/10.1096/fj.00-0122com
Rabinowitz JD, White E (2010) Autophagy and metabolism. Science 330:1344–1348. https://doi.org/10.1126/science.1193497
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT et al (2017) Molecular definitions of autophagy and related processes. EMBO J 36:1811–1836. https://doi.org/10.15252/embj.201796697
Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D et al (2005) Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25:1025–1040. https://doi.org/10.1128/MCB.25.3.1025
Ravanan P, Srikumar IF, Talwar P (2017) Autophagy: the spotlight for cellular stress responses. Life Sci 188:53–67. https://doi.org/10.1016/j.lfs.2017.08.029
Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS (2011) Fatty acids suppress autophagic turnover in B-cells. J Biol Chem 286:42534–42544. https://doi.org/10.1074/jbc.M111.242412
González-Rodríguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillón J, Lo Iacono O et al (2014) Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis 5:e1179. https://doi.org/10.1038/cddis.2014.162
Mir SUR, George NM, Zahoor L, Harms R, Guinn Z, Sarvetnick NE (2015) Inhibition of autophagic turnover in β-cells by fatty acids and glucose leads to apoptotic cell death. J Biol Chem 290:6071–6085. https://doi.org/10.1074/jbc.M114.605345
Trudeau KM, Colby AH, Zeng J, Las G, Feng JH, Grinstaff MW, Shirihai OS (2016) Lysosome acidification by photoactivated nanoparticles restores autophagy under lipotoxicity. J Cell Biol 214:25–34. https://doi.org/10.1083/jcb.201511042
Liu D, Ke Z, Luo J (2017) Thiamine deficiency and neurodegeneration: the interplay among oxidative stress, endoplasmic reticulum stress, and autophagy. Mol Neurobiol 54:5440–5448. https://doi.org/10.1007/s12035-016-0079-9
Qian M, Fang X, Wang X (2017) Autophagy and inflammation. Clin Transl Med 6:24. https://doi.org/10.1186/s40169-017-0154-5
Palmer BF, Clegg DJ (2015) The sexual dimorphism of obesity. Mol Cell Endocrinol 402:113–119. https://doi.org/10.1016/j.mce.2014.11.029
Chowen JA, Argente-Arizón P, Freire-Regatillo A, Argente J (2018) Sex differences in the neuroendocrine control of metabolism and the implication of astrocytes. Front Neuroendocrinol 48:3–12. https://doi.org/10.1016/j.yfrne.2017.05.003
Ruiz-Palmero I, Simon-Areces J, Garcia-Segura LM, Arevalo MA (2011) Notch/Neurogenin 3 signalling is involved in the neuritogenic actions of oestradiol in developing hippocampal neurones. J Neuroendocrinol 23:355–364. https://doi.org/10.1111/j.1365-2826.2011.02110.x
Grassi D, Bellini MJ, Acaz-Fonseca E, Panzica G, Garcia-Segura LM (2013) Estradiol and testosterone regulate arginine-vasopressin expression in SH-SY5Y human female neuroblastoma cells through estrogen receptors-α and -β. Endocrinology 154:2092–2100. https://doi.org/10.1210/en.2012-2137
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45–e445. https://doi.org/10.1093/nar/29.9.e45
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP (2004) Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol Lett 26:509–515. https://doi.org/10.1023/B:BILE.0000019559.84305.47
Portovedo M, Ignacio-Souza LM, Bombassaro B, Coope A, Reginato A, Razolli DS, Torsoni MA, Torsoni AS et al (2015) Saturated fatty acids modulate autophagy’s proteins in the hypothalamus. PLoS One 10:e0119850. https://doi.org/10.1371/journal.pone.0119850
Yamato M, Shiba T, Yoshida M, Ide T, Seri N, Kudou W, Kinugawa S, Tsutsui H (2007) Fatty acids increase the circulating levels of oxidative stress factors in mice with diet-induced obesity via redox changes of albumin. FEBS J 274:3855–3863. https://doi.org/10.1111/j.1742-4658.2007.05914.x
Alsahli A, Kiefhaber K, Gold T, Muluke M, Jiang H, Cremers S, Schulze-Späte U (2016) Palmitic acid reduces circulating bone formation markers in obese animals and impairs osteoblast activity via C16-ceramide accumulation. Calcif Tissue Int 98:511–519. https://doi.org/10.1007/s00223-015-0097-z
Benito-Cuesta I, Diez H, Ordoñez L, Wandosell F (2017) Assessment of autophagy in neurons and brain tissue. Cells 6:25. https://doi.org/10.3390/cells6030025
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata JI et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149–1163. https://doi.org/10.1016/j.cell.2007.10.035
Blommaart EF, Luiken JJ, Blommaart PJ, Van Woerkom GM, Meijer AJ (1995) Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J Biol Chem 270:2320–2326. https://doi.org/10.1074/jbc.270.5.2320
Fitzwalter BE, Thorburn A (2015) Recent insights into cell death and autophagy. FEBS J 282:4279–4288. https://doi.org/10.1111/febs.13515
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11:381–389. https://doi.org/10.1038/sj.cdd.4401373
González-Giraldo Y, Garcia-Segura LM, Echeverria V, Barreto GE (2018) Tibolone preserves mitochondrial functionality and cell morphology in astrocytic cells treated with palmitic acid. Mol Neurobiol 55:4453–4462. https://doi.org/10.1007/s12035-017-0667-3
Liu L, Martin R, Chan C (2013) Palmitate-activated astrocytes via serine palmitoyltransferase increase BACE1 in primary neurons by sphingomyelinases. Neurobiol Aging 34:540–550. https://doi.org/10.1016/j.neurobiolaging.2012.05.017
Su X, Chu Y, Kordower JH, Li B, Cao H, Huang L, Nishida M, Song L et al (2015) PGC-1α promoter methylation in Parkinson’s disease. PLoS One 10:e0134087. https://doi.org/10.1371/journal.pone.0134087
Frago LM, Canelles S, Freire-Regatillo A, Argente-Arizón P, Barrios V, Argente J, Garcia-Segura LM, Chowen JA (2017) Estradiol uses different mechanisms in astrocytes from the hippocampus of male and female rats to protect against damage induced by palmitic acid. Front Mol Neurosci 10:330. https://doi.org/10.3389/fnmol.2017.00330
Santos-Galindo M, Acaz-Fonseca E, Bellini MJ, Garcia-Segura LM (2011) Sex differences in the inflammatory response of primary astrocytes to lipopolysaccharide. Biol Sex Differ 2:7. https://doi.org/10.1186/2042-6410-2-7
Loram LC, Sholar PW, Taylor FR, Wiesler JL, Babb JA, Strand KA, Berkelhammer D, Day HEW et al (2012) Sex and estradiol influence glial pro-inflammatory responses to lipopolysaccharide in rats. Psychoneuroendocrinology 37:1688–1699. https://doi.org/10.1016/j.psyneuen.2012.02.018
Astiz M, Acaz-Fonseca E, Garcia-Segura LM (2014) Sex differences and effects of estrogenic compounds on the expression of inflammatory molecules by astrocytes exposed to the insecticide dimethoate. Neurotox Res 25:271–285. https://doi.org/10.1007/s12640-013-9417-0
De Marinis E, Acaz-Fonseca E, Arevalo MA, Ascenzi P, Fiocchetti M, Marino M, Garcia-Segura LM (2013) 17β-Oestradiol anti-inflammatory effects in primary astrocytes require oestrogen receptor β-mediated neuroglobin up-regulation. J Neuroendocrinol 25:260–270. https://doi.org/10.1111/jne.12007
Azcoitia I, Sierra A, Veiga S, Garcia-Segura LM (2003) Aromatase expression by reactive astroglia is neuroprotective. Ann N Y Acad Sci 1007:298–305. https://doi.org/10.1196/annals.1286.028
Pilitsis JG, Diaz FG, O’Regan MH, Phillis JW (2002) Differential effects of phospholipase inhibitors on free fatty acid efflux in rat cerebral cortex during ischemia-reperfusion injury. Brain Res 951:96–106. https://doi.org/10.1016/S0006-8993(02)03142-6
Fraser T, Tayler H, Love S (2010) Fatty acid composition of frontal, temporal and parietal neocortex in the normal human brain and in Alzheimer’s disease. Neurochem Res 35:503–513. https://doi.org/10.1007/s11064-009-0087-5
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM et al (2009) Autophagy regulates lipid metabolism. Nature 458:1131–1135. https://doi.org/10.1038/nature07976
Singh R, Cuervo AM (2012) Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol 2012:282041–282012. https://doi.org/10.1155/2012/282041
Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, Copple BL, Ding WX (2011) Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther 339:487–498. https://doi.org/10.1124/jpet.111.184341
Cnop M, Abdulkarim B, Bottu G, Cunha DA, Igoillo-Esteve M, Masini M, Turatsinze JV, Griebel T et al (2014) RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 63:1978–1993. https://doi.org/10.2337/db13-1383
Akoumi A, Haffar T, Mousterji M, Kiss RS, Bousette N (2017) Palmitate mediated diacylglycerol accumulation causes endoplasmic reticulum stress, Plin2 degradation, and cell death in H9C2 cardiomyoblasts. Exp Cell Res 354:85–94. https://doi.org/10.1016/j.yexcr.2017.03.032
Jung TW, Hong HC, Hwang HJ, Yoo HJ, Baik SH, Choi KM (2015) C1q/TNF-related protein 9 (CTRP9) attenuates hepatic steatosis via the autophagy-mediated inhibition of endoplasmic reticulum stress. Mol Cell Endocrinol 417:131–140. https://doi.org/10.1016/j.mce.2015.09.027
Yoshii SR, Mizushima N (2017) Monitoring and measuring autophagy. Int J Mol Sci 18:1–13. https://doi.org/10.3390/ijms18091865
Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, Johansen T (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–614. https://doi.org/10.1083/jcb.200507002
Filimonenko M, Stuffers S, Raiborg C, Yamamoto A, Malerød L, Fisher EMC, Isaacs A, Brech A et al (2007) Functional multivesicular bodies are required for autophagic clearance of protein aggregates associated with neurodegenerative disease. J Cell Biol 179:485–500. https://doi.org/10.1083/jcb.200702115
B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G et al (2013) The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41:7683–7699. https://doi.org/10.1093/nar/gkt563
Song C, Mitter SK, Qi X, Beli E, Rao HV, Ding J, Ip CS, Gu H et al (2017) Oxidative stress-mediated NFκB phosphorylation upregulates p62/SQSTM1 and promotes retinal pigmented epithelial cell survival through increased autophagy. PLoS One 12:1–26. https://doi.org/10.1371/journal.pone.0171940
Acknowledgements
We thank Elisa Baides Rosell and Nieves Lopez Andradas for excellent technical assistance.
Funding
This work was supported by grants from Ministerio de Economía, Industria y Competitividad (MINECO), Spain (grant numbers BFU2014–51836-C2-1-R and BFU2017-82754-R); Centro de Investigación Biomédica en Red de Fragilidad y Envejecimiento Saludable (CIBERFES), Instituto de Salud Carlos III, Madrid, Spain; and Fondos FEDER.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All the procedures involving animals used in this study followed the European Commission guidelines (2010/63/UE) and Spanish regulation (RD 53/2013) on the protection of animals for experimental use. These procedures were approved by our institutional animal care and use committee (Comité de Ética de Experimentación Animal del Instituto Cajal) and the Consejería del Medio Ambiente y Territorio (Comunidad de Madrid, PROEX 200/14).
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic Supplementary Material
ESM 1
(DOCX 1536 kb)
Rights and permissions
About this article

Cite this article
Ortiz-Rodriguez, A., Acaz-Fonseca, E., Boya, P. et al. Lipotoxic Effects of Palmitic Acid on Astrocytes Are Associated with Autophagy Impairment. Mol Neurobiol 56, 1665–1680 (2019). https://doi.org/10.1007/s12035-018-1183-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1183-9