
Abstract
Disease modeling has become challenging in the context of amyotrophic lateral sclerosis (ALS), as obtaining viable spinal motor neurons from postmortem patient tissue is an unlikely possibility. Limitations in the animal models due to their phylogenetic distance from human species hamper the success of translating possible findings into therapeutic options. Accordingly, there is a need for developing humanized models as a lead towards identifying successful therapeutic possibilities. In this study, human embryonic stem cells—BJNHem20—were differentiated into motor neurons expressing HB9, Islet1, and choline acetyl transferase using retinoic acid and purmorphamine. These motor neurons discharged spontaneous action potentials with two different frequencies (< 5 and > 5 Hz), and majority of them were principal neurons firing with < 5 Hz. Exposure to cerebrospinal fluid from ALS patients for 48 h induced several degenerative changes in the motor neurons as follows: cytoplasmic changes such as beading of neurites and vacuolation; morphological alterations, viz., dilation and vacuolation of mitochondria, curled and closed Golgi architecture, dilated endoplasmic reticulum, and chromatin condensation in the nucleus; lowered activity of different mitochondrial complex enzymes; reduced expression of brain-derived neurotrophic factor; up-regulated neurofilament phosphorylation and hyperexcitability represented by increased number of spikes. All these changes along with the enhanced expression of pro-apoptotic proteins—Bax and caspase 9—culminated in the death of motor neurons.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Degeneration of specific neuronal class of central nervous system leads to different diseases such as Amyotrophic Lateral Sclerosis (ALS), Parkinson’s disease, Alzheimer’s disease, and Huntington’s disease. ALS is a disease with onset at adulthood with an exception of few juvenile cases attributed to mutations in specific genes [1, 2]. ALS selectively affects the motor neurons of the cortex, brainstem, and spinal cord. Based on the etiology, ALS cases are classified into familial (FALS) and sporadic (SALS) cases. Ten percent are the FALS cases that have a dominant inheritance pattern, whereas the remaining 90% have unknown etiology [3].
The mechanisms underlying ALS are multifactorial and not yet fully understood. Current understanding of the ALS pathogenesis results from animal models or animal-derived cell culture systems. Transgenic mice and rats carrying the mutation in superoxide dismutase 1 (mSOD1) gene, rat embryonic spinal cord cultures, and neuroblastoma × spinal cord hybrid cell line-34 (NSC-34) exposed to cerebrospinal fluid (CSF) from ALS patients (ALS-CSF) or transfected with mSOD1 and rats infused with ALS-CSF are the commonly used ALS models [4,5,6,7]. Other models include (i) Wobbler mice with spontaneous mutation in vsp4 gene, (ii) rats expressing human TAR DNA-binding protein 43 (TDP-43)/ fused in sarcoma (FUS) mutations/M337V mutation in the neurofilament heavy chain promoter, and (iii) drosophila with mutations in SOD1/TDP43/FUS [8,9,10,11,12,13,14].
A recent publication from our laboratory also has clearly shown that ALS-CSF induced cytoplasmic mis-localization of TDP43 and FUS proteins in the NSC-34 motor neuron-like cell line [15]. These models have aided us in understanding the pathogenesis, progression and delineating different stages of the disease to a considerable extent. However, in many instances, these animal models may not faithfully reproduce the human syndrome majorly due to species differences. These models are constrained by the lack of disease reproducibility, response to therapeutic molecules, and extrapolation to human syndrome. For example, olesoxime and dexpramipexole which were effective in mouse models failed in phase III clinical trial emphasizing the need for better models of human origin [16, 17].
The use of postmortem-derived motor neurons for primary culture is not a feasible option due to the degenerative nature of the disease. Human Embryonic Stem Cells (hESCs) offer a renewable source for generating sufficient quantities of neurons, which could be successfully grown in culture. Motor neurons have been derived from mouse Embryonic Stem Cells (mESCs) using retinoic acid (RA) and Sonic hedgehog (SHH), which plays a critical role in in vivo development of motor neurons [18]. Motor neurons are also derived from hESCs and induced Pluripotent Stem Cells (iPSCs) obtained from fibroblasts of ALS patients by somatic cell reprogramming [19,20,21]. Patient-specific iPSCs are useful in derivation of neural phenotypes carrying disease-specific mutations and display characteristic disease features. However, to understand the pathogenesis of SALS, it would demand a larger repertoire of iPSC-derived motor neurons (iPSC-MNs) from several patients to arrive at a common conclusion; even the outcomes of these studies should be assessed cautiously taking into account the epigenetic changes during the somatic reprograming process. In addition, rodent models developed using CSF from sporadic ALS patients in our laboratory have helped us to recapitulate the pathophysiology of ALS [7, 22]. The aim of this study was to develop humanized in vitro model of SALS using ALS-CSF. In this line, we have differentiated hESCs into spinal motor neurons (hESC-MNs), characterized them by physiological read outs, and studied their response to ALS-CSF to understand the pathways involved in ALS-CSF-induced motor neuron degeneration.
Methodology
Procurement and Maintenance of Human Embryonic Stem Cells
This study was approved by the Institutional Ethics Committee and Steering Committee of Stem Cell Task Force, Department of Biotechnology, Government of India. Material access agreement was signed with Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR) and the human embryonic stem cell line—BJNhem20 (Bangalore JNCASR human embryonic stem cells-20) was procured [23]. BJNHem20 was grown on inactivated mouse (Carworth Farm 1 strain) embryonic fibroblasts (feeder layers) and maintained in stem cell culture medium, containing Knockout Dulbecco modified Eagle medium, 20% Knockout serum, 1× minimum essential medium—non-essential amino acids, 1× Glutamax I, 1× penicillin streptomycin solution, and 5 ng/mL basic fibroblast growth factor. After 3–5 days, hESC colonies were passaged by dissociating with Accutase at 37 °C for 5 min. Fresh medium was added, and the cell suspension was centrifuged at 1500 rpm for 5 min. The pellet was re-suspended in fresh medium and seeded on to fresh feeder layer for further experiments. Alternatively, hESCs were frozen in liquid nitrogen for future use.
Motor Neuron Derivation
Motor neurons were derived by using the established protocol with slight modifications [24]. Briefly, hESC colonies were removed from feeder layer as mentioned earlier. Dissociated cells were pelleted and transferred to a petri dish with the medium containing Dulbecco’s modified Eagle medium/nutrient mixture F-12, 1× Glutamax I, 1× penicillin streptomycin solution, and 20 ng/mL bFGF along with inhibitors 10 μM SB435142 and 0.2 μM LDN193189 for the induction of neural fate. The resulting embryoid bodies were treated with RA (1 μM) on day 5 followed by the SHH agonist-purmorphamine (1 μM) on day 7. On day 17, the medium was replaced by neurobasal medium containing DMEM/F12, 1× B27, 1× penicillin streptomycin solution, 1× Glutamax I, and 10 ng/mL growth factors [Brain-Derived Neurotrophic Factor (BDNF), Ciliary Neurotrophic Factor (CNTF), Glial-Derived Neurotrophic Factor (GDNF), and Insulin-like Growth Factor 1 (IGF1)]. The neurospheres were dissociated with TrypLE on day 20 and seeded on culture dishes coated with poly-l-ornithine (PLO)/laminin. These cells were maintained in culture for 30–40 days with regular observation under the microscope.
CSF Exposure
CSF samples from clinically diagnosed sporadic ALS patients [25] and patients suffering from benign intracranial hypertension (non-ALS CSF) were collected with informed consent (CSF details are provided in Table 1). A total of five ALS-CSF samples showing toxicity on hESC-MNs assessed by MTT, and five NALS-CSF were used in this study. In MTT assay, each CSF sample was tested in duplicates. hESCs-MNs at day 30 of post-differentiation were grown on culture plates coated with PLO/laminin. On day 33, the cells were exposed to CSF (10% v/v), and after 48 h, the cells were processed for further analysis. The different study groups were as follows: NC, hESC-MNs grown under normal condition; NALS, hESC-MNs exposed to non-ALS CSF; ALS, hESC-MNs exposed to ALS-CSF. hESC-MNs were exposed to pooled CSF samples [26], and three independent biological replicates were used for statistical analysis. Three different CSF samples in duplicates were tested for electrophysiology experiments and they were consistent.
Quantitative Real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed to measure the relative mRNA levels of the genes expressed by hESCs and hESC-MNs. Total RNA was isolated from hESC/hESC-MNs using RNeasy plus mini kit (Qiagen), as per the manufacturer’s instructions. The relative quantification of mRNA levels was determined using a two-step strategy. In the first step, complementary DNA (cDNA) was generated from RNA derived from the cells using high-capacity cDNA reverse transcription kit (Applied Biosystems, USA). Subsequently, PCR reaction was performed using Takara, SYBR® Premix Ex Taq™ II, and qRT-PCR was carried out in Applied Biosystems 7500. The cycling conditions were as follows: holding stage—95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Reactions were performed in duplicates for each sample, and the values were normalized to internal control, 18S rRNA. The forward and reverse primer sequences are provided in Table 2. The relative gene expression was analyzed by ΔΔCt method [27].
Immunocytochemistry
hESCs/hESC-MNs were grown on feeder layers/PLO/laminin-coated coverslips. The cells were fixed with 4% paraformaldehyde for 15–30 min at room temperature and permeabilized by incubating in 0.1 M phosphate-buffered saline (PBS) (pH 7.4) containing 0.3% Triton X-100 (staining buffer) for 15 min at room temperature. Non-specific staining was blocked by incubating with 3% Bovine Serum Albumin (BSA) in staining buffer for 45 min. Later, the cells were incubated with primary antibodies for 24 h at 4 °C and washed thrice in staining buffer. Subsequently, cells were incubated overnight at 4 °C with appropriate secondary antibodies conjugated with fluorescein isothiocyanate (FITC)/Cy5/Cy3 (1:200). The coverslips were washed with staining buffer and mounted. The details of the antibodies are given in Table 3. Fluorescent images were captured using a confocal laser scanning microscope (Leica TCS-SL; Leica, Wetzlar, Germany/Flow view 10i, Olympus Corporation, Japan).
Flow Cytometry
hESC-MNs (study groups) were harvested using enzymatic dissociation as described earlier. The cells were fixed and permeabilized with BD Cytofix/Cytoperm kit at 4 °C, and they were washed twice with washing buffer containing 0.5% saponin (w/v). The cells were blocked with 2% BSA in 0.1 M PBS containing 0.5% saponin for 15 min at room temperature and incubated with primary antibodies for 3 h at room temperature. After washing to remove unbound antibodies, cells were incubated with FITC-labeled secondary antibody for 1 h. Cells incubated without primary antibodies served as negative controls. Labeled cells were analyzed using FACS ARIA III (Becton Dickinson), and the data was analyzed using FlowJo software (Tree star Inc.). The expression of various markers was analyzed on the differentiated cells by applying a gate to exclude the cellular debris on the FSC-A (forward scatter area) and SSC-A (side scatter area) [28].
Cell Viability (MTT) Assay
Cell viability was measured by a colorimetric assay using reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). Briefly, ~ 2 × 104 cells were seeded per well of 96-well tissue culture plates on day 30 of differentiation and maintained in 200 μL of neurobasal medium at 37 °C with 5% CO2. On day 33, cells were exposed to ALS/non ALS-CSF (10% v/v CSF). After 48 h, 40 μL of MTT (5 mg/mL) was added to each well and incubated at 37 °C for 2 h. Following incubation, spent medium was replaced by 200 μL of DMSO and mixed well by pipetting to dissolve the tetrazolium salts. The absorbance was measured at 570 nm in an ELISA reader (Tecan, GmBH, Germany). The difference in absorbance between the study groups was calculated for deriving percentage reduction in cell viability. A total of ten CSF samples, five from each experimental group in duplicates, were used in MTT assay.
Lactate Dehydrogenase Assay
Cytotoxicity was quantified by measuring leakage of cytosolic lactate dehydrogenase (LDH) due to plasma membrane damage. LDH activity was determined spectrophotometrically by monitoring the oxidation of lactate to pyruvate, coupled to the reduction of nicotinamide adenine dinucleotide (NAD+) to nicotinamide adenine dinucleotide (NADH). Briefly, 40 μL of culture supernatant from NC/NALS-CSF/ALS-CSF-exposed hESC-MNs was added to an assay buffer containing 0.27 mL KH2PO4 (0.1 M, pH 7.4), 0.01 mL sodium pyruvate (2.5 mg/mL), and 0.005 mL NADH (5 mg/mL). The absorbance was measured at 340 nm, for 3 min using an ELISA plate reader (Tecan, GmBH, Germany). The rate of increase in the formation of reduced NADH is directly proportional to the LDH activity. The difference ΔA/min was calculated, and the results were represented as a percent activity compared to control [29].
Electron Microscopy
hESC-MNs maintained under normal conditions and those exposed to CSF were fixed with 2.5% glutaraldehyde for 2 h. Cells were harvested and centrifuged at 1500 rpm for 5 min. The cell pellet was postfixed with 1% osmium tetroxide for 1.5 h at room temperature and dehydrated with different grades of ethanol 70, 80, 90, 95 and 100% each for 1 h. Clearing was done with propylene oxide at room temperature for 15 min, following which the cells were processed for infiltration, using liquid resin araldite CY212 (Sigma-Aldrich, USA) with propylene oxide (1:1) overnight on a rotator. The following day, the solution was changed to 2:1 of araldite:propylene oxide and kept on the rotator for 1 h. The cell pellet was kept in pure araldite for around 5 h at room temperature. Cells were polymerized with dry heat at 60 °C for 48 h. Ultrathin sections of 70 nm were cut using a diamond knife and collected on copper grids. To obtain contrast within biological material, atomic density was introduced by staining the cells with heavy metals, 1% uranyl acetate in 95% ethanol for 1–2 h followed by lead citrate for 5–7 min. The stained grids were washed, dried, and viewed under the transmission electron microscope (FEI, TECNAI G2 Spirit BioTwin, Netherlands) [7].
Extracellular Voltage Recordings
Spontaneous action potentials exerted by the hESC-MNs were recorded using Multi Electrode Array system (MEA) (Multi Channel Systems, Reutlingen, Germany) [30]. Motor neurons at a density of ~ 1 × 105 were grown on MEA plates, and spontaneous action potentials were recorded at various time points (day 23, day 27, day 30, and day 33) of differentiation. Recording was carried out from three representative cultures at each time point. MC-Rack (ver 3.5) software was used to collect spontaneous network activity data. For CSF exposure study, hESC-MNs were exposed to NALS and ALS-CSF on day 33 of differentiation, and the changes in spontaneous activity were recorded after 48 h. Signals from all 60 electrodes were simultaneously sampled. Signals were digitized at a rate of 20 KHz and high-pass filtered (cut-off frequency of 200 Hz). Spike and burst detection was performed off-line using Spike2 software. Spontaneous action potentials that crossed a threshold of 15 μV and voltage threshold by five times the standard deviation were taken for analysis. Auto-correlograms were constructed using NeuroExplorer software.
Mitochondrial Complex Assays/Electron Transport Chain Assay
Cell pellets from different (NC, NALS, and ALS) groups of hESC-MNs were collected and sonicated in 1× PBS, and total protein content was estimated using Bradford’s method. An aliquot of cell pellet was suspended in hypotonic solution containing 25 mM potassium phosphate (pH 7.2) with 5 mM magnesium chloride. Freeze-thaw cycle was followed thrice to release the cellular contents. The resulting cell homogenate was used to determine the mitochondrial complex activity. Citrate synthase (CS) an enzyme of the Krebs cycle was used as an internal control. The activity of CS was determined by the release of –SH group from CoA-SH by using Ellman reagent 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB) which absorbs at 412 nm [31, 32].
NADH-Q Oxidoreductase (Complex I) Assay
Complex I (CI) enzyme assay was carried out as previously described [33, 34]. In brief, the assay was initiated by adding cell homogenate (15μg/10 μL suspended in hypotonic buffer) to 190 μL of the reaction mix containing 0.1 M Tris-HCl (pH 7.2), 500 μM EDTA, 1% BSA, 200 μM NADH, and 200 μM decylubiquinone with or without 150 μM rotenone, in the presence of 2 mM sodium cyanide (NaCN) with 0.002% DCIP as a secondary electron acceptor. The decrease in the absorbance at 600 nm was recorded as a measure of the rate of enzyme reaction at 37 °C for 20 min, and specific activity was calculated as below:
The difference between the specific activities with and without rotenone was used to assess the activity specific to mitochondrial CI.
Succinate-Q Reductase (Complex II Assay)
Complex II assay was initiated by the addition of cell homogenate (30 μg/10 μL suspended in hypotonic buffer) to 190 μL of the reaction mix containing 250 mM potassium phosphate (pH 7.2), 50 mM magnesium chloride, 10 mM of succinate, 0.1% BSA, 3 μM rotenone, 1 μM of antimycin, 0.3 mM of potassium cyanide (KCN), 80 μM of DCIP, and 50 μM decylubiquinone. The decrease in the absorbance at 600 nm was recorded as a measure of the rate of enzyme reaction at 37 °C for 20 min, and specific activity was calculated.
Q-Cytochrome c Oxidoreductase (Complex III Assay)
Complex III assay was initiated by adding 10 μL of cell homogenate (30 μg sample suspended in hypotonic buffer) to 190 μL of reaction mix containing 50 mM potassium phosphate (pH 7.4), 25 mg/mL BSA, 30 mM n-dodecyl-b-D-maltoside, 1 mg/mL rotenone, 1 M KCN, 2.5 mM cytochrome c, and 35 μM reduced decylubiquinone. The enzyme-catalyzed reduction of cytochrome c was measured for 3 min at 550 nm (extinction coefficient for cytochrome c = 18.7 mM−1 cm−1).
Cytochrome c Oxidase (Complex IV Assay)
Complex IV activity was measured as previously described [35]. In brief, activity was measured by monitoring decrease in absorbance due to oxidation of reduced cytochrome c at 550 nm. The reaction mixture contained 50 mM potassium phosphate (pH 7.2), 0.1% BSA, 2.5 mM dodecyl maltoside, and 10 μg of cell homogenate. The reaction was initiated by the addition of reduced cytochrome c (40 μM), and the kinetics was monitored for 5 mins. Activity was calculated using the molar extinction coefficient of cytochrome c (18.7 mM−1 cm−1).
Statistical Analysis
Data from the experiments were subjected to one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test, Student’s t test, or non-parametric Kruskal-Wallis test followed by Mann-Whitney test. Data was expressed as mean ± SD or median (range) between control and experimental groups.
Results
Human Embryonic Stem Cells in Culture
hESCs grown on iMEFs exhibited stem cell characteristics such as higher nucleus to cytoplasm ratio, compact colony with closely packed cells (Fig. 1a, b). hESCs expressed the transcription factors indispensible for maintaining pluripotency such as octamer-binding transcription factor 4 (Oct4), Nanog Homeobox (Nanog), and SRY (sex-determining region Y)-box 2 (Sox2) in the nucleus (Fig. 1c–f).

Human embryonic stem cells-BJNHem20: a, b Representative phase contrast photomicrograph of typical undifferentiated human stem cell colony with closely packed cells (red asterisk) cultured on inactivated mouse embryonic fibroblast feeders (red arrows). Scale bar—a 50 and b 20 μm. Representative confocal photomicrograph of human embryonic stem cells expressing pluripotency markers in the nucleus: c Oct4 (green), d Nanog (red), e Sox2 (blue), and f merge
Motor Neurons Derived from Human Embryonic Stem Cells
Embryoid bodies, the floating spheres/aggregates, of cells were formed on day 3 of differentiation (Fig. 2a). Subsequently, these embryoid bodies were induced with RA and purmorphamine for motor neuron formation. The resulting neurospheres were dissociated and plated on PLO/laminin-coated culture dishes on 20th day of differentiation. These cells adhered to substratum and displayed typical morphology of neurons such as large cell bodies and prominent neuritic processes (Fig. 2b, c). They made interconnections and the complexity of arborization increased with continued growth in culture (Fig. 2g, h). These cells were positive for the neuronal marker, βIII tubulin, and motor neuron marker Choline Acetyl Transferase (ChAT) at day 23 of differentiation as identified by immunocytochemistry. Further, these neurons also expressed MAP2 at day 33 of maturation (Fig. 2d-f & i–l).

Motor neurons derived from human embryonic stem cells: a Representative phase contrast photomicrograph of three-dimensional aggregates—embryoid bodies—formed from human embryonic stem cells on day 3 of differentiation. b, c Representative phase contrast and d–f confocal images of human embryonic stem cell-derived process-bearing neurons at day 23 of differentiation; these neurons expressed ChAT (red), βIII tubulin (blue), and f merge. g, h Representative phase contrast (day 27) and i–l confocal (day 33) images of human embryonic stem cell-derived motor neurons. The neurons established interconnections and the complexity of arborization increased with maturation along with expression of βIII tubulin (green), ChAT (red), MAP2 (blue), and l Merge. Scale bars are indicated
Oct4 mRNA transcribed by hESCs was down-regulated in the differentiated motor neurons indicating loss of pluripotency (Fig. 3a). Motor neuron-specific markers such as Olig2, HB9, and ChAT mRNA were expressed exclusively by the hESC-MNs that were absent in hESCs (Table 4, Fig. 3b). In addition, expression of ChAT, βIII tubulin, HB9, and Islet1 in hESC-MNs was identified using flow cytometry; the gated homogenous population showed ChAT 21 ± 0.70%, βIII tubulin 21.3 ± 0.85%, HB9 15.2 ± 0.85%, and Islet1 15.1 ± 0.35%. These proteins were absent in the hESCs indicating the lack of motor neuron-specific proteins in the undifferentiated state (Fig. 3c–f). Both hESCs and hESC-MNs expressed Forkhead box protein 1 (Foxp1) mRNA. However, there was a considerable fold increase of Foxp1 in hESC-MNs (5.03 ± 0.20) compared to that in hESCs (Fig. 3a).

Expression of motor neuron markers and ultrastructure of hESC-MNs: a qRT-PCR showed down-regulation of Oct4 and up-regulation of FoxP1 in hESC-MNs in comparison to undifferentiated cells. b Olig2, HB9, and ChAT were expressed by hESC-MNs and absent in the undifferentiated hESCs. Gene amplification represented as Ct values. Data were analyzed using Student’s t test; ***p < 0.001, **p < 0.01 (n = 3). c–f Flow cytometry analysis showed the expression of motor neuron-specific markers in hESC-MNs with c ChAT 21 ± 0.70%, d βIII tubulin 21.3 ± 0.85%, e HB9 15.2 ± 0.85%, and f Islet1 15.1 ± 0.354%. Staining in hESCs was like negative control (background cells incubated without primary antibody) indicating the absence of these proteins. g–j Representative electron photomicrographs of hESC-MNs: g (a), (b), and (c) are neurons with the perikaryon carrying different organelles: mitochondria, asterisk; ER, blue arrows; plasma membrane, red arrow; GA, white arrow; nuclear membrane, white arrow head. h Cytoplasm with the rough ER with loosely spread intracytoplasmic basophilic masses called Nissl substances (black arrows), characteristic of neurons and free ribosomes (red arrows). i Neurofilaments (yellow arrow). j (a), (b), and (c) are different neurons; a single mitochondrion was observed in the neuronal process (yellow arrow)
The ultrastructure of hESC-MNs showed plasma membrane enclosing the perikaryon carrying different healthy organelles such as mitochondria, endoplasmic reticulum (ER), Golgi apparatus (GA), and nucleus (Fig. 3g). The cytoplasm had loosely distributed intracytoplasmic basophilic Nissl substances representing the characteristic feature of neurons (Fig. 3h). The neurons expressed neurofilaments (Fig. 3i), and the neuronal processes also had mitochondria (Fig. 3j).
Human Embryonic Stem Cell-Derived Motor Neurons Were Electrically Active
Motor neurons are electrically active cells; hence, to further characterize the hESC-MNs for their electrophysiological properties, extracellular voltage signals were recorded at different days following differentiation, viz., day 23, day 27, day 30, and day 33. Differentiated neurons discharged spontaneous action potentials as early as day 23. Figure 4a depicts hESC-MNs cultured on the MEA plate, and Fig. 4b–g shows the spontaneous action potentials generated by hESC-MNs. The amplitude of the spikes was almost 100 ± 18 μV. From among the 60 channels that were recorded, spontaneous firing was observed in 12 ± 3 channels on day 23 of differentiation. However, by day 33, electrical activity was observed from almost all electrodes and 1000–4000 spikes were observed. We observed neurons firing at two different ranges of frequencies: < 5 and > 5 Hz, indicating the heterogeneity of the cell population consisting of principal and uncharacterized neurons respectively. The population consisted of 84% of neurons firing at a frequency of < 5 Hz, suggestive of the presence of a larger proportion of principal neurons (Fig. 4l, m). The existence of two distinct populations was further confirmed by auto-correlogram analysis that showed the peaks generated due to firing of principal neurons having a fast exponential dacay while (Fig. 4n, o), while the rate of decay of unclassified neurons was slow (Fig. 4p, q) [30]. The number of spikes as well as the mean spike frequency increased over time from day 27 to day 33 (represented in Table 5), and the highest was on day 33 (***p < 0.001 day 23 vs day 33; day 27 vs day 33; day 30 vs day 33) (Fig. 4d–g). The scatter plot (Fig. 4h–k) shows the number of spikes elicited by the principal neurons at different days of maturation, suggesting progressive maturation of hESC-MNs over time in culture. Further, experiments with CSF exposure were carried out on day 33 hESC-MNs.

Electrical activity recorded from hESC-MNs: a Stem cell-derived neurons grown on the multielectrode array plate. b Magnified view of electrical activity phenotype of 500-ms duration showing regular spikes. The figure depicts the action potentials extracted using spike sorter (green-action potentials and black-background). c Superimposed waveform of principal neuron. d–g Representative electrical activity of neurons at different days of maturation: d very few spikes discharged on day 23, e increased spike number on day 27, f spike trains on day 30, g spike trains and bursts on day 33. h–k Scatter plots of the number of spikes (frequency < 5 Hz) discharged by hESC-MNs at different days of maturation in culture (day 23, day 27, day 30, and day 33 respectively). The number of spikes and the mean firing frequency were less on day 23, day 27, and day 30 (h–j), and it was significantly increased on day 33 (k). Data analyzed using Wilcoxon signed-rank test (***p < 0.001 with respect to day 33, n = 102). l Scatter plot depicting number of spikes and mean spike frequency exerted by hESC-derived neurons at day 33. A larger population of the neurons fired with a frequency of < 5 Hz (highlighted) indicating principal neurons and a negligible number of neurons fired with frequency of > 5 Hz (n = 4). m Percentage of neurons with firing frequency < 5 Hz was 84%, assumed to be motor neurons based on the qualitative assessment of motor neuron-specific markers. Data was analyzed using Student’s t test; ***p < 0.001. n–q Auto-correlograms revealing the presence of two different types of neurons: n, o principal neurons and p, q uncharacterized neurons derived from hESC at day 33 of differentiation
Altered Morphology and Reduced Viability of hESC-Derived Motor Neurons Following Exposure to ALS-CSF
To determine the toxicity of ALS-CSF, hESC-MNs at day 33 were exposed to NALS-CSF/ALS-CSF for 48 h. Phase contrast microscopy revealed that ALS-CSF induced beading of neurites and vacuolation (Fig. 5c, d), whereas hESC-MNs exposed to NALS-CSF did not differ from NC (Fig. 5a, b). MTT assay results indicated that while NALS-CSF exposure did not produce any cytotoxicity in the hESC-MNs (~ 94% viability), ALS-CSF exposed cells showed only 63.7% viability (***NC vs ALS p < 0.001; ###NALS vs ALS p < 0.001, Fig. 5e). Another measure of cytotoxicity is LDH release, assessed by an assay for LDH activity in the culture supernatant following CSF exposure. After 48 h of ALS-CSF exposure, we observed a significant increase in LDH activity in the culture supernatant, when compared to both NC and NALS-CSF exposed cells, suggesting an increase in the anaerobic glycolysis induced by ALS-CSF (***NC vs ALS p < 0.001; ###NALS vs ALS p < 0.001) (Fig. 5f).

a–d Representative phase contrast photomicrograph of hESC-MNs. a NC—cells maintained under normal condition. b NALS—cells exposed to non-ALS-CSF. c, d ALS—cells exposed to ALS-CSF. Notice the ALS-CSF-induced vacuolation (c, red arrow) and beading of neurites (d, yellow arrows) in the hESC-MNs. e ALS-CSF reduced the viability of hESC-MNs in culture: Viability assessed by MTT assay showed significant reduction in the ALS-exposed group compared to that in the control. The values are expressed as percentage MTT reduction compared to normal control (***NC vs ALS p < 0.001, ###NALS vs ALS p < 0.001, n = 5). f Increased LDH leakage in the hESC-MNs exposed to ALS-CSF: LDH leakage was assessed in the spent media of the NC, NALS, and ALS groups. LDH release is represented as % activity compared to control (***NC vs ALS p < 0.001, ###NALS vs ALS p < 0.001, n = 5). g–i Reduced expression of ChAT in the ALS-CSF-exposed hESC-MNs: g qRT-PCR showed down-regulation of ChAT mRNA in the ALS-CSF-exposed hESC-MNs compared to that in controls (n = 3) (***NC vs ALS p < 0.001, ###NALS vs ALS p < 0.001). h Representative histogram of flow cytometry analysis and i quantification data of gated population of cells showing reduced ChAT-positive neurons in the ALS-CSF group compared to those in the other control groups (*NC vs ALS p < 0.05, ##NALS vs ALS p < 0.01). j–l Reduced expression of BDNF in the ALS-CSF-exposed hESC-MNs: j qRT-PCR showed down-regulation of BDNF mRNA in the ALS group compared to that in the controls (***NC vs ALS p < 0.001, ###NALS vs ALS p < 0.001, n = 3). k Representative histogram of flow cytometry analysis and l quantification data showing reduced BDNF-positive neurons in the ALS group compared to those in the other control groups (**NC vs NALS p < 0.01, *** NC vs ALS p < 0.001, ###NALS vs ALS p < 0.001). Data analyzed using one-way ANOVA followed by Tukey’s post hoc test
Reduced Expression of ChAT and BDNF in ALS-CSF-Exposed hESC-Derived Motor Neurons
ChAT mRNA levels and protein concentration were significantly reduced by ALS-CSF which remained unchanged in NALS group. While ALS-CSF induced a − 1.34 ± 0.08-fold down-regulation of ChAT mRNA, NALS-CSF did not alter ChAT mRNA levels (1.1 ± 1.12)(***NC vs ALS p < 0.001, ###NALS vs ALS p < 0.001) (Fig. 5g). The ChAT-positive motor neurons as identified by flow cytometry were NC = 20.05 ± 3.75%, NALS = 22.25 ± 0.92%, and ALS = 4.05 ± 0.78%, hence correlating the mRNA level (*NC vs ALS p < 0.05; ##NALS vs ALS p < 0.01, Fig. 5h, i).
BDNF mRNA level was significantly reduced in the ALS-CSF-exposed hESC-MNs compared to that in controls. mRNA fold change was NC = 1 ± 0.37, NALS = 1.06 ± 0.57, ALS = − 1.96 ± 0.67 (***NC vs ALS p < 0.001; ###NALS vs ALS p < 0.001) (Fig. 5j). Flow cytometry analysis also revealed a significant down-regulation of BDNF-positive neurons in the ALS-CSF group; the values were as follows: NC = 7.9 ± 0.42, NALS = 10.7 ± 0.28, ALS = 1.85 ± 0.35 (***NC vs ALS p < 0.001; ###NALS vs ALS p < 0.001; **NC vs NALS p < 0.01) (Fig. 5k, l).
Ultrastructural Changes Induced by ALS-CSF
To understand the ultrastructural changes in the present ALS model, we carried out electron microscopic studies. Normal (control) hESC-MNs and NALS-CSF-exposed hESC-MNs had healthy plasma and nuclear membranes, with intact double layers and normal cytoplasmic features. The nucleus was compact with homogenously distributed chromatin (Fig. 6g, h). The cytoplasm had several mitochondria which were oval or round with prominent cristae (Fig. 6a, b). GA had flattened cisterns (Fig. 6d, e), and ER was well developed with poly ribosomal rosettes (Fig. 6g, h). ALS-CSF-exposed cells, on the other hand, had dilated ER (Fig. 6l), with cytoplasmic thinning or disruption (Fig. 6k). Mitochondria were also swollen with electrolucent matrix and occasional vacuolation (Fig. 6c). Interestingly, GA bore electron dense cisternae, which were condensed, curled, and had a closed appearance (Fig. 6f). In addition, the presence of nuclear chromatin condensation, deformed and discontinuous nuclear membrane, and disruption of karyoplasm and nucleoli along with cytoplasmic vacuolation suggested the presence of apoptotic cells (Fig. 6i, j, f).

Representative electron photomicrographs of hESC-MNs showing organelle changes following ALS-CSF exposure: a Normal control; normal mitochondria, red arrows. b NALS; normal mitochondria, red arrows. c ALS; swollen mitochondria, red arrows; vacuolated mitochondria, yellow arrows. d Normal control; normal GA, red arrows. e NALS; normal GA, red arrows. f ALS, abnormal Golgi wherein the cisterns were condensed; round and closed, red arrows; vacuolation in the cytoplasm, yellow arrow. g Normal control; normal rough ER (red arrows), intact nuclear membrane (yellow arrow), and nucleus with uniformly distributed chromatin (red asterisk). h NALS; normal rough ER (red arrows), intact nuclear membrane (yellow arrow), and nucleus with uniformly distributed chromatin (red asterisk). i–l ALS: i discontinuous nuclear membrane (yellow arrow); condensed chromatin in the nucleus (red asterisk); j disruption of karyoplasm and nucleoli; k cytoplasmic disruption (yellow arrow); l dilated rough ER (red arrows)
Reduced Mitochondrial Complex Activities
To understand if the ultrastructural alterations observed within the mitochondria are correlated with functional deficits, we measured the activities of the respiratory complexes (I, II, III, and IV) in CSF-exposed hESC-MNs. Enzyme activities of the respiratory complexes were normalized to the activity of an internal control citrate synthase, an enzyme of the Krebs cycle. The activities of complex I, II, III, and IV were lowered by ~ 40, ~ 30, ~ 30, and ~ 60% respectively in ALS group compared to those in controls. Complex I, II, and IV activities were significantly lowered in ALS-exposed motor neurons. Complex III activity showed a decreasing trend in ALS-exposed hESC-MNs compared to that in other groups (Fig. 7a–d); Complex I-*NC vs ALS p < 0.05; Complex II-*NC vs ALS p < 0.05; #NALS vs ALS p < 0.05; $NC vs NALS p < 0.05; Complex IV-*NC vs ALS p < 0.05).

a–d Lowered mitochondrial respiratory complex activities in ALS-CSF-exposed hESC-MNs: a, b, c, and d represent complex I, II, III, and IV activities. The activities were normalized to citrate synthase activity. The activities of complexes I, II, III, and IV were lowered by ~ 40, ~ 30, ~ 30, and ~ 60% respectively in the ALS group compared to those in the control (*NC vs ALS, #NALS vs ALS, $NC vs NALS p < 0.05, n = 3). e–j Altered expression of cytoskeletal proteins in the hESC-MNs exposed to ALS-CSF: e representative histogram of flow cytometry analysis showing βIII tubulin-positive neurons and f quantification data showing unchanged βIII tubulin-positive neurons in the ALS-CSF group compared to those in the other control groups; g representative histogram of flow cytometry analysis showing MAP2-positive neurons and h quantification data showing increasing trend of MAP2-positive neurons in the ALS-CSF group compared to those in the other control groups. i Representative histogram of flow cytometry analysis and j quantification data showing significant increase in phosphorylated neurofilaments in ALS-CSF group compared to those in the other control groups (*NC vs ALS, #NALS vs ALS, p < 0.05, n = 3). Data were analyzed using one-way ANOVA followed by Tukey’s post hoc test
ALS-CSF Induced Changes in Cytoskeletal Proteins
The number of βIII tubulin-positive cells remains unaltered in the ALS-CSF-exposed hESC-MNs compared to that in the control groups, and the values were NC = 46.6 ± 0.85, NALS = 57.35 ± 0.92, and ALS = 57.15 ± 0.35 (Fig. 7e, f). The number of MAP2-positive neurons remain unchanged, and the values were NC = 47.05 ± 2.90, NALS = 51.55 ± 0.64, and ALS = 54.95 ± 1.48. However, there was an increasing trend in the ALS-CSF group (Fig. 7g, h). Neurofilament phosphorylation determined by flow cytometry using SMI-31 antibody was significantly up-regulated in the ALS-CSF-exposed motor neurons compared to that in other control groups, and the values were NC = 68.9 ± 1.98, NALS = 71.95 ± 0.78, and ALS = 78.85 ± 1.20 (*NC vs ALS p < 0.05; #NALS vs ALS p < 0.05, Fig. 7i, j).
ALS-CSF Induced Apoptosis in hESC-Derived Motor Neurons
Motor neurons positive for BCL2, Bax, and caspase 9 were investigated using flow cytometry to determine whether the degeneration of motor neurons is due to the altered levels of these pro- and anti-apoptotic proteins. BCL2 positivity was slightly increased in the ALS group, which was not significant (NC = 26.8 ± 3.96, NALS = 32 ± 2.12, and ALS = 30.25 ± 1.98) (Fig. 8a, b). Bax positivity was significantly increased in the ALS-CSF group compared to that in the control groups (NC = 13.5 ± 0.07, NALS = 17.25 ± 0.35, and ALS = 23 ± 1.98; **NC vs ALS p < 0.01; #NALS vs ALS p < 0.05) (Fig. 8c, d). The ratio of BCL2/Bax showed a decreasing trend in the ALS-CSF group (Fig. 8g). Caspase 9-positive motor neurons were significantly increased in the ALS/NALS-CSF-exposed group compared to those in the control group; however, ALS-CSF-induced caspase 9 level in the hESC-MNs was significantly higher than that in the NALS group (NC = 5.9 ± 0.0, NALS = 8.7 ± 1.13, and ALS = 13.6 ± 0.14; **NC vs ALS p < 0.01; #NALS vs ALS p < 0.05; *NC vs NALS p < 0.05) (Fig. 8e, f).

Altered expression of apoptotic proteins in the hESC-MNs exposed to ALS-CSF: a Representative histogram of flow cytometry analysis and b quantification data showing unaltered BCL2 levels in the ALS-CSF group compared to those in the other control groups. c Representative histogram of flow cytometry analysis and d quantification data showing up-regulated Bax-positive motor neurons in the ALS-CSF group compared to those in the other control groups (**NC vs ALS p < 0.01, #NALS vs ALS p < 0.05, n = 3). e Representative histogram of flow cytometry analysis and f quantification data showing increased caspase 9-positive motor neurons in the ALS-CSF- and NALS-CSF-exposed groups (*NC vs NALS p < 0.05, **NC vs ALS p < 0.01, #NALS vs ALS p < 0.05, n = 3). g Bar graph showing decreasing trend in the BCL2/Bax ratio in the ALS-CSF group compared to that in controls. Data were analyzed using one-way ANOVA followed by Tukey’s post hoc test (n = 3)
ALS-CSF Modulated the Spontaneous Activity of hESC-Derived Motor Neurons
After the electrophysiological characterization of hESC-MNs, they were exposed to ALS-CSF for 48 h and MEA recordings were performed to observe alterations in the spontaneous activity patterns. Within the neurons firing with < 5 Hz representing principal neurons, the number of spikes was significantly high in the ALS-CSF-exposed neurons [avg. 1800, range 285–3412; n = 220] indicating hyperexcitability, when compared to that in the NC [avg. 1444, range 108–3305; n = 155] and NALS-CSF groups [avg. 1536, range 111–3436; n = 140] (***NC vs ALS p < 0.001; ### NALS vs ALS p < 0.001, Fig. 9).

Hyperexcitability induced by ALS-CSF exposure: a–c Scatter plot showing the number of spikes exerted by principal neurons in different study groups (frequency < 5 Hz). The spiking pattern was different in the CSF-exposed groups; however, there was no significant difference between the normal control and NALS groups. The number of spikes was significantly increased in the ALS-CSF group indicating hyperexcitability. d Bar graph representing increased number of spikes (with < 5 Hz) in the hESC-MNs following ALS-CSF exposure. Data were analyzed using Kruskal-Wallis test. Mann-Whitney U tests were done as post hoc test and Bonferroni corrected p values (0.05/3 = 0.017) (***NC vs ALS p < 0.001; ###NALS vs ALS p < 0.001)
Discussion
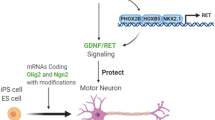
The advent of ESCs has opened new vistas of research, thanks to their ability to self-renew and differentiate into different cell types. Beating cardiomyocytes and retinal pigmented epithelial cells were derived from BJNHem20 [23, 36]. However, ours is the first study to attempt motor neuron derivation from these cells (Fig. 10). We have established and propagated these stem cells on iMEFs; their pluripotency was confirmed by the presence of Oct4, Nanog, and Sox2. We further adapted an established protocol for differentiating hESCs into motor neurons [24].

Schematic representation of motor neuron derivation from human embryonic stem cells and their vulnerability to ALS-CSF toxicity: Human embryonic stem cells (BJNHem20) were grown on inactivated mouse embryonic fibroblasts. Motor neurons were successfully derived using retinoic acid and purmorphamine. Cells displayed typical neuronal morphology, and they were positive for motor neuron markers such as Olig2, HB9, Islet1, and ChAT. These motor neurons matured in culture over a period and discharged spontaneous action potentials. ALS-CSF exposure for 48 h induced degenerative changes such as organelle dysfunction, reduced BDNF expression, altered expression of cytoskeletal proteins, and increased phosphorylation of neurofilaments and hyperexcitability. Such changes along with the increased expression of pro-apoptotic molecules culminated in the death of motor neurons. These results conform to the neurodegenerative changes observed in our earlier studies in rodent models of sporadic ALS
The differentiation protocol transformed hESCs into motor neurons. Their phenotype was confirmed by the expression of pan-neuronal markers βIII tubulin and MAP2 along with motor neuron-specific markers Olig2, HB9, and islet1 which are required for fate determination and survival of motor neurons. HB9 knockout in mice causes abnormal migratory pattern, topological disorganization of motor columns, and defective target innervation indicating the improper differentiation of motor neurons [37, 38]. Islet1 imparts membrane-specific electrical signatures and acts as a switch between motor neuron and interneuron fate [39,40,41]. Foxp1 positivity of hESC-MNs confirmed that they are limb-innervating motor neurons [42,43,44]. Cholinergic transmission is involved in the production of rhythmic bursts during spinal cord development [45, 46]. hESC-MNs expressed ChAT and discharged spontaneous single spike activity that was observed around 3 weeks of differentiation. Spike trains and bursts were observed at 4–5 weeks of culture; however in another study, the electrical maturation was observed only after 7 weeks of culture [47]. Further, neurons differentiated in this study were majorly principal neurons discharging spikes with frequency < 5 Hz [30].
The toxic effect of ALS-CSF was assessed on the mature hESC-MNs. Beading of neurites and vacuolations in cytoplasm, the early signs of neurodegeneration were observed in hESC-MNs upon exposure to ALS-CSF. This finding is similar to our earlier studies carried out in the rodent models of ALS [22, 48]. Vacuolation of cytoplasm was observed, a hallmark in the mouse spinal cord cultures and transgenic mouse lines carrying SOD1 mutation [49, 50]. Loss of ChAT-positive α-motor neurons was found in the spinal cord of ALS patients and rodent models of ALS [22, 51,52,53]. Similarly, ALS-CSF diminished the expression of ChAT in hESC-MNs and reduced their viability significantly.
Increased microtubule dynamics with altered axonal transport are reported in mSOD1 mice [54]. Mutant SOD1 physically interacts with tubulin forming aggregates, resistant to constitutive degradation [55]. MAP2 helps the microtubules to from long polymers maintaining the morphological plasticity [56, 57]. Abnormal/hyperdynamics of microtubules occurs early before symptom onset in mSOD1 mice [58]. ALS-CSF exposure did not affect the expression of βIII tubulin in hESC-MNs. However, an increasing trend in MAP2 level was found in ALS-CSF-exposed neurons indicating altered expression profile of this key cytoskeletal protein.
ALS-CSF has been reported to cause aberrant neurofilament phosphorylation in the soma of ventral horn motor neurons of embryonic rat and chick spinal cord cultures; the phosphorylated neurofilaments were aggregated and ubiquitinated in NSC-34 cells [22, 48, 59]. Mutations in the neurofilament gene are reported in SALS, and their expression was five to ten times elevated compared to that in controls [60, 61]. In accordance with these reports, we have also observed a steady increase in the phosphorylated neurofilaments in hESC-MNs exposed to ALS-CSF. The rate of transport of neurofilaments was reduced in mice overexpressing human NF-H [62, 63]. The presence of abundant number of phosphorylated neurofilaments in addition to altered microtubule dynamics could burden the transport machinery, thus affecting the functionality and survival of motor neurons.
Increased reactive oxygen species, mitochondrial depolarization, and reduced complex activities were detected in the spinal cords of ALS-CSF-injected rats [26]. The current data on structural alterations in mitochondria and impaired mitochondrial complex activities in hESC-MNs exposed to ALS-CSF are in agreement with that found in the lumbar spinal cord of the SALS patients, mSOD1 mice, and ALS-CSF-injected rats [26, 64]. Electron leakage due to attenuated complex activities increases oxidative stress [65]. Energy production is considerably reduced in cells carrying mSOD1 [66]. Thus, the defective respiratory chain complexes induced as a result of ALS-CSF exposure cannot cope up with the high energy demands of motor neurons.
Dysregulation of calcium homeostasis following mitochondrial defects causes disruption of ER-mediated stress response [67]. The failure of unfolded protein response (UPR) or abnormal sustained activation of UPR leads to induction of cell death signaling cascade [68, 69]. Ultrastructural studies revealed dilated ER in hESC-MNs following ALS-CSF exposure. Morphological changes such as rough ER dilation, fragmentation, distortion of cisternae, and larger ribosomes were evident in postmortem samples of ALS patients, SOD1 mice, and SALS models [7, 70,71,72].
Under pathological conditions, perturbed protein synthesis in ER leads to Golgi fragmentation [73, 74]. Lesion and disruption of Golgi have been reported in the anterior horn cells and motor cortex of ALS patients, in spinal motor neurons of mSOD1 mice, and also in spinal cords of rats infused with ALS-CSF [6, 75,76,77,78]. Golgi changes coincide with the accumulation of phosphorylated neurofilaments, wherein accumulation of massive phosphorylated neurofilaments induced focal accumulation or clustering of GA [79]. Further, polymerization/depolymerization of microtubules influence the Golgi organization [80]. Accordingly, the observed imbalance in the cytoskeletal components due to ALS-CSF is likely to contribute to peculiar accumulation and end to end swirling of Golgi affecting their functionality in hESC-MNs.
Cortical hyperexcitability is found in both FALS and SALS; it is considered as an early feature that precedes clinical onset [81, 82]. Cortical neurons with mSOD1 in culture elicited action potentials even with less current injection compared to control suggesting reduced time and threshold potential in mSOD1 neurons. Similarly, rate of firing was increased in the spinal motor neurons in culture carrying mSOD1 [83, 84]. iPSC-MNs and motor neurons from brain slice preparation of pre-symptomatic mSOD1 mice showed an increase in the ratio of slope of the frequency vs injected current (F-I) emphasizing intrinsic hyperexcitability of motor neurons in ALS [85, 86]. Similarly, hESC-MNs also showed hyperexcitability by discharging increased number of spikes following ALS-CSF exposure, even in the absence of any injected current. This is in agreement with our earlier observations of ALS-CSF-induced alterations in local field potentials and an increased rate of firing by rat primary motor cortical neurons [87, 88]. Defects in Na+ and K+ currents, increased calcium influx, and mitochondrial defects are related to hyperexcitability [89,90,91,92]. Our earlier studies showed perturbed expression of Nav1.6 and Kv1.6 in the spinal motor neurons of rats infused with ALS-CSF and also the rat spinal cord cultures exposed to ALS-CSF [93].
BDNF required for the survival of motor neurons was significantly down-regulated in ALS-CSF-exposed hESC-MNs supporting our earlier studies. It has been previously shown from our laboratory that ALS-CSF induced aggregation of phosphorylated neurofilaments, reduced ChAT expression, and increased apoptosis could be reverted with exogenous BDNF supplementation [52, 94]. Autocrine secretion of BDNF is required for axon differentiation and growth; it acts in a self-amplifying manner by elevating cyclic AMP levels [95]. BDNF increased the survival of axotomized motor neurons, synaptic plasticity, and long-term potentiation [96]. Alteration in the synthesis and activity of BDNF signaling was reported in various neurodegenerative diseases [97,98,99]. As coordinated interaction between the different organelles is crucial for the proper functioning of neurons, morphological and functional deterioration of the organelles along with impaired respiratory chain activity and reduced BDNF expression found in ALS-CSF group can compromise the survival of hESC-MNs following exposure to ALS-CSF.
Mitochondrial degeneration and electron transport chain impairment induced by ALS-CSF persuaded us to explore the activation of downstream apoptotic pathway. BCL2 is involved in repressing cell death, whereas Bax in contrary accelerates cell death. Increased expression of Bax and DNA fragmentation was detected in the spinal cords of ALS patients [100]. Consistent with this, we also found an up-regulated expression of Bax in ALS-CSF group with an unchanged BCL2 expression. The ratio of BCL2/Bax showed a decreasing trend following exposure to ALS-CSF. Bax is known to permeabilize the mitochondrial outer membrane and release cytochrome c which in turn can activate caspase 9 [101,102,103,104]. Compromised trophic support with increased Bax accentuated apoptotic response with a marked upsurge of caspase 9, as seen in this study. Elevated levels of caspase 9 were also documented in the serum of ALS patients. Increased caspase 9 in turn can cleave caspase 7 and caspase 3 executing apoptosis [103, 105,106,107]. The presence of above mentioned changes along with organelle pathology suggests the commencement of apoptosis in hESC-MNs following ALS-CSF exposure. We also observed an increased caspase 9 level in NALS-CSF group which could be attributed to reactive oxygen species and free radicals in patients with intracranial hypertension. There are reports on rodents with increased intracranial pressure being associated with increased expression of proteins that are hallmarks of apoptosis including elevated caspase levels and DNA damage. A field study has reported oxidative stress response to acute hypobaric hypoxia and its association with increased intracranial pressure [108, 109]. However, ALS-CSF showed a significant increase in caspase 9 levels compared to NALS group.
ALS-CSF has been used to develop ALS phenotype in rodent models for more than two decades, and the toxicity of ALS-CSF in inducing neurodegenerative changes has been well established. To identify the toxic factors in ALS-CSF, mass spectroscopic analysis was performed, and we reported significant increase in the levels of chitotriosidase-1 (CHIT1) a potent inflammatory protein in ALS-CSF (tenfold increase compared to that in control CSF) [110]. CHIT1 was up-regulated in microglia that were transformed into phagocytic phenotype releasing pro-inflammatory cytokines following ALS-CSF exposure. Exposure of microglia to recombinant CHIT1 induced changes like those observed with ALS-CSF exposure [111]. Recombinant CHIT1 when intrathecally injected in newborn rats significantly reduced ChAT expression (manuscript under preparation). Experiments are being carried out to unveil the mechanisms downstream to CHIT1 activation.
Conclusion
In this study, we have successfully derived motor neurons from BJNHem20 and characterized those using specific markers and electrophysiological function. These motor neurons were vulnerable to the toxic effects of ALS-CSF, which induced an array of neurodegenerative changes such as reduced viability, organelle dysfunction, alteration in BDNF expression, perturbed cytoskeletal proteins, hyperexcitability, and increased apoptotic proteins, like the changes observed in ALS patients as well as in animal models of ALS. Accordingly, our humanized model comprehensively captures the molecular pathways and their complex interplay in the pathogenesis of ALS, thereby validating the utility of this model for future studies in this field.
References
Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A et al (2004) DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74:1128–1135
Orlacchio A, Babalini C, Borreca A, Patrono C, Massa R, Basaran S, Munhoz RP, Rogaeva EA et al (2010) SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 133:591–598
Rowland LP, Shneider NA (2001) Amyotrophic lateral sclerosis. N Engl J Med 344:1688–1700
Frey, D., Schneider, C., Xu, L., Borg, J., Spooren, W., and Caroni, P. (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci: Off J Soc Neurosci 20, 2534–2542.
Julien J-P (2001) Amyotrophic lateral sclerosis: unfolding the toxicity of the misfolded. Cell 104:581–591
Ramamohan PY, Gourie-Devi M, Nalini A, Shobha K, Ramamohan Y, Joshi P, Raju TR (2007) Cerebrospinal fluid from amyotrophic lateral sclerosis patients causes fragmentation of the Golgi apparatus in the neonatal rat spinal cord. Amyotroph Lateral Scler: Off Publ World Fed Neurol Res Group Motor Neuron Dis 8:79–82
Vijayalakshmi K, Alladi PA, Ghosh S, Prasanna VK, Sagar BC, Nalini A, Sathyaprabha TN, Raju TR (2011) Evidence of endoplasmic reticular stress in the spinal motor neurons exposed to CSF from sporadic amyotrophic lateral sclerosis patients. Neurobiol Dis 41:695–705
Huang C, Zhou H, Tong J, Chen H, Liu YJ, Wang D, Wei X, Xia XG (2011) FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet 7:e1002011
Kashlan, O.N., Kashlan, B.N., Oh, S.S., McGinley, L.M., Chen, K.S., Kupfer, R., Erman, A., Sakowski, S.A., and Feldman, E.L. (2015). Histological bulbar manifestations in the ALS rat. Neurodegener Dis 15, 121–126.
Lin MJ, Cheng CW, Shen CK (2011) Neuronal function and dysfunction of Drosophila dTDP. PLoS One 6:e20371
Miguel L, Avequin T, Delarue M, Feuillette S, Frebourg T, Campion D, Lecourtois M (2012) Accumulation of insoluble forms of FUS protein correlates with toxicity in Drosophila. Neurobiol Aging 33(1008):e1001–e1015
Schmitt-John T (2015) VPS54 and the wobbler mouse. Front Neurosci 9:381
Watson, M.R., Lagow, R.D., Xu, K., Zhang, B., and Bonini, N.M. (2008). A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J Biol Chem 283, 24972–24981.
Zhou H, Huang C, Chen H, Wang D, Landel CP, Xia PY, Bowser R, Liu YJ et al (2010) Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet 6:e1000887
Shantanu S, Vijayalakshmi K, Shruthi S, Sagar BKC, Sathyaprabha TN, Nalini A, Raju TR, Alladi PA (2017) VEGF alleviates ALS-CSF induced cytoplasmic accumulations of TDP-43 and FUS/TLS in NSC-34 cells. J Chem Neuroanat 81:48–52
Cudkowicz M, Bozik ME, Ingersoll EW, Miller R, Mitsumoto H, Shefner J, Moore DH, Schoenfeld D et al (2011) The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat Med 17:1652–1656
Sunyach C, Michaud M, Arnoux T, Bernard-Marissal N, Aebischer J, Latyszenok V, Gouarne C, Raoul C et al (2012) Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology 62:2346–2352
Wichterle H, Lieberam I, Porter JA, Jessell TM (2002) Directed differentiation of embryonic stem cells into motor neurons. Cell 110:385–397
Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G et al (2008) Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321:1218–1221
Sances S, Bruijn LI, Chandran S, Eggan K, Ho R, Klim JR, Livesey MR, Lowry E et al (2016) Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat Neurosci 19:542–553
Zhang SC (2006) Neural subtype specification from embryonic stem cells. Brain Pathol(Zurich, Switzerland) 16:132–142
Vijayalakshmi K, Alladi PA, Sathyaprabha TN, Subramaniam JR, Nalini A, Raju TR (2009) Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces degeneration of a cultured motor neuron cell line. Brain Res 1263:122–133
Inamdar MS, Venu P, Srinivas MS, Rao K, VijayRaghavan K (2009) Derivation and characterization of two sibling human embryonic stem cell lines from discarded grade III embryos. Stem Cells Dev 18:423–433
Amoroso MW, Croft GF, Williams DJ, O'Keeffe S, Carrasco MA, Davis AR, Roybon L, Oakley DH et al (2013) Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J Neurosci: Off J Soc Neurosci 33:574–586
Brooks BR, Miller RG, Swash M, Munsat TL (2000) El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 1:293–299
Sharma A, Varghese AM, Vijaylakshmi K, Sumitha R, Prasanna VK, Shruthi S, Chandrasekhar Sagar BK, Datta KK et al (2016) Cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients induces mitochondrial and lysosomal dysfunction. Neurochem Res 41:965–984
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods (San Diego, Calif). 25:402–408
Toli D, Buttigieg D, Blanchard S, Lemonnier T, Lamotte d'Incamps B, Bellouze S, Baillat G, Bohl D et al (2015) Modeling amyotrophic lateral sclerosis in pure human iPSc-derived motor neurons isolated by a novel FACS double selection technique. Neurobiol Dis 82:269–280
Wroblewski F, Ladue JS (1955) Lactic dehydrogenase activity in blood. Proc Soc Exp Biol Med 90:210–213
Sabitha KR, Sanjay D, Savita B, Raju TR, Laxmi TR (2016) Electrophysiological characterization of Nsc-34 cell line using microelectrode array. J Neurol Sci 370:134–139
Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, Stoltenborg-Hogenkamp BJ, Rodenburg RJ (2007) Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem 53:729–734
Kirby DM, Thorburn DR, Turnbull DM, Taylor RW (2007) Biochemical assays of respiratory chain complex activity. Methods Cell Biol 80:93–119
Mythri RB, Jagatha B, Pradhan N, Andersen J, Bharath MM (2007) Mitochondrial complex I inhibition in Parkinson's disease: how can curcumin protect mitochondria? Antioxid Redox Signal 9:399–408
Trounce IA, Kim YL, Jun AS, Wallace DC (1996) Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol 264:484–509
Rustin P, Chretien D, Bourgeron T, Gerard B, Rotig A, Saudubray JM, Munnich A (1994) Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta 228:35–51
Mariappan I, Maddileti S, Joseph P, Siamwala JH, Vauhini V (2015) Enriched cultures of retinal cells from BJNhem20 human embryonic stem cell line of Indian origin. Invest Ophthalmol Vis Sci 56:6714–6723
Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, Sockanathan S (1999) Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron 23:659–674
Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, Pfaff SL (1999) Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron 23:675–687
Hutchinson SA, Eisen JS (2006) Islet1 and Islet2 have equivalent abilities to promote motoneuron formation and to specify motoneuron subtype identity. Development 133:2137–2147
Moreno RL, Ribera AB (2014) Spinal neurons require Islet1 for subtype-specific differentiation of electrical excitability. Neural Dev 9:19
Pfaff SL, Mendelsohn M, Stewart CL, Edlund T, Jessell TM (1996) Requirement for LIM homeobox gene Isl1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell 84:309–320
Jessell TM (2000) Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet 1:20–29
Landmesser L (1978) The distribution of motoneurones supplying chick hind limb muscles. J Physiol 284:371–389
Rousso DL, Gaber ZB, Wellik D, Morrisey EE, Novitch BG (2008) Coordinated actions of the forkhead protein Foxp1 and Hox proteins in the columnar organization of spinal motor neurons. Neuron 59:226–240
Hanson MG, Landmesser LT (2004) Normal patterns of spontaneous activity are required for correct motor axon guidance and the expression of specific guidance molecules. Neuron 43:687–701
Miller, R.G., Mitchell, J.D., Lyon, M., and Moore, D.H. (2007). Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). The Cochrane database of systematic reviews, CD001447.
Johnson MA, Weick JP, Pearce RA, Zhang SC (2007) Functional neural development from human embryonic stem cells: accelerated synaptic activity via astrocyte coculture. J Neurosci 27:3069–3077
Nagaraja TN, Gourie-Devi M, Nalini A, Raju TR (1994) Neurofilament phosphorylation is enhanced in cultured chick spinal cord neurons exposed to cerebrospinal fluid from amyotrophic lateral sclerosis patients. Acta Neuropathol 88:349–352
Bendotti C, Tortarolo M, Suchak SK, Calvaresi N, Carvelli L, Bastone A, Rizzi M, Rattray M et al (2001) Transgenic SOD1 G93A mice develop reduced GLT-1 in spinal cord without alterations in cerebrospinal fluid glutamate levels. J Neurochem 79:737–746
Durham HD, Roy J, Dong L, Figlewicz DA (1997) Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol 56:523–530
Oda Y, Imai S, Nakanishi I, Ichikawa T, Deguchi T (1995) Immunohistochemical study on choline acetyltransferase in the spinal cord of patients with amyotrophic lateral sclerosis. Pathol Int 45:933–939
Shruthi S, Sumitha R, Varghese AM, Ashok S, Chandrasekhar Sagar BK, Sathyaprabha TN, Nalini A, Kramer BW et al (2017) Brain-derived neurotrophic factor facilitates functional recovery from ALS-cerebral spinal fluid-induced neurodegenerative changes in the NSC-34 motor neuron cell line. Neurodegener Dis 17:44–58
Tsang YM, Chiong F, Kuznetsov D, Kasarskis E, Geula C (2000) Motor neurons are rich in non-phosphorylated neurofilaments: cross-species comparison and alterations in ALS. Brain Res 861:45–58
Farah CA, Nguyen MD, Julien JP, Leclerc N (2003) Altered levels and distribution of microtubule-associated proteins before disease onset in a mouse model of amyotrophic lateral sclerosis. J Neurochem 84:77–86
Kabuta T, Kinugawa A, Tsuchiya Y, Kabuta C, Setsuie R, Tateno M, Araki T, Wada K (2009) Familial amyotrophic lateral sclerosis-linked mutant SOD1 aberrantly interacts with tubulin. Biochem Biophys Res Commun 387:121–126
Kaech S, Ludin B, Matus A (1996) Cytoskeletal plasticity in cells expressing neuronal microtubule-associated proteins. Neuron 17:1189–1199
Pryer NK, Walker RA, Skeen VP, Bourns BD, Soboeiro MF, Salmon ED (1992) Brain microtubule-associated proteins modulate microtubule dynamic instability in vitro. Real-time observations using video microscopy. J Cell Sci 103(Pt 4):965–976
Fanara P, Banerjee J, Hueck RV, Harper MR, Awada M, Turner H, Husted KH, Brandt R et al (2007) Stabilization of hyperdynamic microtubules is neuroprotective in amyotrophic lateral sclerosis. J Biol Chem 282:23465–23472
Rao MS, Devi MG, Nalini A, Shahani N, Raju TR (1995) Neurofilament phosphorylation is increased in ventral horn neurons of neonatal rat spinal cord exposed to cerebrospinal fluid from patients with amyotrophic lateral sclerosis. Neurodegener: J Neurodegener Disord, Neuroprot Neuroregener 4:397–401
Manetto V, Sternberger NH, Perry G, Sternberger LA, Gambetti P (1988) Phosphorylation of neurofilaments is altered in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 47:642–653
Schymick JC, Talbot K, Traynor BJ (2007) Genetics of sporadic amyotrophic lateral sclerosis. Hum Mol Genet 16:R233–R242
Cote F, Collard JF, Julien JP (1993) Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell 73:35–46
Marszalek, J.R., Williamson, T.L., Lee, M.K., Xu, Z., Hoffman, P.N., Becher, M.W., Crawford, T.O., and Cleveland, D.W. (1996). Neurofilament subunit NF-H modulates axonal diameter by selectively slowing neurofilament transport. J Cell Biol 135, 711–724.
Kong J, Xu Z (1998) Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci 18:3241–3250
Wei YH (1998) Oxidative stress and mitochondrial DNA mutations in human aging. Proc Soc Exp Biol Med 217:53–63
Beretta S, Sala G, Mattavelli L, Ceresa C, Casciati A, Ferri A, Carri MT, Ferrarese C (2003) Mitochondrial dysfunction due to mutant copper/zinc superoxide dismutase associated with amyotrophic lateral sclerosis is reversed by N-acetylcysteine. Neurobiol Dis 13:213–221
Grosskreutz J, Van Den Bosch L, Keller BU (2010) Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 47:165–174
Rutkowski, D.T., Arnold, S.M., Miller, C.N., Wu, J., Li, J., Gunnison, K.M., Mori, K., Sadighi Akha, A.A., Raden, D., and Kaufman, R.J. (2006). Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol 4, e374.
Schroder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569:29–63
Lautenschlaeger J, Prell T, Grosskreutz J (2012) Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 13:166–177
Oyanagi K, Yamazaki M, Takahashi H, Watabe K, Wada M, Komori T, Morita T, Mizutani T (2008) Spinal anterior horn cells in sporadic amyotrophic lateral sclerosis show ribosomal detachment from, and cisternal distention of the rough endoplasmic reticulum. Neuropathol Appl Neurobiol 34:650–658
Tobisawa S, Hozumi Y, Arawaka S, Koyama S, Wada M, Nagai M, Aoki M, Itoyama Y et al (2003) Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem Biophys Res Commun 303:496–503
Sutterlin C, Hsu P, Mallabiabarrena A, Malhotra V (2002) Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell 109:359–369
Warren G, Malhotra V (1998) The organisation of the Golgi apparatus. Curr Opin Cell Biol 10:493–498
Fujita Y, Okamoto K (2005) Golgi apparatus of the motor neurons in patients with amyotrophic lateral sclerosis and in mice models of amyotrophic lateral sclerosis. Neuropathology 25:388–394
Fujita Y, Okamoto K, Sakurai A, Amari M, Nakazato Y, Gonatas NK (1999) Fragmentation of the Golgi apparatus of Betz cells in patients with amyotrophic lateral sclerosis. J Neurol Sci 163:81–85
Gonatas NK, Stieber A, Gonatas JO (2006) Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J Neurol Sci 246:21–30
Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC (1996) The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci U S A 93:5472–5477
Uesugi M, Okamoto K, Tanaka M, Gonatas NK (2002) Masses of phosphorylated neurofilaments are associated with abnormal Golgi apparatus of anterior horn neurons of beta, beta’-iminodipropionitrile-intoxicated rats. Neuropathology 22:61–65
Sandoval, I.V., Bonifacino, J.S., Klausner, R.D., Henkart, M., and Wehland, J. (1984). Role of microtubules in the organization and localization of the Golgi apparatus. J Cell Biol 99, 113s–118s.
Vucic S, Kiernan MC (2006) Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 129:2436–2446
Vucic S, Nicholson GA, Kiernan MC (2008) Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 131:1540–1550
Pieri M, Albo F, Gaetti C, Spalloni A, Bengtson CP, Longone P, Cavalcanti S, Zona C (2003) Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett 351:153–156
Pieri M, Carunchio I, Curcio L, Mercuri NB, Zona C (2009) Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol 215:368–379
Devlin AC, Burr K, Borooah S, Foster JD, Cleary EM, Geti I, Vallier L, Shaw CE et al (2015) Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun 6:5999
Fuchs A, Kutterer S, Muhling T, Duda J, Schutz B, Liss B, Keller BU, Roeper J (2013) Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Physiol 591:2723–2745
Sankaranarayani R, Nalini A, Rao Laxmi T, Raju TR (2010) Altered neuronal activities in the motor cortex with impaired motor performance in adult rats observed after infusion of cerebrospinal fluid from amyotrophic lateral sclerosis patients. Behav Brain Res 206:109–119
Sankaranarayani R, Raghavan M, Nalini A, Laxmi TR, Raju TR (2014) Reach task-associated excitatory overdrive of motor cortical neurons following infusion with ALS-CSF. J Neural Transmission (Vienna, Austria: 1996) 121:49–58
Bostock H, Sharief MK, Reid G, Murray NM (1995) Axonal ion channel dysfunction in amyotrophic lateral sclerosis. Brain 118(Pt 1):217–225
Ellis DZ, Rabe J, Sweadner KJ (2003) Global loss of Na,K-ATPase and its nitric oxide-mediated regulation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 23:43–51
Heath PR, Shaw PJ (2002) Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 26:438–458
Kanai K, Kuwabara S, Misawa S, Tamura N, Ogawara K, Nakata M, Sawai S, Hattori T et al (2006) Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain 129:953–962
Gunasekaran R, Narayani RS, Vijayalakshmi K, Alladi PA, Shobha K, Nalini A, Sathyaprabha TN, Raju TR (2009) Exposure to cerebrospinal fluid of sporadic amyotrophic lateral sclerosis patients alters Nav1.6 and Kv1.6 channel expression in rat spinal motor neurons. Brain Res 1255:170–179
Deepa P, Shahani N, Alladi PA, Vijayalakshmi K, Sathyaprabha TN, Nalini A, Ravi V, Raju TR (2011) Down regulation of trophic factors in neonatal rat spinal cord after administration of cerebrospinal fluid from sporadic amyotrophic lateral sclerosis patients. J Neural Transmission (Vienna, Austria: 1996) 118:531–538
Cheng PL, Song AH, Wong YH, Wang S, Zhang X, Poo MM (2011) Self-amplifying autocrine actions of BDNF in axon development. Proc Natl Acad Sci U S A 108:18430–18435
Gooney M, Lynch MA (2001) Long-term potentiation in the dentate gyrus of the rat hippocampus is accompanied by brain-derived neurotrophic factor-induced activation of TrkB. J Neurochem 77:1198–1207
Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA (2000) Reduced BDNF mRNA expression in the Parkinson's disease substantia nigra. Exp Neurol 166:127–135
Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW (1991) BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 7:695–702
Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM et al (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 293:493–498
Ekegren T, Grundstrom E, Lindholm D, Aquilonius SM (1999) Upregulation of Bax protein and increased DNA degradation in ALS spinal cord motor neurons. Acta Neurol Scand 100:317–321
Eskes R, Antonsson B, Osen-Sand A, Montessuit S, Richter C, Sadoul R, Mazzei G, Nichols A et al (1998) Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J Cell Biol 143:217–224
Eskes R, Desagher S, Antonsson B, Martinou JC (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol 20:929–935
Guegan C, Vila M, Rosoklija G, Hays AP, Przedborski S (2001) Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci 21:6569–6576
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59
Ilzecka J (2012) Serum caspase-9 levels are increased in patients with amyotrophic lateral sclerosis. Neurol Sci 33:825–829
Pasinelli, P., Houseweart, M.K., Brown, R.H., Jr., and Cleveland, D.W. (2000). Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 97, 13901–13906.
Vukosavic S, Stefanis L, Jackson-Lewis V, Guegan C, Romero N, Chen C, Dubois-Dauphin M, Przedborski S (2000) Delaying caspase activation by Bcl-2: a clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 20:9119–9125
Dal Cengio Leonardi A, Keane NJ, Hay K, Ryan AG, Bir CA, VandeVord PJ (2013) Methodology and evaluation of intracranial pressure response in rats exposed to complex shock waves. Ann Biomed Eng 41:2488–2500
Tan, H., Yang, W., Wu, C., Liu, B., Lu, H., Wang, H., and Yan, H. (2017). Assessment of the role of intracranial hypertension and stress on hippocampal cell apoptosis and hypothalamic-pituitary dysfunction after TBI. Sci Rep 7, 3805.
Varghese AM, Sharma A, Mishra P, Vijayalakshmi K, Harsha HC, Sathyaprabha TN, Bharath SM, Nalini A et al (2013) Chitotriosidase—a putative biomarker for sporadic amyotrophic lateral sclerosis. Clin Proteomics 10:19
Mishra PS, Vijayalakshmi K, Nalini A, Sathyaprabha TN, Kramer BW, Alladi PA, Raju TR (2017) Etiogenic factors present in the cerebrospinal fluid from amyotrophic lateral sclerosis patients induce predominantly pro-inflammatory responses in microglia. J Neuroinflammation 14:251
Acknowledgements
We are grateful to Prof. Inamdar, JNCASR, for providing the human embryonic stem cell line-BJNHem20. We are thankful to Ms. Jessena Ponmalar for her assistance in conducting mitochondrial assays and Dr. Mariamma Philip for the statistical analysis related to electrophysiology experiments.
Funding
This study was supported by National Institute of Mental Health and Neurosciences, Bengaluru, India. Senior Research Fellowship for SR was funded by Indian Council of Medical Research (ICMR) (File No. 81/03/2013/SCRT/BMS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
This study was approved by the Institutional Ethics Committee and Steering Committee of Stem Cell Task Force, Department of Biotechnology, Government of India.
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Sumitha, R., Manjunatha, V.M., Sabitha, R.K. et al. Cerebrospinal Fluid from Patients with Sporadic Amyotrophic Lateral Sclerosis Induces Degeneration of Motor Neurons Derived from Human Embryonic Stem Cells. Mol Neurobiol 56, 1014–1034 (2019). https://doi.org/10.1007/s12035-018-1149-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1149-y