
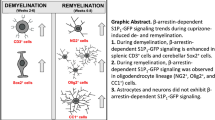
Abstract
In multiple sclerosis patients, demyelination is prominent in both the white and gray matter. Chronic clinical deficits are known to result from acute or chronic injury to the myelin sheath and inadequate remyelination. The underlying molecular mechanisms of remyelination and its failure remain currently unclear. Recent studies have recognized G protein-coupled receptor 17 (GPR17) as an important regulator of oligodendrocyte development and remyelination. So far, the relevance of GPR17 for myelin repair was mainly tested in remyelinating white matter lesions. The relevance of GPR17 for gray matter remyelination as well as remyelination of chronic white matter lesions was not addressed so far. Here, we provide a detailed characterization of GPR17 expression during experimental de- and remyelination. Experimental lesions with robust and limited endogenous remyelination capacity were established by either acute or chronic cuprizone-induced demyelination. Furthermore, remyelinating lesions were induced by the focal injection of lysophosphatidylcholine (LPC) into the corpus callosum. GPR17 expression was analyzed by complementary techniques including immunohistochemistry, in situ hybridization, and real-time PCR. In control animals, GPR17+ cells were evenly distributed in the corpus callosum and cortex and displayed a highly ramified morphology. Virtually all GPR17+ cells also expressed the oligodendrocyte-specific transcription factor OLIG2. After acute cuprizone-induced demyelination, robust endogenous remyelination was evident in the white matter corpus callosum but not in the gray matter cortex. Endogenous callosal remyelination was paralleled by a robust induction of GPR17 expression which was absent in the gray matter cortex. Higher numbers of GPR17+ cells were as well observed after LPC-induced focal white matter demyelination. In contrast, densities of GPR17+ cells were comparable to control animals after chronic cuprizone-induced demyelination indicating quiescence of this cell population. Our findings demonstrate that GPR17 expression induction correlates with acute demyelination and sufficient endogenous remyelination. This strengthens the view that manipulation of this receptor might be a therapeutic opportunity to support endogenous remyelination.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is an inflammatory disease of the central nervous system (CNS) associated with the development of large demyelinated plaques, oligodendrocyte destruction, and axonal degeneration. This pathology is paralleled by the activation of astrocytes and microglia as well as the recruitment of peripheral immune cells to the site of tissue injury. The feature, which distinguishes MS from other inflammatory and neurodegenerative disorders, is the formation of confluent plaques of primary demyelination. The sequence of molecular events leading to oligodendrocyte loss and consequently demyelination are not fully understood, but different stressors are known which can induce oligodendrocyte degeneration including oxidative stress, mitochondrial dysfunction, nitric oxide, protein misfolding, or inflammatory cytokine exposure [1, 2]. Of note, viable oligodendrocytes and an intact myelin sheath are indispensable for neuronal health. Oligodendrocytes provide nutritional support to neurons [3], fast axonal transport depends on oligodendrocytes [4], and mice deficient for mature myelin proteins display severe neurodegeneration [5].
Remyelination is one of the best-documented and most robust examples of tissue repair in the human adult CNS. Approximately 20–30% of postmortem MS tissues demonstrate remyelination, which can occur early and late during the course of the disease [6, 7]. Steps involved in this regenerative approach include activation and proliferation of oligodendrocyte progenitor cells (OPCs), migration of these OPCs towards the demyelinated axons, and OPC-axon interactions resulting in OPC differentiation and remyelination. Of note, the source of such OPCs might be manifold including progenitor cells dispersed in the brain parenchyma or located within neurogenic niches such as the subventricular zone [8]. Although the underlying mechanisms are poorly understood, remyelination is widespread in some MS patients, while in others it is sparse. A differentiation block of OPCs appears to be a major determinant of remyelination failure in chronic MS lesions [9]. Understanding why a relatively robust regenerative process should lose momentum is an important prerequisite for developing an effective therapeutic approach. Furthermore, while recent studies clearly showed that remyelination of the gray and white matter might be differentially regulated in both, MS [10] and its toxin-induced demyelination animal models [10,11,12], the underlying mechanisms are poorly understood.
Recent studies have recognized G protein-coupled receptors (GPCRs) as important regulators of oligodendrocyte development and maturation. At the most basic level, all GPCRs are characterized by the presence of seven membrane-spanning α-helical segments separated by alternating intracellular and extracellular loop regions. GPR17, in particular, is a G protein-coupled receptor that has been identified as regulator of remyelination [13,14,15,16]. Although the actual activating ligands for and some functions of this receptor are disputed, GPR17 has been reported to be activated by cysteinyl leukotrienes [17] and by purines such as ATP [18] or UDP [15]. GPR17 expression is almost absent in early OPCs, gradually increases in more mature precursors, reaches a plateau in immature/pre-oligodendrocytes, and then gradually decreases during terminal differentiation. In line with these findings, GPR17 is co-expressed with the early oligodendrocyte marker NG2 and markers of pre/immature oligodendrocyte phenotype but is downregulated in cells expressing myelin proteins such as myelin basic protein [18, 19]. It is, thus, believed that GPR17 is required for initiating the differentiation of OPCs but has to be downregulated to allow cells to undergo terminal maturation.
The consequence of GPR17 activation for oligodendrocyte differentiation and in consequence (re-)myelination appears to be context dependent. GPR17 transgenic mice with persistent GPR17 overexpression in specifically oligodendrocytes showed failure of proper developmental myelination, and in vitro Gpr17 overexpression inhibited oligodendrocyte differentiation and maturation. Conversely, Gpr17 knockout mice showed early onset of oligodendrocyte myelination suggesting that the loss of GPR17 function accelerates OPC differentiation and myelination [20]. In line with this finding, chemical activation of GPR17 inhibited the maturation of primary oligodendrocytes from heterozygous but not GPR17−/− mice in culture, as well as in cerebellar slices [21, 22], whereas inhibition of GPR17 activity with pranlukast promoted OPC differentiation in vitro. Beyond, the deletion of Gpr17, either globally or specifically in the oligodendrocyte lineage, resulted in an earlier onset of remyelination in a model of focally lysophosphatidylcholine-induced demyelination [13]. The hypothesis that deactivation of GPR17 is needed for oligodendrocyte maturation is further supported by the recent finding that GPR17 desensitization by G-protein receptor kinase phosphorylation and subsequent internalization are necessary events for terminal differentiation of OPCs [22]. Taken together, these data suggest that GPR17 acts to negatively regulate terminal oligodendrocyte differentiation and myelination. In contrast to these results, it has been reported that GPR17 activation by natural agonists can promote OPC differentiation in vitro [16, 19].
The most commonly used animal models used to study MS-related aspects of remyelination are toxin models. In principal, toxin-mediated demyelination with subsequent remyelination can be induced by either focal injection or systemic administration of the toxin. While focal demyelination is usually induced by injection of lysolecithin (also called lysophosphatidylcholine; LPC), the copper chelator cuprizone is used for systemic intoxication [23, 24]. LPC acts as a membrane detergent, resulting in rapid myelin sheath disassembly and, finally, demyelination in a matter of days. In the cuprizone model, demyelination is due to a metabolic insult of oligodendrocytes. Cuprizone-induced primary oligodendrocyte apoptosis leads to almost complete demyelination in a matter of weeks (i.e., 5–6 weeks). In both models, endogenous remyelination is robust and occurs within weeks after induction of experimental demyelination [25, 26]. In contrast, endogenous remyelination is limited after prolonged cuprizone exposure (i.e., chronic demyelination) [24, 26].
In the current study, we aimed to analyze expression of GPR17 in different MS animal models and relate its expression to de- and remyelination.
Material and Methods
Animals
Seven- to 8-week-old C57BL/6 male and 8-week-old female mice were purchased from Janvier Labs, Le Genest-Saint-Isle, France. Microbiological monitoring was performed according to the Federation of European Laboratory Animal Science Associations recommendations. Animals were randomly allocated to the different experimental groups. A maximum of five animals were housed per cage (cage area 435 cm2). Animals were kept under standard laboratory conditions (12 h light/dark cycle, controlled temperature 23 °C ± 2 °C and 55% ± 10% humidity) with access to food and water ad libitum. It was assured that researches and technicians did not use any light during the night cycle period. Nestlets were used for environmental enrichment. All cuprizone experiments were formally approved by Regierung Oberbayern (Bayern, Germany; reference number 55.2-154-2532-73-15). All lysophosphatidylcholine-induced experiments were performed according to the guidelines of the European Directive 2010/63/EU and Belgian legislation. The ethical committee for animal experimentation from UCB Biopharma SPRL (LA1220040 and LA2220363) approved the experimental protocols.
Induction of Experimental Demyelination
Systemic demyelination was induced by feeding male mice with ground standard rodent chow (Ssniff, Soest, Germany) containing 0.25% cuprizone [bis(cyclohexanone)oxaldihydrazone; Sigma-Aldrich Inc., St Louis, MO, USA] for the indicated treatment period. Control groups were fed standard rodent chow. In summary, 71 mice were used for the cuprizone experiments.
Focal demyelination was induced by stereotactic injection of the membrane detergent lysophosphatidylcholine (LPC; n = 9). All surgical procedures were performed under general anesthesia by intraperitoneal injections of ketamine (50 mg/kg body weight; Nimatek, Eurovet Animal Health B.V.) and medetomidine (0.5 mg/kg body weight; Domitor Orion Corporation). Mice recovery was accelerated with one injection of 2.5 mg/kg atipamezole (Antisedan, Orion Corporation). In order to prevent dehydration, mice were subcutaneously injected with 500 μL of Ringer’s solution (B. Braun, Germany). For the stereotactic surgery, mice were fixed in a standard stereotaxic frame (model 900 and mouse adaptor model 922; Kopf CA, USA). The scalp was incised, and two drill holes were made at the following coordinates relative to the bregma to target the corpus callosum in both hemispheres: anterior-posterior − 1.10 mm, medial-lateral ± 0.90 mm, and dorsal-ventral − 1.45 from the skull (mm) [27, 28]. A 1% LPC solution (two administrations of 0.75 μL of 10 μg/μL LPC from bovine brain; Ref L1381, Cas Number 9008-30-4, Sigma-Aldrich; Germany in saline) was injected with a syringe needle (1702 (25 μL), 33 gauges, 8 mm length; Hamilton, Bonaduz, Switzerland), protected from light with aluminum, and connected to a syringe pump (Model Legato 130, KD Scientific), at a flow rate of 0.2 μL/min. Control animals (n = 10) received 0.75 μL of saline as a vehicle solution instead. To ensure complete diffusion and avoid spillage into neighboring structures, the needle was left in place for 2.5 min before being slowly retracted. The animals were monitored daily after surgery and finalized after 5 days by transcardial perfusion.
Induction of experimental autoimmune encephalomyelitis (EAE) in female mice was induced by injection of an emulsion of MOG35–55 peptide dissolved in complete Freund’s adjuvant followed by injection of pertussis toxin in PBS according to the manufacturer’s recommendations (Hooke Laboratories, USA). Disease severity was scored as published previously [29]. The animals were monitored daily and finalized 14 days after immunization (i.e., at peak of the disease).
Tissue Preparation
For histological and immunohistochemical studies (IHC), preparation of tissues was performed as previously described [30]. In brief, mice were transcardially perfused with ice-cold PBS followed by a 3.7% paraformaldehyde solution (PFA; pH 7.4). After overnight post-fixation in the same fixative, the brains were dissected and embedded in paraffin, and coronal or medio-sagittal 5-μm-thick sections were prepared for immunohistochemistry or in situ hybridization. The spinal cords were incubated in EDTA (ethylenediaminetetraacetic acid) for 48 h at 37 °C (changed once) prior to paraffin embedding. Coronal slices were analyzed at the level 265 (i.e., ventral hippocampus) according to the mouse brain atlas published by Sidman et al. (http://www.hms.harvard.edu/research/brain/atlas.html). For gene expression studies, mice were transcardially perfused with ice-cold PBS and corpus callosum, cortex, and spinal cord tissues were manually dissected and snap-frozen in liquid nitrogen. Samples were then kept at − 80 °C until further processing.
Immunohistochemistry (IHC) and (Densitometric) Analysis
For immunohistochemistry, sections were rehydrated and, if necessary, antigens were unmasked with heating in Tris/EDTA (pH 9.0) or citrate (pH 6.0) buffer. After washing in PBS, sections were incubated overnight (4 °C) with anti-proteo-lipid antibodies to detect myelin ([PLP] 1:5000), or with anti-G protein-coupled receptor 17 antibodies to detect pre-myelinating oligodendrocytes ([GPR17] 1:200). The next day, slides were incubated either with biotinylated secondary antibodies [(i) horse anti-mouse IgG, 1:50; (ii) goat anti-rabbit IgG, 1:50] for 1 h and then with peroxidase-coupled avidin–biotin complex (ABC-HRP kit; order number PK-6100, RRID AB 2336819, Vector Laboratories, Burlingame, USA) and treated with 3,3′-diaminobenzidine (order number K3468, DAKO, Hamburg, Germany) as a peroxidase substrate. Some slides were incubated with the EnVision System-HRP Labeled Polymer [(iii) goat anti-mouse; (iv) goat anti-rabbit]. A detailed list of applied antibodies are given in Table 1 and Table 2.
Stained and processed sections were digitalized using a Nikon Eclipse 80i microscope (Nikon, Nikon Instruments, Germany) equipped with a DS-2MV camera. The open source program ImageJ 1.48v (NIH, Bethesda, MD, USA) was used to evaluate staining intensity using semi-automated densitometrical evaluation after threshold setting. In brief, acquired images were converted to gray scale images, and a global thresholding algorithm was used for dividing each image into two classes of pixels (black and white, i.e., binary conversion). Global thresholding works by choosing a value cutoff, such that every pixel less than that value is considered one class, while every pixel greater than that value is considered the other class. Relative staining intensity was then quantified in binary converted images, and results are presented as percentage area.
Stained sections of a different cohort of mice were additionally scanned using the Nikon Eclipse E200 microscope (Nikon Instruments, Germany) equipped with a Basler acA1920-40um camera (Basler AG, Ahrensburg, Germany) and a manual scanning software (manualWSI software, Microvisioneer, München, Germany). Cell numbers were quantified after manually delineating the region of interest (ROI) using the program ViewPoint (PreciPoint GmbH, Freising, Germany).
Luxol Fast Blue (LFB) Periodic Acid–Schiff (PAS) Stain
For validation of the myelination data, intact and damaged myelin was additionally visualized using LFB/PAS histochemical stains. To this end, slides were deparaffinized in 4 × 5 min xylene, rinsed 3 × 3 min in 100% ethanol, followed by 2 × 5 min in 96% ethanol. Sections were then subsequently incubated in LFB solution (0.1 g Luxol fast blue (order number 7709, Carl Roth, Germany) in 100 mL 96% ethanol plus 500 μL acetic acid (order number 3738, Carl Roth, Germany), overnight at 60 °C. On the next day, sections were dipped in 96% followed by water and processed in a lithium carbonate solution (0.05 g lithium carbonate [order number 1.05680.0250, Merck, Germany] in 100 mL aqua dist.). Sections were further differentiated in 70% ethanol for a few seconds and rinsed in water. Afterwards, oxidation was performed in periodic acid (0.5 g periodic acid [order number 1.00524.0025, Merck, Germany] in 100 mL aqua dist.). Sections were rinsed followed by incubation in Schiff’s reaction (order number 1.09033.0500, Merck, Germany) for 15 min, then rinsed in warm tap water for 5 min and counterstained with hematoxylin (order number 1.04302.0025, Merck, Germany) for 1 min. Sections were dehydrated and subsequently mounted in Depex (order number 18243, Serva, Germany) for further analyses.
Immunofluorescence Double Labeling
For immunofluorescence double labeling, sections were rehydrated, unmasked with Tris/EDTA buffer (pH 9.0) and heating, blocked with PBS containing 2% heat-inactivated fetal calf serum ([FCS], order number A15-152, PAA, Germany) and 1% bovine serum albumin ([BSA], order number 0163, Carl Roth, Germany), and incubated overnight (4 °C) with the combination of primary antibodies diluted in blocking solution. Anti-GPR17 (1:100) was either combined with anti-OLIG2 (1:1000) to visualize oligodendrocytes, with anti-GFAP (1:1000) to visualize astrocytes, with anti-PDGF receptor alpha (1:100) to visualize oligodendrocyte progenitor cells, or with anti-NeuN (1:2000) to visualize neurons. After washing, sections were incubated in a combination of fluorescent anti-rabbit secondary antibodies (1:500, Alexa Fluor) and fluorescent anti-mouse secondary antibodies (1:500, Alexa Fluor), or fluorescent anti-rat secondary antibodies (1:500, Alexa Fluor) both diluted in blocking solution. Subsequently, sections were incubated with Hoechst 33342 solution (1:10000, order number H3570, Life Technologies, USA) diluted in PBS for the staining of cell nuclei. A detailed list of applied antibodies is given in Table 1 and Table 2.
To rule out unspecific binding of the fluorescent secondary antibodies to primary antibodies, appropriate negative controls were performed by first incubating sections with the primary antibodies and subsequently incubating these sections with switched fluorescent secondary antibody. Unspecific secondary antibody binding to the tissue itself was checked by performing negative controls by incubating sections with each of the fluorescent secondary antibodies alone (data not shown). Stained and processed sections were documented with the Leica microscope DMI6000B (Leica Microsystems, Germany). Cell numbers were quantified after manually delineating the corpus callosum using the open source program ImageJ 1.48v (NIH, Bethesda, MD, USA). For a clearer demonstration, colors were digitally adjusted using ImageJ, in order to use an identical color code in the representative figures.
In Situ Hybridization
A commercial fluorescence in situ hybridization kit (ViewRNA® in situ hybridization tissue assay, Affymetrix-Panomics, order number QVT0012) was used for single labeling of formalin-fixed, paraffin-embedded tissues, according to the manufacturer’s recommendations. Protease digestion time was adjusted to 20 min. The probe for Gpr17 was purchased from Affymetrix (Affymetrix-Panomics, VB1-13617 Type 1). Fluorescence images were captured using the Leica microscope DMI6000B (Leica Microsystems), and cell numbers were quantified using open source program ImageJ 1.48v (NIH, Bethesda, MD, USA) after manually outlining the corpus callosum.
Gene Expression Analyses
Gene expression levels were analyzed using cDNA from the isolated tissues, with real-time reverse transcription-PCR (Bio-Rad, Germany) using SensiMix SYBR and Fluorescein (Bioline, Germany). Primer sequences and individual annealing temperatures are shown in Table 3. Results were normalized to ß-actin as the reference gene and target gene expression was calculated using the ∆∆Ct method. Melting curves were analyzed and PCR products were routinely separated by gel electrophoresis to determine the specificity of the PCR reaction (data not shown).
Statistical Analysis
Statistical analyses were performed using Prism 5 (GraphPad Software Inc., San Diego, CA, USA). All data are given as arithmetic means ± SEMs. A p value of ≤ 0.05 was considered to be statistically significant. Applied statistical tests are given in the respective figure legends. Normal distribution was assumed. No outliers were excluded from the analyses.
Results
In the cuprizone model, demyelination is robust after a 5-week intoxication period. After termination of intoxication, endogenous remyelination occurs and is complete within 3 to 4 weeks. In a first step, we investigated changes of Gpr17 expression during acute demyelination and early remyelination. To this end, one cohort of mice was sacrificed after 5 weeks cuprizone intoxication, whereas another was sacrificed after 7 days recovery and compared to control animals (see Fig. 1 (A) for the experimental setup of this part of the study). Gpr17 mRNA expression levels were analyzed by rt RT-PCR and in situ hybridization, whereas GPR17 protein expression was investigated by immunohistochemistry. To verify de- and remyelination, respectively, mid-sagittal sections were processed for anti-PLP immunohistochemistry. Anatomical landmarks in the mid-sagittal plane are shown in Fig. 1 (B). As demonstrated in Fig. 1 (C, C’), profound demyelination of the medial (mCC) and caudal (cCC) part of the corpus callosum was readily visible after 5 weeks cuprizone intoxication. Demyelination was as well present within the rostral part of the corpus callosum (rCC), even though loss of anti-PLP staining was incomplete there. In line with previous reports [31], demyelination was as well extensive in the gray matter cortex (see star in Fig. 1 (C)). Other regions were resistant to the 5 weeks cuprizone intoxication period such as the anterior commissure (ac), or diverse midbrain structures (mid). After 7 days of recovery, semi-maximal remyelination was evident in the previously demyelinated structures. While pronounced recovery of anti-PLP staining loss was evident in the caudal corpus callosum (co 96.39 ± 1.37%; 5 weeks 3.005 ± 2.325%; 5 weeks + 7 days 70.02 ± 16.36%), recovery of anti-PLP staining intensity was minor in the gray matter cortex (co 37.4 ± 5.49%; 5 weeks 2.235 ± 0.925%; 5 weeks + 7 days 6.38 ± 0.63%) (see Fig. 1 (D)). Demyelination and early remyelination of the corpus callosum was as well observed in LFB/PAS-stained sections (data not shown).

Cuprizone-induced de- and remyelination. (A) Schematic depicting the experimental setup. Group I was sacrificed after 5 weeks normal chow (control), group II after acute demyelination (5 weeks cuprizone; 0.25%), and group III after early remyelination (5 weeks cuprizone, followed by 7 days recovery). (B) Anatomical landmarks in the mid-sagittal plane: cCC caudal corpus callosum, mCC medial corpus callosum, rCC rostral corpus callosum, mid midbrain, ac anterior commissure. (C, C’) Expression of PLP, visualized by anti-PLP immunohistochemistry. (D) Quantification of anti-PLP staining intensity by densitometric analysis (two biological replicates each). Note the severe demyelination of the caudal and medial corpus callosum and gray matter cortex (star in C). As well, note the ongoing remyelination in the CC, but not in the cortex region
Next, we aimed to correlate de- and remyelination with GPR17 expression. Anti-GPR17 immunohistochemistry in control animals revealed numerous GPR17+ cells evenly distributed within the white matter corpus callosum and gray matter cortex. Especially in the cortex, GPR17+ cells displayed a multipolar morphology with multiple, fine-branched processes, reminiscent of pre-myelinating oligodendrocytes (see insert in Fig. 2 (A)). In the white matter corpus callosum, GPR17+ cells as well displayed fine processes; however, these appeared to be aligned in parallel to axonal fiber bundles. After 5 weeks cuprizone intoxication, a profound accumulation of GPR17+ cells was found, especially in the rostral and caudal part of the corpus callosum (arrows), less so in its medial portion (see Fig. 2 (B)). This accumulation persisted during the recovery period (see Fig. 2 (C)). Densitometric evaluation revealed increased anti-GPR17 staining intensity in the corpus callosum of cuprizone-treated mice (co 18.12 ± 3.015%; 5 weeks 81.16 ± 5.77%; 5 weeks + 7 days 74.71 ± 9.2%). In contrast, GPR17 staining intensity was not increased in the demyelinated and remyelinating cortex (co 72.54 ± 26.99%; 5 weeks 49.39 ± 23.37%; 5 weeks + 7 days 84.98 ± 8.17%) (see Fig. 2 (D)). For a more detailed analysis, we quantified numbers of GPR17+ cells in the gray matter cortex. As shown in Fig. 2 (E), numbers of GPR17+ cells slightly decreased after 5 weeks cuprizone intoxication (co 19.5 ± 1.5 cells/mm2; 5 weeks 14.5 ± 2.5 cells/mm2) and remained at this reduced level after 7 days remyelination (5 weeks + 7 days 15.5 ± 2.5 cells/mm2). Since GPR17 expression in oligodendrocyte progenitor cells has been shown to be downregulated during terminal oligodendrocyte differentiation [19], an additional cohort of mice was analyzed after a 11-day remyelination period. As demonstrated in Fig. 2 (F, G), numerous GPR17+ cells were found in the corpus callosum at week 5, but numbers were significantly lower after prolonged remyelination. Comparable to our observation at day 7, while there was sufficient remyelination of the white matter corpus callosum, remyelination of the gray matter cortex was delayed at day 11.

Cuprizone-induced demyelination leads to GPR17+ cell accumulation in the corpus callosum but not in the cortex region. Expression of GPR17, visualized by anti-GPR17 immunohistochemistry, in control (A), 5 weeks (B), and 5 weeks cuprizone, followed by 7 days recovery groups (C, sagittal plane). Inserts in the upper row depict the morphology of a gray and white matter GPR17+ cell, respectively. (D) Quantification of anti-GPR17 staining intensity by densitometric analysis in the white matter corpus callosum and gray matter cortex (two biological replicates). (E) Quantification of GPR17+ cell numbers in the gray matter cortex (two biological replicates). Note the induction of GPR17 in the corpus callosum, but not in the cortex region. (F, G) Representative anti-PLP and anti-GPR17 immunohistochemistry of the midline of the corpus callosum (coronal plane) after 5 weeks cuprizone (F) and 5 weeks cuprizone followed by 11 days recovery (G). (H) Quantification of anti-GPR17 staining intensity by densitometric analysis in the white matter corpus callosum (at least six biological replicates each). CC corpus callosum, Cx cortex. Differences between groups were statistically tested using a two-tailed t test; ***p ≤ 0.001
To verify our results obtained on the protein level, Gpr17 mRNA expression was additionally analyzed in a parallel cohort of mice. As shown in Fig. 3 (A), rt RT-PCR analysis with isolated corpus callosum tissues revealed Gpr17 expression induction after 5 weeks cuprizone intoxication (co 100 ± 19.57% vs 5 weeks 611.3 ± 180.6%). Increased Gpr17 mRNA expression levels remained stable during the 7 days recovery period (5 weeks + 7 days 526.2 ± 251.4%). Minor expression induction was as well found in isolated cortex tissues (co 100 ± 11.14%; 5 weeks 147 ± 21.57%; 5 weeks + 7 days 120.7 ± 8.719%); however, this induction failed to reach statistical significant levels. In line with these observations, higher numbers of Gpr17+ cells were as well observed in the corpus callosum by in situ hybridization (co 76 ± 27.77 cells/mm2; 5 weeks 254.5 ± 88.15 cells/mm2; 5 weeks + 7 days 190.5 ± 17.32 cells/mm2) (see Fig. 3 (B, C)). To verify that GPR17+ cells belong to the oligodendrocyte lineage, sections were processed for OLIG2/GPR17, GFAP/GPR17, and NeuN/GPR17 immunofluorescence double labeling (see Fig. 4). As shown in Fig. 4 (A), and in line with our results presented above, densities of OLIG2+/GPR17+ cells (yellow) were low (34 ± 1 cells/mm2) in control animals, but high at week 5 (142 ± 9 cells/mm2) and after 7 days of remyelination (137.5 ± 28.5 cells/mm2). Densities of OLIG2+/GPR17− cells (red) were higher compared to the OLIG2+/GPR17+ cell population, most likely representing OLIG2+ mature oligodendrocytes (co 1203 ± 118 cells/mm2; 5 weeks 623.5 ± 119.5 cells/mm2; 5 weeks + 7 days 1318 ± 183 cells/mm2). Of note, OLIG2−/GPR17+ cells (green) were virtually absent at all investigated time points (co 3 ± 3 cells/mm2; 5 weeks 7.5 ± 7.5 cells/mm2; 5 weeks + 7 days 1.5 ± 1.5). In line with these findings, almost no GFAP+/GPR17+ cells were found in the corpus callosum of control and cuprizone-intoxicated mice (see Fig. 4 (B)). Double labeling experiments for NeuN and GPR17 showed that some cells in the cortex stained positive for both antigens (see Fig. 4 (C)). Of note, none of these GPR17/NeuN double-positive cells displayed the characteristic morphology of OPCs.

Cuprizone-induced demyelination induces Gpr17 mRNA expression in the corpus callosum but not in the cortex region. (A) Gpr17 mRNA expression in the isolated corpus callosum of control, 5 weeks cuprizone, and 5 weeks cuprizone followed by 7 days recovery groups (at least three biological replicates). Differences between groups were statistically tested using one-way ANOVA with the obtained p values corrected for multiple testing using Dunnett’s post hoc. (B) Quantification of Gpr17+ cell numbers in the corpus callosum visualized by in situ hybridization (two biological replicates). (C) Representative images of Gpr17 in situ hybridization experiments in the mid-sagittal plane. Dotted line, border of the caudal corpus callosum; blue, Hoechst; red, Gpr17

GPR17+ cells belong to the oligodendrocyte lineage. Representative double immunofluorescence stains of the caudal corpus callosum of control, 5 weeks cuprizone, and 5 weeks cuprizone followed by 7 days recovery groups (left side) and quantification of distinct cell populations (right side; two biological replicates each). The dotted line delineates the border of the caudal corpus callosum towards the neocortex; white arrows highlight GPR17+ cells expressing as well OLIG2. Green, GPR17; red, OLIG2 (A), GFAP (B), NeuN (C); blue, Hoechst. Note that virtually all GPR17+ cells as well express the oligodendrocyte marker protein OLIG2 (A). Almost no GPR17+ cells express the astrocytic GFAP marker protein (B). Further, note that some neocortical NeuN+ neurons express GPR17 (see arrowheads)
Next, we investigated at what time point GPR17 expression induction occurs in the cuprizone model. To this end, another cohort of mice was fed cuprizone for up to 5 weeks, processed at different time points for immunohistochemistry, and compared to control animals. As shown in Fig. 5 (A), a profound demyelination of the medial part of the corpus callosum was readily visible after 5 weeks cuprizone intoxication. Demyelination was not evident at week 1 or 3. Densitometric evaluation of the anti-GPR17 staining intensity revealed a significant increase of GPR17 expression in the corpus callosum at week 5 (co 31.76 ± 5.392% vs 5 weeks 80.92 ± 8.332%; p ≤ 0.001) (see Fig. 5 (C)). In line with this finding, a significant higher number of GPR17+ cells were found in the corpus callosum (co 45.98 ± 3.517 cells/mm2 vs 5 weeks 205.4 ± 33.62 cells/mm2; p ≤ 0.001) but not in the cortex region (co 21.28 ± 1.829 cells/mm2 vs 5 weeks 25.23 ± 1.368 cells/mm2) after 5 weeks cuprizone intoxication (Fig. 5 (D, E)).

GPR17 induction in the course of cuprizone-induced demyelination. (A) Representative anti-PLP stains of control mice and mice intoxicated with cuprizone for 1, 3, and 5 weeks. (B) Representative anti-GRP17 stains of control mice and mice intoxicated with cuprizone for 1, 3, and 5 weeks. High-power views show the midline of the corpus callosum (mCC). (C) Quantification of anti-GPR17 staining intensity by densitometric analysis in the white matter corpus callosum (at least four biological replicates). (D) Quantification of GPR17+ cell numbers in the corpus callosum (at least four biological replicates). (E) Quantification of GPR17+ cell numbers in the cortex (at least four biological replicates). Differences between groups were statistically tested using one-way ANOVA with the obtained p values corrected for multiple testing using Dunnett’s post hoc test; *p ≤ 0.05 or ***p ≤ 0.001
Next, we were interested whether induction of GPR17 expression is restricted to the cuprizone model, or is as well evident in another model of experimental demyelination with robust remyelination. To this end, we induced focal demyelination by stereotactic injection of the membrane detergent lysophosphatidylcholine (LPC) into the corpus callosum [32, 33]. Five days after bilateral lesion induction (indicated by orange lines in Fig. 6 (A)) with either vehicle or LPC, brains were processed for immunohistochemistry. As shown in Fig. 6 (A), LFB/PAS stains revealed profound demyelination of the midline of the caudal corpus callosum in LPC-injected (stars) but not vehicle-injected mice (co 99.4 ± 0.4% vs LPC 4.333 ± 0.333%; p ≤ 0.001) (see Fig. 6 (C)). Vehicle-injected mice showed small GPR17+ cells with thin and ramified processes (see Fig. 6 (B)). Densitometric quantification of GPR17 staining intensity revealed a highly significant increase of GPR17 expression within the LPC-injected compared to the vehicle-injected corpus callosum (co 9.208 ± 1.063% vs LPC 39.28 ± 3.769%; p ≤ 0.001) (see Fig. 6 (B)).

Focal demyelination leads to GPR17+ cell accumulation. (A) Representative LFB/PAS staining of vehicle (upper row) or LPC-injected (lower row) mice. The orange lines illustrate the theoretical position of the needles during the bilateral stereotactic LPC injection. The midline of the corpus callosum is shown on the right in higher magnification. Note the complete demyelination of the midline of the corpus callosum (stars and high-power view). (B) Expression of GPR17 in vehicle (upper picture) and LPC-injected (lower picture) mice, visualized by anti- GPR17 immunohistochemistry. (C) Quantification of LFB/PAS staining intensity by blinded evaluation of the white matter corpus callosum (at least nine biological replicates). (D) Quantification of anti-GPR17 staining intensity by densitometric analysis in the white matter corpus callosum (at least nine biological replicates). Differences between groups were statistically tested using unpaired t test; ***p ≤ 0.001. Note the intense accumulation of GPR17-expressing cells in the demyelinated corpus callosum
Next, we wanted to investigate GPR17 expression in an experimental setting with limited endogenous remyelination capacity. To this end, a separate cohort of animals was intoxicated with cuprizone for 5 weeks (i.e., acute demyelination) or 12 weeks (i.e., chronic demyelination), and numbers of GPR17 cells were quantified in the midline of the corpus callosum and compared to control animals. As demonstrated in Fig. 7 (A), severe demyelination was evident at weeks 5 and 12. In line with our previous findings, numbers of GPR17+ cells in the corpus callosum were significantly increased after a 5-week cuprizone intoxication period (co 44 ± 9.597 vs 5 weeks 245 ± 16.66 cells/mm2; p ≤ 0.001). The number of GPR17+ cells was low after chronic cuprizone-induced demyelination (12 weeks 68.80 ± 2.107 cells/mm2). To clarify if low GPR17 expression in chronic lesions is due to loss of OPCs, subsequent sections were stained for the OPC marker protein platelet-derived growth factor receptor alpha (PDGFRα). As demonstrated in Fig. 7 (C), densities of PDGFRα-expressing cells were low in control animals, high after acute, and intermediate after chronic cuprizone-induced demyelination, respectively. Statistical comparison revealed significant lower densities of PDGFRα+ cells after chronic (112.2 ± 34.35 cells/mm2) compared to acute demyelination (340 ± 37.07 cells/mm2; p ≤ 0.01).

GPR17 expression in acute and chronic cuprizone lesions. Representative anti-PLP (A), anti-GPR17 (B), and anti-PDGFRα stains of control mice and mice intoxicated with cuprizone for 5 or 12 weeks. (D) Quantification of GPR17+ cell numbers in the white matter corpus callosum (five biological replicates each). Differences between groups were statistically tested using one-way ANOVA with the obtained p values corrected for multiple testing using Dunnett’s post hoc test; ***p ≤ 0.001. (E) Quantification of PDGFRα+ cell numbers in the white matter corpus callosum (five biological replicates each). Differences between groups (5 and 12 weeks) were statistically tested using unpaired t test; **p ≤ 0.01
Finally, we wanted to investigate GPR17 expression in an autoimmune model of MS, namely, experimental autoimmune encephalomyelitis (EAE). As demonstrated in Fig. 8, in both spinal cord sections of control and EAE mice, GPR17+ cells what appeared to be motoneurons were found in the ventral horn (see arrowheads in Fig. 8 (C, G)). Similar to what we found in the forebrain, GPR17+ cells with a multipolar morphology and multiple, fine-branched processes were found in the gray and white matter spinal cord (see arrowhead in Fig. 8 (D)). It appeared that the densities of such GPR17+, small, ramified cells was higher in inflammatory white matter spinal cord lesions of EAE mice; however, no induction of Gpr17 mRNA expression was observed by rt RT-PCR in a separate cohort of animals (co 100 ± 18.04%; EAE 81.57 ± 10.05%; data not shown).

GPR17 expression in the spinal cord of EAE mice. Representative LFB/PAS stains of spinal cord sections from control (A) and EAE (E) mice. Arrowheads in (E) highlight inflammatory spinal cord white matter lesions. Representative anti-GRP17 stains of spinal cord sections from control (B) and EAE (F) mice. (C, G) show the ventral spinal cord gray matter in higher magnification. Arrowheads highlight GPR17+ motorneurons. (D, H) show the spinal cord white matter in higher magnification. Arrowheads highlight GPR17+ oligodendrocyte progenitor cells. The dotted line in (H) delineates the border of an active inflammatory white matter lesion. Note that densities of GPR17+ cells appear increased in such areas
Discussion
Because most therapeutic strategies for MS target the immune system and are less effective or even ineffective in the progressive phase of the disease, preservation of oligodendrocytes or increasing their remyelinating capacity is a promising strategy for novel MS treatments. A first step towards developing potential promyelinating therapies is to identify genes whose expression is affected during demyelination and subsequent remyelination. In this study, we used two commonly applied remyelination models, namely, the cuprizone and the LPC model, to study expression of the G-protein-coupled receptor Gpr17 on the mRNA and protein level. GPR17 was previously found to be expressed in OPCs and premature oligodendrocytes [19], and it has been proposed to have a role in OPC differentiation and developmental myelination [14, 20, 34]. These studies showed that GPR17 inhibits OPC differentiation in vitro and delays myelination during development in vivo. Furthermore, GPR17 activation inhibited oligodendrocyte survival in the LPC model [13]. The same authors showed that inhibition of GPR17 promoted oligodendrocyte differentiation and remyelination. Pharmacological inhibition of GPR17 could, therefore, be considered as a potential therapeutic approach for enhancing myelin repair.
In a first step, we investigated the expression of GPR17 in the cuprizone model. After 5 to 6 weeks of cuprizone intoxication, specific parts of the corpus callosum are almost completely demyelinated, a process called “acute demyelination.” Acute demyelination is followed by spontaneous remyelination during subsequent weeks when mice are fed normal chow. In contrast, endogenous remyelination is highly restricted when cuprizone administration is prolonged (12 weeks or longer), a process called “chronic demyelination” [24, 26]. In line with the results from other groups, our immunohistochemical stains using anti-GPR17 antibodies showed that GPR17+ cells are present in similar numbers in the white matter, corpus callosum, and gray matter cortex [35]. Consistent with previous reports, by immunohistochemistry, we observed two distinct cortical GPR17+ cell populations: first, non-neuronal cells labeled with single intracellular spots or a staining on the whole cell membrane. These cells were characterized by a small, heavily stained cell soma and a network of highly branched cellular processes. The processes extended into all directions with no obvious preferential spatial orientation. Although not formally proven in this study, we propose that these GPR17+ cells resemble NG2-glia [34]. Second, we observed faintly stained GPR17+ cells what appeared to be (pyramidal) neurons. Expression of GPR17 in neocortical neurons was further verified by immunofluorescence double labeling experiments using anti-NeuN antibodies (see Fig. 4). Beyond, cells what appeared to be spinal cord motoneurons strongly expressed GPR17 (see Fig. 8). Whether or not such observed neuronal staining pattern are specific remains to be clarified in future studies. In this context, a recent study should be mentioned demonstrating that GPR17 can drive the fate of neural precursor cells by instructing precursors towards the neuronal lineage [36]. That the applied antibody principally is valid was verified by our follow-up gene expression studies: both in situ hybridization and rt RT-PCR analyses verified Gpr17 expression induction in the demyelinated corpus callosum. It is tempting to speculate about the potential function of these stellate GPR17+ cells. The morphology is somewhat reminiscent of resting, ramified microglia cells [37]. With their highly branched processes and protrusions, microglial cells continuously screen the extracellular space in a seemingly random fashion and at a high turnover rate [38]. A similar function might fulfill GPR17+ OPC. With their highly developed network of cellular processes, GPR17+ OPC might survey their environment for a potential demyelinating insult to initiate OPC activation, proliferation, and finally remyelination. In fact, it has been suggested that GPR17 may act as a “sensor” that is activated upon brain injury and may play a key role in orchestrating local remodeling/repair responses [16]. In an elegant study, Vigano and colleagues were able to show, by using a GPR17-iCreER(T2) mouse line for fate mapping studies, that virtually all GPR17 expressing cells co-express the oligodendrocyte-specific transcription factor OLIG2. No GFP+ neurons, astrocytes, or microglia were detected [34]. Comparably, we observed that almost all GPR17+ cells are OLIG2+ and do not express GFAP. GPR17-expressing cells therefore most likely resemble adult OPCs. Due to their comparable densities in the white matter corpus callosum and gray matter cortex region, the potential for endogenous remyelination of both areas should be comparable.
In this context, it is interesting to note that the expression of GPR17 is induced in the white matter corpus callosum, but not in the gray matter cortex after experimental demyelination. Our data are in line with the findings from another cuprizone study showing accumulation of NG2+ cells in the corpus callosum but not in the cortex [39]. The same group demonstrated a lower remyelination potential of the cortex compared to the white matter corpus callosum [11].
But why should cortical remyelination be less effective or slower, respectively? Using the cuprizone model, it has recently been shown that the temporal dynamics of OPC differentiation varies significantly between white and gray matter regions. While OPCs rapidly repopulate the corpus callosum and mature into myelinating oligodendrocytes, OPC differentiation in the gray matter occurs much more slowly, resulting in a delay in remyelination relative to the corpus callosum [12]. Xing and colleagues were recently able to show that in cuprizone-challenged mice, substantial numbers of neural precursor cells migrate into the demyelinated corpus callosum and, there, contribute to oligodendrogenesis [8]. Delay of cortical remyelination might, therefore, simply be due to a failure of neural precursor cells to migrate the long distance from the subventricular zone into the cortex. To what extent GPR17+ cells differentially contribute to white versus gray matter remyelination is currently unknown. However, our observation of GPR17 induction in the white but not gray matter suggests an important role of GPR17 for delayed cortical remyelination in this model.
MS was long considered a disease of the white matter, but recent magnetic resonance imaging (MRI) and pathology studies have revealed extensive gray matter pathology in MS patients [40]. Since both white and gray mater demyelination significantly contribute to disease burden in MS, it is of vital importance to understand the factors that may hinder or aide successful remyelination in these regions. Obviously, mechanisms operant during white matter remyelination are not necessarily active during gray matter demyelination.
Another interesting outcome of this study is that while GPR17 induction is pronounced at week 5 (i.e., acute demyelination), numbers of GPR17+ cells did not significantly differ from control animals after a 12-week cuprizone intoxication period (i.e., chronic demyelination). Whether this is due to a loss of OPCs in chronic lesions or due to loss of GPR17 expression by OPC remains to be clarified in future studies. However, our finding that lower densities of GPR17+ cells in chronic lesions is paralleled by lower densities of PDGFRα+ OPCs suggests that OPCs are less abundant in chronic lesions, where remyelination is insufficient.
Of note, GPR17 might not simply be involved in terminal OPC differentiation but potentially regulates oligodendrocyte physiology and pathology in a broader context. Ou and colleagues [13], for example, were able to demonstrate that overexpression or activation of GPR17 in vitro resulted in increased LPC-induced oligodendrocyte apoptosis, whereas GPR17 inhibition reduced oligodendrocyte apoptosis, respectively. At the cellular scale, oligodendrocyte damage can principally spread “centrifugally” (i.e., from the oligodendrocyte cell body to myelin) or “centripetally” (i.e., from myelin to the soma) [41]. In the LPC model and in the inflammatory experimental autoimmune encephalomyelitis model, changes in oligodendrocyte morphology proceed from the myelin sheath to the cell body [41], whereas in the cuprizone model, oligodendrocyte damage spreads centrifugally. It would, thus, be interesting to see whether pharmacological manipulation of GPR17 ameliorates cuprizone-induced apoptosis and whether interference with the GPR17 signaling cascade modulates remyelination in the cuprizone model.
Naturally occurring GPR17 agonists are cysteinyl leukotrienes (CysLTs) such as LTD4 or LTC4 and uracil nucleotides such as UDP, UDP-glucose, and UDP-galactose [16]. It has been suggested that uracil nucleotides can be released from glia cells [42, 43]. Microglia and astrocyte activation are characteristic histopathological hallmarks of MS and its animal models [44,45,46,47]. In this context, one might speculate that microglia and astrocyte communicate the presence of brain damage via GPR17 activation to OPCs and by this cell–cell communication network initiate early regenerative processes such as remyelination.
In summary, we were able to demonstrate the expansion of the GPR17+ OPC pool in two different models of toxin-induced demyelination and that this expansion, at least in the cuprizone model, is restricted to the white matter corpus callosum. These data support the interest to evaluate if the pharmacological modulation of the GPR17 signaling cascade could accelerate the remyelination process as already suggested in GPR17 null mice.
References
Aboul-Enein F, Lassmann H (2005) Mitochondrial damage and histotoxic hypoxia: a pathway of tissue injury in inflammatory brain disease? Acta Neuropathol 109(1):49–55. https://doi.org/10.1007/s00401-004-0954-8
Smith KJ, Lassmann H (2002) The role of nitric oxide in multiple sclerosis. Lancet Neurol 1(4):232–241
Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM et al (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485(7399):517–521. https://doi.org/10.1038/nature11007
Edgar JM, McLaughlin M, Yool D, Zhang SC, Fowler JH, Montague P, Barrie JA, McCulloch MC et al (2004) Oligodendroglial modulation of fast axonal transport in a mouse model of hereditary spastic paraplegia. J Cell Biol 166(1):121–131. https://doi.org/10.1083/jcb.200312012
Uschkureit T, Sporkel O, Stracke J, Bussow H, Stoffel W (2000) Early onset of axonal degeneration in double (plp-/-mag-/-) and hypomyelinosis in triple (plp-/-mbp-/-mag-/-) mutant mice. J Neurosci 20(14):5225–5233
Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, Sorensen PS, Bruck W et al (2006) Remyelination is extensive in a subset of multiple sclerosis patients. Brain J Neurol 129(Pt 12):3165–3172. https://doi.org/10.1093/brain/awl217
Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, Mandrekar J, Bramow S et al (2015) Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol 78(5):710–721. https://doi.org/10.1002/ana.24497
Xing YL, Roth PT, Stratton JA, Chuang BH, Danne J, Ellis SL, Ng SW, Kilpatrick TJ et al (2014) Adult neural precursor cells from the subventricular zone contribute significantly to oligodendrocyte regeneration and remyelination. J Neurosci 34(42):14128–14146. https://doi.org/10.1523/jneurosci.3491-13.2014
Kuhlmann T, Miron V, Cui Q, Wegner C, Antel J, Bruck W (2008) Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain J Neurol 131(Pt 7):1749–1758. https://doi.org/10.1093/brain/awn096
Strijbis EMM, Kooi EJ, van der Valk P, Geurts JJG (2017) Cortical remyelination is heterogeneous in multiple sclerosis. J Neuropathol Exp Neurol 76(5):390–401. https://doi.org/10.1093/jnen/nlx023
Gudi V, Moharregh-Khiabani D, Skripuletz T, Koutsoudaki PN, Kotsiari A, Skuljec J, Trebst C, Stangel M (2009) Regional differences between grey and white matter in cuprizone induced demyelination. Brain Res 1283:127–138. https://doi.org/10.1016/j.brainres.2009.06.005
Baxi EG, DeBruin J, Jin J, Strasburger HJ, Smith MD, Orthmann-Murphy JL, Schott JT, Fairchild AN et al (2017) Lineage tracing reveals dynamic changes in oligodendrocyte precursor cells following cuprizone-induced demyelination. Glia 65(12):2087–2098. https://doi.org/10.1002/glia.23229
Ou Z, Sun Y, Lin L, You N, Liu X, Li H, Ma Y, Cao L et al (2016) Olig2-targeted G-protein-coupled receptor Gpr17 regulates oligodendrocyte survival in response to lysolecithin-induced demyelination. J Neurosci 36(41):10560–10573. https://doi.org/10.1523/jneurosci.0898-16.2016
Simon K, Hennen S, Merten N, Blattermann S, Gillard M, Kostenis E, Gomeza J (2016) The orphan G protein-coupled receptor GPR17 negatively regulates oligodendrocyte differentiation via Galphai/o and its downstream effector molecules. J Biol Chem 291(2):705–718. https://doi.org/10.1074/jbc.M115.683953
Coppi E, Maraula G, Fumagalli M, Failli P, Cellai L, Bonfanti E, Mazzoni L, Coppini R et al (2013) UDP-glucose enhances outward K(+) currents necessary for cell differentiation and stimulates cell migration by activating the GPR17 receptor in oligodendrocyte precursors. Glia 61(7):1155–1171. https://doi.org/10.1002/glia.22506
Lecca D, Trincavelli ML, Gelosa P, Sironi L, Ciana P, Fumagalli M, Villa G, Verderio C et al (2008) The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS One 3(10):e3579. https://doi.org/10.1371/journal.pone.0003579
Ciana P, Fumagalli M, Trincavelli ML, Verderio C, Rosa P, Lecca D, Ferrario S, Parravicini C et al (2006) The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J 25(19):4615–4627. https://doi.org/10.1038/sj.emboj.7601341
Ceruti S, Vigano F, Boda E, Ferrario S, Magni G, Boccazzi M, Rosa P, Buffo A et al (2011) Expression of the new P2Y-like receptor GPR17 during oligodendrocyte precursor cell maturation regulates sensitivity to ATP-induced death. Glia 59(3):363–378. https://doi.org/10.1002/glia.21107
Fumagalli M, Daniele S, Lecca D, Lee PR, Parravicini C, Fields RD, Rosa P, Antonucci F et al (2011) Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. J Biol Chem 286(12):10593–10604. https://doi.org/10.1074/jbc.M110.162867
Chen Y, Wu H, Wang S, Koito H, Li J, Ye F, Hoang J, Escobar SS et al (2009) The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat Neurosci 12(11):1398–1406. https://doi.org/10.1038/nn.2410
Hennen S, Wang H, Peters L, Merten N, Simon K, Spinrath A, Blattermann S, Akkari R et al (2013) Decoding signaling and function of the orphan G protein-coupled receptor GPR17 with a small-molecule agonist. Sci Signal 6(298):ra93. https://doi.org/10.1126/scisignal.2004350
Daniele S, Trincavelli ML, Fumagalli M, Zappelli E, Lecca D, Bonfanti E, Campiglia P, Abbracchio MP et al (2014) Does GRK-beta arrestin machinery work as a “switch on” for GPR17-mediated activation of intracellular signaling pathways? Cell Signal 26(6):1310–1325. https://doi.org/10.1016/j.cellsig.2014.02.016
Kipp M (2016) Remyelination strategies in multiple sclerosis: a critical reflection. Expert Rev Neurother 16(1):1–3. https://doi.org/10.1586/14737175.2016.1116387
Kipp M, Clarner T, Dang J, Copray S, Beyer C (2009) The cuprizone animal model: new insights into an old story. Acta Neuropathol 118(6):723–736. https://doi.org/10.1007/s00401-009-0591-3
Huang JK, Jarjour AA, Oumesmar BN, Kerninon C, Williams A, Krezel W, Kagechika H, Bauer J et al (2011) Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci 14(1):45–53. https://doi.org/10.1038/nn.2702
Slowik A, Schmidt T, Beyer C, Amor S, Clarner T, Kipp M (2015) The sphingosine 1-phosphate receptor agonist FTY720 is neuroprotective after cuprizone-induced CNS demyelination. Br J Pharmacol 172(1):80–92. https://doi.org/10.1111/bph.12938
Paxinos G, Franklin KBJ (2001) Mouse brain in stereotaxic coordinates. 2nd ed edn. Academic, San Diego, Calif. ; London,
Paxinos G, Watson C The rat brain in stereotaxic coordinates Elsevier Academic Press
Ruther BJ, Scheld M, Dreymueller D, Clarner T, Kress E, Brandenburg LO (2017) Combination of cuprizone and experimental autoimmune encephalomyelitis to study inflammatory brain lesion formation and progression. 65 (12):1900–1913. doi:https://doi.org/10.1002/glia.23202
Clarner T, Janssen K, Nellessen L, Stangel M, Skripuletz T, Krauspe B, Hess FM, Denecke B et al (2015) CXCL10 triggers early microglial activation in the cuprizone model. J Immunology (Baltimore, Md: 1950) 194(7):3400–3413. https://doi.org/10.4049/jimmunol.1401459
Skripuletz T, Lindner M, Kotsiari A, Garde N, Fokuhl J, Linsmeier F, Trebst C, Stangel M (2008) Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. Am J Pathol 172(4):1053–1061. https://doi.org/10.2353/ajpath.2008.070850
Hoflich KM, Beyer C, Clarner T, Schmitz C, Nyamoya S, Kipp M, Hochstrasser T (2016) Acute axonal damage in three different murine models of multiple sclerosis: a comparative approach. Brain Res 1650:125–133. https://doi.org/10.1016/j.brainres.2016.08.048
Sahel A, Ortiz FC, Kerninon C, Maldonado PP, Angulo MC, Nait-Oumesmar B (2015) Alteration of synaptic connectivity of oligodendrocyte precursor cells following demyelination. Front Cell Neurosci 9:77. https://doi.org/10.3389/fncel.2015.00077
Vigano F, Schneider S, Cimino M, Bonfanti E, Gelosa P, Sironi L, Abbracchio MP, Dimou L (2016) GPR17 expressing NG2-glia: oligodendrocyte progenitors serving as a reserve pool after injury. Glia 64(2):287–299. https://doi.org/10.1002/glia.22929
Boda E, Vigano F, Rosa P, Fumagalli M, Labat-Gest V, Tempia F, Abbracchio MP, Dimou L et al (2011) The GPR17 receptor in NG2 expressing cells: focus on in vivo cell maturation and participation in acute trauma and chronic damage. Glia 59(12):1958–1973. https://doi.org/10.1002/glia.21237
Boccazzi M, Lecca D, Marangon D, Guagnini F, Abbracchio MP, Ceruti S (2016) A new role for the P2Y-like GPR17 receptor in the modulation of multipotency of oligodendrocyte precursor cells in vitro. Purinergic Signal 12(4):661–672
Morrison H, Young K, Qureshi M, Rowe RK, Lifshitz J (2017) Quantitative microglia analyses reveal diverse morphologic responses in the rat cortex after diffuse brain injury. Sci Rep 7(1):13211. https://doi.org/10.1038/s41598-017-13581-z
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318. https://doi.org/10.1126/science.1110647
Koutsoudaki PN, Hildebrandt H, Gudi V, Skripuletz T, Skuljec J, Stangel M (2010) Remyelination after cuprizone induced demyelination is accelerated in mice deficient in the polysialic acid synthesizing enzyme St8siaIV. Neuroscience 171(1):235–244. https://doi.org/10.1016/j.neuroscience.2010.08.070
Rudick RA, Trapp BD (2009) Gray-matter injury in multiple sclerosis. N Engl J Med 361(15):1505–1506. https://doi.org/10.1056/NEJMcibr0905482
Romanelli E, Merkler D, Mezydlo A, Weil MT, Weber MS, Nikic I, Potz S, Meinl E et al (2016) Myelinosome formation represents an early stage of oligodendrocyte damage in multiple sclerosis and its animal model. Nat Commun 7:13275. https://doi.org/10.1038/ncomms13275
Lazarowski ER, Shea DA, Boucher RC, Harden TK (2003) Release of cellular UDP-glucose as a potential extracellular signaling molecule. Mol Pharmacol 63(5):1190–1197
Kreda SM, Seminario-Vidal L, Heusden C, Lazarowski ER (2008) Thrombin-promoted release of UDP-glucose from human astrocytoma cells. Br J Pharmacol 153(7):1528–1537. https://doi.org/10.1038/sj.bjp.0707692
Skripuletz T, Hackstette D, Bauer K, Gudi V, Pul R, Voss E, Berger K, Kipp M et al (2013) Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain J Neurol 136(Pt 1):147–167. https://doi.org/10.1093/brain/aws262
Grosse-Veldmann R, Becker B, Amor S, van der Valk P, Beyer C, Kipp M (2016) Lesion expansion in experimental demyelination animal models and multiple sclerosis lesions. Mol Neurobiol 53(7):4905–4917. https://doi.org/10.1007/s12035-015-9420-y
van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L, Gerritsen W, Kooi EJ et al (2012) Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflammation 9:156. https://doi.org/10.1186/1742-2094-9-156
Lassmann H, van Horssen J, Mahad D (2012) Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 8(11):647–656. https://doi.org/10.1038/nrneurol.2012.168
Acknowledgements
This study was supported by UCB BioPharma (Braine L’Alleud, Belgium), the Dr. Robert Pfleger Stiftung (M.K.), and the Deutsche Forschungsgemeinschaft (KI 1469/8-1). The technical support, H. Helten, P. Ibold, A. Baltruschat, B. Aschauer, JM. Frequin, and M. Caruso, are acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Main Points
1. Cortical remyelination is delayed in the cuprizone model.
2. GPR17 expression is induced in the white but not gray matter.
3. GPR17 expression is induced in acute, but not chronic lesions.
Electronic supplementary material
Supplementary figure 1
Results of anti-GPR17 antibody optimization. Representative pictures illustrate results of standard heat induced epitome retrieval (HIER) of brain tissues stained with anti-GRP17 antibodies. Tissue sections were either cooked in Tris/EDTA buffer (pH 9.0) or citrate (pH 6.0) buffer for 10 or 20 min, receptively. In parallel, one slide was not subjected to HIER. Arrowheads indicate GPR17+ cells in the cortex. (GIF 482 kb)
Rights and permissions
About this article

Cite this article
Nyamoya, S., Leopold, P., Becker, B. et al. G-Protein-Coupled Receptor Gpr17 Expression in Two Multiple Sclerosis Remyelination Models. Mol Neurobiol 56, 1109–1123 (2019). https://doi.org/10.1007/s12035-018-1146-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1146-1