
Abstract
Prolongation of the T2 relaxation time, an increase in T2-weighted signal intensity (T2-SI), and a decrease in the apparent diffusion coefficient (ADC) calculated from diffusion-weighted images (DWI) on magnetic resonance imaging (MRI) are conventional indicators of the vasogenic (interstitial) or cytotoxic (cellular) cerebral edema that develops after ischemic stroke. However, these parameters obtained on stroke imaging have not given us a precise threshold at which we can determine the viability or vulnerability of the tissue, allowing us to decide on an intervention that will help reversible tissue in the acute phase. Here, we introduce a new indicator—the essential diffusion coefficient or EDC, calculated from the T2-SI and ADC—that permits detection of irreversible brain damage after induction of experimental, focal cerebral ischemia. Our three-vessel occlusion (3-VO) method (Yang et al. Eur Neurol 71:4–18, 2014) was applied to investigate early changes on 7-T MRI. In the 3-VO model, which targets only a part of the cortex, animals seldom die at least within 24 h. The T2-SI and the ADC value were monitored, starting at 60 min after reperfusion, and every 30–60 min, for 10 h after the induction of focal ischemia. The region of interest (ROI) was set in each of the following: (1) the ischemic core (the dead zone); (2) the medial border area (the dying/dead mixed zone, including the ischemic penumbra); (3) the lateral border area (the surviving zone after the ischemic stress, where the rCBF is above the threshold for death); and (4) The intact area (outside the ischemic zone). The diagnosis was made by histological analysis performed 24 h after reperfusion. Significant increases in the T2-SI were observed, in ROI-1 at 1 h, in ROI-2 at 2.5 h, and in ROI-3 at 4 h post-reperfusion (1.10, 1.11, or 1.11; > 1.10, respectively, p < 0.001). Significant reductions in the ADC were also observed in ROI-1, ROI-2, and ROI-3, at 1 h post-reperfusion (0.55, 0.52, or 0.58; < 0.60, respectively, p < 0.001), indicating that both types of cerebral edema develop simultaneously in the acute phase. In the EDC analysis, from 5.0 h post-reperfusion, the value in the dying/dead zone (ROI-1 and ROI-2) was consistently reduced to < 50%, showing repeated, significant differences from the value in the surviving zone (ROI-3). A reduction in the EDC to below 50% indicated irreversible tissue damage, with transformation to cerebral infarction. We could detect a sign of cerebral infarction (initial necrosis-like irreversible lesion) as early as 5.25 h after the onset of ischemia. Although the biological time that depends on the body weight must be different between mice and humans, the earliest irreversible tissue damage or tissue destruction (to have achieved the risk of hemorrhagic transformation) that progressed after invisible or silent cell death in the ultra-acute phase, seems to occur at a similar time point.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The detection of an early, irreversible change in brain tissue after ischemic stress is extremely important in the acute phase, as with this information, we can intervene more safely to increase the perfusion pressure in the affected region before cerebral infarction occurs. However, recent advances in noninvasive stroke imaging have not provided a satisfactory threshold parameter that permits this distinction [1,2,3]. Among the multiple modalities utilized in the clinical setting, magnetic resonance imaging (MRI) measures changes in the condition of protons in the water molecules that generate cerebral edema, changes that are modified as the integrity of the ischemic lesion deteriorates. MRI is more sensitive than computed tomography (CT) and helps us to detect earlier changes after the insult of focal ischemia.
Formation of cerebral edema, an increase in the net uptake of cations and water into brain tissue, is initiated when neuronal cells are still alive as an active response to the ischemic stress, well before breakdown of the blood-brain barrier (BBB) occurs [4]. The T2-weighted image (T2-WI) obtained using a spin-echo pulse sequence relies on the transverse relaxation of the net magnetization vector, which demonstrates differences in regional T2-relaxation time. The accumulation of free water (extravasation) in the interstitial brain tissue, which is referred to as vasogenic edema, can be detected by an elevated T2-weighted signal intensity (T2-SI), which appears as a bright area on T2-WI.
Since areas with elevated T2-SI at 24 h after the onset of stroke match the lesions of cerebral infarction, and it is known that the anatomical size of the infarcted lesion usually does not increase between 24 and 72 h [5, 6], “elevated de-novo T2-SI at 24 h ischemia” has been used to diagnose the location and size of a newly completed cerebral infarction. However, the size of a lesion with increased T2-SI expands progressively over the initial 4 days after onset [7,8,9]. Therefore, the monitoring of T2-SI beyond 24 h has been considered to illustrate not a lesion with irreversible tissue damage, but the size of the area of vasogenic edema that expands beyond an infarcted lesion [10, 11].
In terms of the earliest development of vasogenic edema after the onset of an ischemic stroke, T2-SI monitoring cannot detect any abnormality earlier than 2 to 3 h after thread-induced focal ischemia [12, 13]. On the other hand, diffusion-weighted imaging (DWI), which exploits the random motion of water molecules corresponding to a narrowing of the interstitial space, a decrease in the fraction occupied by the extracellular space, cellular swelling, or blebbing, is more sensitive in the detection of early cellular or intra-cellular changes than is T2-SI [7, 14,15,16]. Elevated intensities on DWI after the occlusion of the middle cerebral artery (MCA) have been found to match the areas with cytotoxic (or cellular) edema, which can be induced by ischemia, hypoxia, or anoxia (energy failure in neural cells) in the brain [17]. Cytotoxic (or cellular) edema can also be detected by characteristic changes that include loss of ATP following the disturbance of energy metabolism, an imbalance in transmembrane ionic gradients [12, 18, 19], and alterations in the electric impedance [20]. Although limited to ex-vivo models, monitoring with iontophoresis (the iontophoretic electrode insertion method) can also detect an ionic abnormality that occurs during cytotoxic (or cellular) edema [21]. In this regard, an MRI scan can detect the deficit of free water perfusion [12, 13, 18, 22] (restriction in the diffusion capacity) of the interstitial free water non-invasively [12, 14, 15, 23], during the process of cellular swelling.
Although both types of cerebral edema elevate DWI signals, the individual pathophysiology was believed to be differentiated by calculating the apparent diffusion coefficient (ADC), which has a good sensitivity and specificity in the detection of cytotoxic edema, when the lesion was more than 24(12) hours and less than 10 days old, before the anatomical breakdown of the cell/membrane architecture [8]. The ADC value, determined by the extent of tissue cellularity and the presence of an intact cell membrane, represents the magnitude of water-molecule diffusion. Cytotoxic edema produces a shrinkage of extracellular spaces due to redistribution of extracellular water into more diffusion-restricted intra-cellular compartments, resulting in a decrease in the mean diffusion capacity of free water and reducing the ADC value [12, 24, 25]. However, alteration in the ADC value is not an indicator of irreversible tissue damage [3], or of pure cytotoxic edema in the acute phase of ischemic stroke.
Brain tissue with a perfusion pressure below the threshold to induce electrical silence on EEG according to the occurrence of neural depolarizations, but above the threshold to induce energy failure, is termed the ischemic penumbra [1, 2]. Neurons in this location can survive only when an effective intervention is introduced within a critical period. The neocortex, supplied by multiple feeding arteries accompanied by rich vascular anastomoses (collaterals), which comprise the vascular reserve (the region resistant against ischemic stresses), is a typical location of the ischemic penumbra. In contrast, the basal ganglia, supplied by perforating end arteries, easily encounter the condition of most severe ischemia, i.e., there is no perfusion phenomenon.
In this study, we used the three-vessel occlusion (3-VO) method, a well-established technique in our laboratory [26,27,28], to study the development of cerebral edema, in relation to the destiny of individual brain regions with different ischemic levels that develop around the ischemic core. The 3-VO method can reduce regional cerebral blood flow (rCBF) to below 30% in the neocortex, and removal of the clips produces adequate reperfusion, via luxury cortical collaterals, overall achieving consistent reproducibility and homogenous volume development in infarcted lesions [27]. Because of the relatively small “infarct variation coefficient (IVC)” value, 15%, in this model, there is no need to exclude animals (according to an outcome where there has been no infarcted lesion) after the induction of ischemia [26, 27]. However, the exclusion of animals is inevitable in the 1- or 2-VO models.
The animal models that mimic ischemic stroke are designed or expected to induce a critical, but still treatable, zone: the ischemic penumbra, between the upper, 30%, and lower thresholds, 10%, in the rCBF, and any neuroprotective intervention should be initiated within the limits of viability of the neurons in this area [1, 29]. In the model used worldwide at present, the intra-luminal thread insertion (ITI) model [30, 31], a thread (foreign body) is inserted into the artery with the intention of occluding the orifice of the MCA with or without the occlusion of the anterior cerebral artery (ACA) in a blind manner, but it is known that the procedure fails to induce ischemia in 13% of cases, and the mortality rate is 20% in the initial 24 h (overall, 33% animal exclusion only in the acute phase), when SD rats were used [32]. When C57BL/6 mice were used, the mortality rate was 67% within 24 h, and the mean survival time was only 14 h after 60-min ITI-occlusion [33]. It has been also reported that 60-min ITI-occlusion exhibited more than 80% mortality within 24 h in C57BL6 mice, probably because of the developmental characteristics on the posterior communicating artery, indicating the difficulty in the induction of cerebral infarction limited in the MCA territory, and evaluating various interventions [34].
In the ITI model, regarding the mechanism of induction of ischemia, treatment with the potent anti-coagulant heparin improved reduced rCBF during and after thread insertion and also reduced the size of the infarcted lesion. In other words [35], blood clot formation triggered around the foreign body (intra-arterial thread with a negatively charged surface activates the coagulation system) is involved in the reduction of rCBF and the induction of ischemia in the ITI model. In contrast, heparin did not reduce the lesion size in our 3-VO model with a direct clip occlusion of the part of MCA-1 crossing over the lateral edge of the olfactory tract, not affecting perforating arteries arising from the proximal MCA [36]. The ineffectiveness of heparin in reducing infarcted lesion volumes in the 3-VO model indicated that the method does not involve irregular and uncontrollable clot formation that causes heterogeneity in the depth of ischemia and the magnitude of reperfusion levels, both of which are accompanied by large IVCs. Importantly, a small craniectomy is not invasive but the prolonged insersion/removal of a thread (foreign body) into/from the artery, in the aspect of the mortality related to the development of thrombosis or multiple embolisms [33, 34]. Because of the minimum risk of animal death after the induction of focal ischemia, the 3-VO model has been used in search of a neuroprotectant [36,37,38,39,40].
The purpose of this study was to find a threshold on MRI that might reveal the irreversible damage or destruction of tissue, allowing us to introduce an acute intervention, salvage the living tissue at risk, and improve the condition of patients with acute ischemic stroke. We monitored the appearance of cerebral edema on MRI, T2-SI (the parameter for vasogenic edema), and ADC (the parameter for “cytotoxic edema”), in areas covering different levels of ischemia, including the surviving zone surrounding the infarcting (already dead and destructing) region, the dying/dead border zone (the ischemic penumbra), and the dead zone (the ischemic core), based on the histological outcome assessed at 24 h after reperfusion.
Method
The Animal
For the induction and development of cerebral infarction, we used 12-week-old, male C57BL/6J mouse (CLEA-Japan, Tokyo, Japan), weighing 26 g. The protocol was designed in accordance with the guidelines and regulations issued by the Animal Care and Use Committee of the National Cerebral and Cardiovascular Center in Japan (Approval ID: 17022). Every effort is made to minimize the number of animals used and their suffering. All mice are allowed food and water ad libitum before and after induction of ischemia.
Induction of Temporary Focal Ischemia
Temporary focal ischemia was induced as described in detail elsewhere, where multiple photographic images and a downloadable video image of the surgical procedure are included [26]. Using micro-surgical techniques with many improvements in the model evolved over multiple generations, we were able to avoid any blood loss during the operation, acute death of the animal, and the “no-infarcted lesion” phenomenon.
Briefly, under anesthesia with halothane, the left common carotid artery (CCA) was occluded using a clip proximal to the carotid bifurcation, which can reduce vascular reserve in the territory of the left external carotid artery and minimize blood loss in the following operation that involves manipulation of the left side of the head. Through a linear, horizontal incision between the outer corner of the eye and the ear, the left temporal muscle was exposed. The fascia of the muscle was cut along the ventral edge of the zygomatico-orbital branch of the facial nerve to avoid postoperative peripheral nerve palsy. The muscle bundles under the fascia were cut horizontally with operative viewing, and the surface of the zygomatic bone was exposed. The capsule of the temporomandibular joint and the temporomandibular articulation of the sphenomandibular ligament (seen under the right edge of the zygomatic arch) was cut/opened. After removing the zygomatic arch, the left mandible was ventrally retracted, which opened the left mandibular joint, and exposed the left temporal cranial bone. After opening a one-millimeter diameter burr hole at the location of the nerve impression generated by the exit zone of the trigeminal nerve, located rostral to the foramen ovale (at the bottom of the operative view), the MCA trunk was cauterized/occluded permanently using a bipolar-coagulator, at the exact location of the lateral edge of the olfactory tract. Clip occlusion of the right CCA, proximal to the carotid bifurcation, completed the 3-VO procedure. Fifteen minutes after the induction of focal ischemia, the CCA clips in the neck were removed to allow reperfusion in the MCA territory through the rich collaterals.
The QR code, used to access the whole operative procedure described above, contained in a 10-min movie (published in [26]).
MRI Scan
MRI scans were performed using a 7-T horizontal bore imaging system (BioSpec 70/30 USR; Bruker BioSpin, Ettlingen, Germany), equipped with a gradient system capable of a maximum gradient amplitude of 669 mT/m and a slew rate of 7989 T/m/s. Radiofrequency transmission was performed using a volume coil with an 86-mm inner diameter. Signals were detected using a four-channel receive-only phased-array surface coil. During the scan, the mouse was anesthetized using isoflurane (3.0% for induction and 1.5–2.0% for maintenance) in room air at 1.0 L/min mixed with O2 at 0.5 L/min. The animal was placed in a prone position, and the head was fixed with a custom-designed head holder inside the magnet. Body temperature was monitored by a rectal thermometer and maintained using a warm water bed and warm air. The heart rate and the respiratory rate were continuously monitored during the repeated scans.
The T2-WIs were acquired using rapid acquisition with relaxation enhancement (RARE) sequences with the following parameters: RARE factor, 8; repetition time (TR)/echo time (TE), 2500/35.14 ms; number of averages, 2; matrix size, 200 × 200; field of view (FOV), 2.0 × 2.0 cm2; in-plane spatial resolution, 100 × 100 μm2; slice thickness, 1.0 mm; gapless; number of slices, 20; and scan time, 2 min 5 s.
The DWI images were acquired using spin-echo echo planar imaging (EPI) sequences with the following parameters: two shot; TR/TE, 10000/28.38 ms; number of averages, 1; matrix size, 100 × 100; FOV, 2.0 × 2.0 cm2; in-plane spatial resolution, 200 × 200 μm2; slice thickness, 1.0 mm; gapless; number of slices, 20; b value, 1000 s/mm2; 30 directions; and scan time, 11 min 40 s. In the DWI, the generalized auto-calibrating partially parallel acquisition (GRAPPA) technique was used with an acceleration factor of 2. The ADC values were calculated on a pixel-by-pixel basis using ParaVison 5.1 (Bruker BioSpin) and then displayed as a parametric map that reflects the degree of diffusion of water molecules through different tissues. Using a dedicated workstation, ADC maps were recorded so that we could select regions of interest (ROIs) on the ADC-map [10, 11].
The MRI scan was started 60 min after the onset of reperfusion, and was repeated every 30 or 60 min for a total of 9 h. Data were analyzed using ImageJ (a Java image processing program inspired by the NIH, USA).
Analysis of the T2-Weighted Signal Intensity and the ADC Value on the MRI Images
The ROIs were set with the following definitions:
-
ROI-1. The ischemic core; the dead and destructing zone, selected from the histological outcome; completely dead tissue at 24 h post-reperfusion.
-
ROI-2. The medial border zone including the ischemic penumbra; the dying/dead intermediate zone, selected from the histological outcome; the mixture of surviving and dead tissue at 24 h post-reperfusion.
-
ROI-3. The lateral border zone surrounding the infarcting (destructing) region; a surviving zone selected from the outcome; viable, lateral to the ischemic penumbra.
-
ROI-4. The intact zone without direct ischemic stress, at least during the 3-VO.
The measurement of T2-SI and ADC value was performed at five locations set in each ROI (Fig. 1). Each T2-SI or each ADC value was normalized by the corresponding contralateral area (non-ischemic location in the right hemisphere), which was expressed as the ratio of ipsilateral to contralateral values.

The location of each ROI. Left: The result of a TTC stain on a brain section. Right: T2-weighted image of the section selected on the left. Four ROIs were set in the left cortex (the reference was set in the corresponding right cortex) as shown on the left. ROI-1, ischemic core, where cells are dead; the dead zone. ROI-2, medial border zone including the ischemic penumbra, where some cells are dying or dead; the mixed zone of dying/dead. ROI-3, lateral border zone excluding the ischemic penumbra; the surviving zone. ROI-4, intact zone, the area without ischemic stress; the non-ischemic zone
Identification of Infarct Lesions and Evaluation of the Tissue Damage
Under deep anesthesia and intra-arterial perfusion using phosphate-buffered saline (PBS) with heparin at 24 h after the induction of ischemia/reperfusion, the blood component (including red cells) in the cerebral vasculature was completely removed. The forebrain was cut coronally from the frontal tip into 1-mm thick slices, and each slice was immersed in a 2% solution of 2,3,5-triphenyltetrazolium chloride (TTC), before measurement of the size of the infarcted lesion [41].
The slice with the largest lesion was selected to analyze the relation between the parameters on MRI and the outcome of the tissue, in which the infarcted zone (necrotic tissue without living cells: white, non-stained area), non-infarcted zone (intact tissue, staining red), or the border zone (with a mixture of living and necrotic cells, and a pinkish stain), was defined.
On the TTC stain at 24 h, the density was analyzed using ImageJ, at five locations (the same with MRI analysis) in each ROI. The density, normalized by the corresponding contralateral area (non-ischemic location in the right hemisphere), was expressed as the ratio of ipsilateral to contralateral values (as the asymmetry or the final survivability index).
The Essential Diffusion Coefficient (EDC) Calculated from the ADC Value and the T2-Weighted Signal Intensity
The EDC was calculated by subtracting the increment in T2-SI ratio (> 1.0) from the ADC ratio, aiming to remove the effect of the simultaneous development of vasogenic edema, counteracting and diluting the vasogenic edema-derived elevation in the ADC.
Statistical Analysis
All data are presented as the means ± SD. Several general linear model procedures including multivariate repeated measures and one-way or two-way ANOVA were applied to compare the variance within the different parameters at the same time point. The difference was considered significant at a threshold of p < 0.05.
Result
The Vital Signs of the Mouse During Surgery or MRI Scans
The respiratory rate, heart rate retrieved from the electrocardiogram (ECG), and the core temperature of the body (deep rectal temperature) were regulated within the physiological range during the 3-VO surgery for 30 min and the subsequent 10 h MRI scans (data not shown).
Detection of Infarcted Lesion and the Cerebral Edema Index
The infarcted lesion volume calculated from the coronal slices, corrected by an edema index of 1.04 measured at 24 h post-reperfusion, was 24.1 mm3. The size was within the normal range after 3-VO performed without multiple MRI scans [26]. The edema index was calculated as the left hemispheric volume divided by the right hemispheric volume, as an indicator of cerebral edema developed in the left hemisphere, which was also in the normal range after the 3-VO [26], confirming that the effect of multiple MRI scans could be disregarded in development and extension of the ischemic lesion.
The TTC Stain Index
Individual densities of the TTC stain at different ROIs are shown in Fig. 2a. The ischemic core (ROI-1), the ischemic penumbra (ROI-2), and the survived (lateral border) zone (ROI-3) showed significant reductions compared to the corresponding contralateral area (control). The reduction levels in ROI-2 was significantly stronger than ROI-3, but significantly weaker than ROI-1, indicating that the death ratio (the final survivability index) of the tissue in each ROI related to ischemia was confirmed to represent the individual significance.

a The densities measured in each ROI after the TTC stain at 24 h post-reperfusion. The density in each ROI was significantly different, indicating that the individual ROIs possessed a quantitatively different survivability of the tissue with a different destiny. Alterations in the T2-SI ratio (T2-SI) after focal ischemia/reperfusion. b The T2-SI increased rapidly after induction of ischemia/reperfusion in every ischemic zone (ROI-1, ROI-2, ROI-3). An increase in the T2-SI in the surviving zone, interpreted as benign vasogenic edema, achieved a significant increase, at 1, 1.5, 2, and 3.5 h and thereafter, compared to the contralateral control. The T2-SI in the medial border zone (including the ischemic penumbra) achieved a significant increase at 2.5 h and thereafter, while the T2-SI in the ischemic core, interpreted as malignant vasogenic edema, achieved a significant increase at 4 h post-reperfusion. c Comparisons between the zone with irreversible tissue damage (including cellular death at 24 h post-reperfusion) and the zone without irreversible tissue damage (no death at 24 h post-reperfusion). There was no significant difference in the T2-SI, except at 3.5 h post-reperfusion, when vasogenic edema was more severe in the surviving zone. p < 0.001 vs. contralateral control (one-way ANOVA)
The T2-SI Ratio
Alterations in the T2-SI ratio after ischemia/reperfusion during the observation period are shown in Fig. 2b. At 1–2 h post-reperfusion, the T2-SI achieved significant differences in the surviving (lateral border) zone alone, with no significant differences in the dying (penumbra or core) zone; increasing from 1.10 to 1.67, compared to the corresponding contralateral area (control). From 2.5 to 3.0 h post-reperfusion, the significant increase in the T2-SI disappeared transiently, but again appeared at 3.5 h post-reperfusion and thereafter. In the initial 2-h reperfusion period, the T2-SI in the surviving zone alone achieved a significant difference compared to the corresponding contralateral control.
In the dying/dead mixed (medial border) zone, the T2-SI significantly and temporarily increased from 2.5 to 3.0 h post-reperfusion; with values of 1.11 and 1.16 respectively, compared to the contralateral control. In the dead (ischemic core) zone, the T2-SI was significantly increased at 4 h post-reperfusion, with a value of 1.11 compared to the control, which lasted throughout the following observation period.
The T2-SI in the non-ischemic intact zone gradually increased, achieving a significant difference of 1.11 at 9 h post-reperfusion, compared to the control.
In Fig. 2c, the differences in the T2-SI are compared between the surviving zone, lateral border area, and the dying/dead zone, the mean value of ischemic core and penumbra. During the initial 2-h reperfusion period, the increase in the T2-SI was relatively greater in the surviving zone than in the dying/dead zone, but the tendency was reversed at the end of observation (during 8–10 h post-reperfusion). At every time point, there were no significant differences between the surviving and dying/dead zones with respect to T2-SI after reperfusion, excepting at 3.5 h when cytotoxic edema entered the secondary increase phase (as shown in the following EDC analysis).
The ADC Ratio
Alterations in the ADC ratio during the observation period are shown in Fig. 3a. The ADC decreased both in the dying/dead and surviving zone, at every time point, with at least two peak reductions at 1 and 5 h post-reperfusion. The pattern of alteration in the dying/dead and surviving zones was similar during the observation period, during which a more marked reduction was seen in the dying/dead zone at 2 h post-reperfusion and thereafter.

Alterations in ADC values after focal ischemia/reperfusion. a The ADC values were significantly reduced in every zone (ROI-1, ROI-2, ROI-3) in the ultra-acute phase and 1 h post-reperfusion; this lasted throughout the observation period. The reduction was severe at 1 and 5 h post-reperfusion, but diminished at 3- and 9 h post-reperfusion. b Comparisons between the zone with irreversible tissue damage (including cellular death at 24 h post-reperfusion) and the zone without irreversible tissue damage (no death at 24 h post-reperfusion). There are significant differences between the two zones, from 2.5 to 8 h post-reperfusion, when cytotoxic edema was more severe in the dying zone. At the end of the observation period, the significant difference disappeared. p < 0.001 vs. contralateral control (one-way ANOVA)
The ADCs in the intact area were significantly decreased, 0.87 at 1.5 h, 0.93 at 4 h, and 0.91 at 7 h post-reperfusion, respectively, compared to the control, demonstrating that cytotoxic (cellular) edema may spread to the non-ischemic intact area intermittently.
In Fig. 3b, the differences in the ADC were compared between the surviving and dying/dead zone. The reduction in the ADC was greater in the dying/dead zone for temporary periods, at 2.5, 3.5, 4, 5, 6, 7, and 8 h post-reperfusion. The reduction after reperfusion returned to a moderate level; > 70% of the control, in the later reperfusion phase during the observation.
The EDC Calculated from the ADC Value and the T2-Weighted Signal Intensity
The alterations in the EDC ratio during the observation period are shown in Fig. 4a. The EDC decreased both in the dying/dead and surviving zone, compared to the corresponding control, at almost every observation time, with at least two peak reductions at 1 and 5 h.

Alterations in EDC ratios after focal ischemia/reperfusion. a The EDC in the dying/dead zone (the ischemic core and the medial border zone) decreased to less than 50% at 1 h, or 5 h post-reperfusion. The reduction was severe at 1, 5, and 10 h post-reperfusion, but diminished at 3 h post-reperfusion. b Comparisons between the different outcomes (cellular death or survival at 24 h post-reperfusion). There were significant differences between the zones, at multiple time points, when cytotoxic edema was more severe in the dying/dead zone. The significant difference persisted until the end of the observation period. p < 0.001 vs. contralateral control (one-way ANOVA)
At 1 h post-reperfusion, a severe reduction of under 50% was observed only in the surviving zone. In contrast, at 5 h post-reperfusion and thereafter, a similar severe reduction below 50% was observed only in the dying/dead zone. During the observation period, the reduction levels in the EDC were stronger in the dying/dead zone, apart from during the ultra-acute phase between 1.0 and 1.5 h post-reperfusion.
In Fig. 4b, the EDC ratios are compared in the surviving and dying/dead zones. The reduction in the EDC was significantly and consistently greater in the dying/dead zone, at 2.5, 5, 6, 7, 9, and 10 h post-reperfusion. The rate of reduction after reperfusion did not return to a moderate level of more than 70% of the control in the EDC analysis, which was in contrast to the gradually diminishing (improving) pattern in the ADC analysis.
Discussion
The T2-SI extracted from T2-WI has been recognized as a marker for an infarcted lesion or vasogenic edema [42] whether reversible or irreversible; the ADC calculated from DWI is a marker for an infarcted lesion or cytotoxic edema [18, 24], which may also be either reversible or irreversible. DWI is used worldwide in a clinical setting for the detection of the earliest ischemic changes in the acute phase of the patients undergoing ischemic stroke. Importantly, however, DWI is always affected by the development of vasogenic edema as well as cytotoxic edema—the phenomenon is termed “T2 shine through” (DWI increases with the development of vasogenic edema, not necessarily from the development of cytotoxic edema). That is, increased DWI detected in the acute phase does not always represent increased water inside the cells, but represents increased interstitial water, or the presence of extravasation of water.
On the DWI/ADC analysis, the development of cytotoxic edema causes an increase in DWI as well as a decline in the ADC value, while the development of vasogenic edema (increased extracellular water that prolongs T2 relaxation time and enhances T2-SI) causes an elevation (normalization or masking) of the reduced ADC. Because of the presence of this controversial phenomenon, “the apparent degree” of cytotoxic edema estimated from reduced ADC values is less than “the essential degree” that would be present if there was simultaneous development of vasogenic edema. In other words, the detection of a significant reduction in the ADC value precisely indicates the presence of an excessive amount of cytotoxic edema that surpasses the vasogenic edema.
In the paradigm of focal ischemia without induced reperfusion, Minematsu et al., using an ITI model with 2.0-T MRI in rats [43], reported that T2-SI did not alter after 30 min of ischemia, but significantly increased after 3 h of ischemia. Later, Chen et al., using a photothrombotic occlusion technique for the proximal MCA with 1.5-T MRI in rats, reported a significant increase in T2-SI at 3 h of ischemia, which reached a maximum at 12 h of ischemia [44]. In their study, the ADC value in the same region showed a consistent decrease during the first 12 h, but not cytotoxic edema, appeared significantly in the acute phase (< 24 h) after permanent focal ischemia.
In the paradigm of focal ischemia with induced reperfusion, Pham et al., using the ITI model with 17.6 (ultra-high)-T MRI in mice, reported T2-SI at “the ischemic region” did not alter at 2 h post-reperfusion after 1 h ischemia, but increased at 24 h post-reperfusion. In their study, the ADC values at the same region decreased significantly with twin peak reductions, observed at 2 and 24 h post-reperfusion [45, 46], indicating that cytotoxic edema, but not vasogenic edema, appeared significantly in the acute phase (< 24 h) after temporary focal ischemia.
In the results described above that used the ITI model, the T2-SI analysis detected the earliest development of vasogenic edema with permanent ischemia at 3 h, while the ADC analysis detected the earliest development of cytotoxic edema, at 2 h post-reperfusion after 1 h ischemia. In other words, neither type of edema appeared earlier than 3 h after the induction of ischemia.
Accordingly, it has been believed that the appearance of cytotoxic edema (primarily appearing after reperfusion) and vasogenic edema (primarily appearing after permanent ischemia) do not overlap, at least in the acute phase of ischemic stroke, the critical time when a medical intervention aimed at increasing regional reperfusion pressure may rescue the brain at risk. It might have been speculated that early development of cytotoxic edema suppresses interstitial spaces mechanically and prevents the development of vasogenic edema, at least in the acute phase. The diluting/erasing effect of the vasogenic edema on the appearance of cytotoxic edema in the ADC value has thus been ignored in the research field as well as in the hospital (at diagnosis).
Since the reperfusion phenomenon, if defined as an early or delayed recovery in the absolute rCBF from critical to non-critical level, is not limited in the paradigm of temporary focal ischemia, but observed in permanent focal ischemia [47], induced reperfusion is considered to enhance the development of cytotoxic edema in the ischemic brain. In the ITI model with reperfusion, however, the intra-arterial thread (foreign body) can induce blood clot formation, and clot-derived cerebral embolization can interfere with reperfusion, so if there is any significant reduction in the ADC value (cellular edema) in the ultra-acute phase, this might be diminished by the clot [48]. In this context, induced luxury reperfusion in the 3-VO model [27], without the clot formation that should prevent reperfusion levels at least to some extent, can act as an enhancer of morphological and pathological changes after ischemia, including the development of reversible (regulatory) or irreversible (malignant/non-regulatory) cellular edema, in addition to rescue the brain at risk effectively.
In studies that tried to find the threshold in some MRI parameter that can distinguish the destiny of brain tissue in various regions—reversible or irreversible—Hoehn-Berlage et al., used a 4.7-T MRI system and a permanent focal ischemia (MCA occlusion) model in rats. They reported that a reduction in ADC below 80% of the contralateral control value at 7 h ischemia was irreversible (destined to be infarcted) [49]. On the other hand, Sakoh et al., using their method to occlude the proximal MCA for 2 h in pigs, demonstrated that ADC values decreased at 1 h post-reperfusion, i.e., 3 h after the induction of focal ischemia, a finding which continued for at least another 5 h (till the end of observation), with parallel reductions in the cerebral metabolic rate of oxygen (CMRO2) [50]. They concluded that lesions with an ADC value more than 80% of the contralateral control were reversible, while < 75% signaled irreversibility, resembling our results obtained in the ADC analysis.
In a multiple embolism (i.e., permanent multiple focal ischemia) model in rats [51], in the zone with the most severe focal ischemia, the ADC value declined within the first 5–10 min after stroke onset in the range to below 70%, lasting for the entire observation period of 2.5 h. Kim et al. [32], using the ITI technique and 3.0-T MRI in rats, reported that a reduction of the ADC to above 70% appeared after 30 min of ischemia but disappeared after the following 30 min of reperfusion, indicating reversibility in the range above 70%. Taken together, the threshold ADC value that distinguishes tissue reversibility from irreversibility is relatively high in the literature, ranging between 70 and 80%.
In the present study, it was demonstrated for the first time that cellular and vasogenic edema can appear together in the acute phase. In the EDC analysis, in which the counteracting/diluting effect of vasogenic edema on cytotoxic edema (appearing as an erasing artifact in the evaluation of cytotoxic edema, beyond the phenomenon, T2 shine through) was abolished by subtracting the T2-SI, the increased part than 1.0, from the ADC ratio, the cellular edema in the surviving zone showed the first peak reduction, < 50%, at 1 h post-reperfusion, and then the EDC increased to > 70% with loss of a significant difference compared to the contralateral control at 3 h post-reperfusion. The EDC ratio in the dying/dead zone; the average of the ischemic core and the medial border zone, showed a secondary peak reduction down to less than 50% at 5 h post-reperfusion, and the significant reduction persisted till the end of the observation after then.
In the comparisons between the EDC ratios in the surviving (ROI-3) and the dying/dead zone (ROI-1 and ROI-2 combined) (Fig. 4b), there were significant differences between the two zones at 5 h post-reperfusion and thereafter, indicating that an EDC reduction below 50% after 5 h post-reperfusion, but not around 1 h post-reperfusion, is an early indicator of regional death. In contrast, there was no significant (and persisting) difference between the surviving and the dying/dead zone in the T2-SI analysis, indicating that the T2-SI in the acute phase, at least until 10 h, cannot be an indicator that distinguishes outcomes. In humans, the acute intervention to trigger thrombolisis and induce subsequent reperfusion has been allowed within 4.5 h after the onset [52], indicating the earliest destruction of arterial walls to cause hemorrhagic transformation could occur soon after the time limit. Although "the biological time (the time in homeothermic animals during life)", known to depend on the body weight, must be different between mice and humans [53, 54], the earliest irreversible tissue damage or tissue destruction, i.e. the initial necrosis (to have achieved the risk of hemorrhagic transformation) that progressed after invisible or silent cell death in the ultra-acute phase, seems to occur at a similar time point.
In terms of the destruction of the blood-brain barrier (BBB) that triggers the development of cerebral edema, a biphasic disruption-phenomenon of BBB had been reported after 3-VO, measured by the chronological alterations in the densities of extravasated compliment factor; C3, in rats [55]. There was a gradual and mild increase in the density (indicating no significant disruption of the BBB) in the acute phase after the induction of permanent focal ischemia, but after the induction of temporary focal ischemia, a robust increase appeared at 30 min post-reperfusion (indicating an immediate and significant disruption of the BBB), which was followed by the secondary increase at 3 h post-reperfusion.
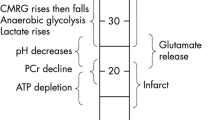
The dual-peak reductions (< 50%) in the EDC ratio currently observed, along with the previous finding on the biphasic disruption-phenomenon of BBB post-reperfusion, indicate the biphasic development of cellular edema post-reperfusion. We postulate that the EDC range, < 50%, with the time factor < 1.5 h, reflects an increased risk of cell death (i.e., the presence of a risk on the tissue destiny), and the EDC range, < 50%, with the time factor > 1.5 h, reflects the histological transformation to cerebral infarction (i.e., the early sign of tissue necrosis). As the alterations in the ADC are reportedly delayed or not significant in the paradigm of the acute permanent focal ischemia, the time factor, > 1.5 h post-reperfusion, or even > 1.5 h post-ischemia without the consideration of (or the information on) reperfusion, is essential in making an EDC-based precise (errorless) diagnosis on the tissue integrity (Fig. 5).

Upper panels: alterations in T2-WI. Lower panels: alterations in ADC values. Right panels: the TTC stain at 24 h post-reperfusion with ROIs; 1 (ischemic core), 2 (medial border zone), and 3 (lateral border zone)
In terms of the physiological mechanism of cerebral edema, spreading depression (SD) temporarily switches the fundamental barrier function of cell membranes off, releasing intra-cellular potassium, and exchanging it with extracellular calcium. In the generation of cellular swelling (cytotoxic) [56, 57], SD suppresses energy metabolism and causes oxygen supply-demand mismatch. Although SD that occurs under normal conditions is safe for brain tissue, SD or clusters of SD occurring in the ischemic penumbra sometimes induce severe regional hypo-perfusion [58], which results in death of the tissue in that area. SD that spreads beyond the territory of ischemia is termed “peri-infarct depolarization” (PID) [59, 60]. SD shares influx of water into neurons driven by the ionic changes across the cellular membrane, which has been considered the principal mechanism of the cytotoxic edema [61]. The cytotoxic edema detected in the surviving zone can be generated not only by ischemic stress but also by ischemia-induced PID. Interestingly, SD or PID can spread into the subcortical tissues [62, 63], generating a transient shuttling of SD between the tissues at different depths [63], which indicates that cerebral edema can expand to including deep brain regions beyond the ischemic lesion.
As regards the molecular mechanism of cerebral edema, an activated Na+/K+ pump (stimulated Na+/K+-ATPase) causes excessive Na+ influx and vasogenic edema, before breakdown of the blood-brain barrier (BBB) [64]. A membrane protein distributed predominantly in the luminal membrane of the BBB, the electroneutral Na+-K+-Cl− cotransporter (NKCC) [65], may participate in the development of cytotoxic edema [66]. Unregulated opening of nonselective cation (NCCa-ATP) channels after depletion of ATP following ischemia can trigger cellular depolarization and cytotoxic edema [67]. Recently, upregulation of the water-transporting protein in the plasma membrane, aquaporin (AQP), which contributes to the water homeostasis in the body, has been postulated in the (active) regulation of cerebral edema, especially around peri-infarct border zones [68, 69].
As for the commonality of the present result among various animals/neurons, the number of neurons targeted is estimated as 4.4 × 105 based on neuronal counting in the cortex [70], the speed of 2–5 mm/min at which SD that rapidly changes intra-cellular ionic/water condition spreads in the brain, i.e., the induction threshold for SD induced by various triggers, and the threshold for death or survival of individual neurons during ischemia are consistent among neurons in various animals [1, 2, 71,72,73]. When the relationship between the level of ischemia and the response of neurons was focused, a reduction in rCBF below 15 to 18 ml/100 g/min causes immediate electro-physiologic dysfunction of neurons (expressed as electrical silence or SD), as observed by flattening of the EEG or abolition of somatosensory evoked potentials in different animals including humans [1, 2]. Irrespective of species or regional differences, a reduction below 15 ml/100 g/min, or < 30% rCBF of pre-ischemic control, is necessary to generate an ischemic penumbra and ischemic depolarization, while more severe ischemia, < 6 ml/100 g/min, or < 10% rCBF, causes immediate depletion of ATP and immediate neuronal death [1, 29, 74]. Thus, the response and vulnerability of neurons, at least to an ischemic stress, does not differ in individual neurons or animals, indicating that an EDC threshold of 50%, as shown in the present study, has the potential to distinguish reversible and irreversible (necrotic or infarcted) tissue damage of brain tissue in the acute phase of human ischemic stroke.
Summary of the Results
-
Vasogenic edema (the significant level in the current analysis; T2-SI > 110%) appeared at 1 h post-reperfusion in the surviving zone (Fig. 2a).
-
Vasogenic edema (T2-SI > 110%) appeared at 2.5 h post-reperfusion in the gradually dying zone (Fig. 2a).
-
Vasogenic edema (T2-SI > 110%) appeared at 4 h post-reperfusion in the rapidly dying zone (Fig. 2a).
-
Vasogenic edema (T2-SI > 110%) appeared at 9 h post-reperfusion in the intact zone (Fig. 2a).
-
Vasogenic edema in the dying zone was not significantly different from the surviving zone, throughout the observation period (Fig. 2b).
-
Cellular edema demonstrated by ADC (the severest level in the current observation, < 60%) appeared at 1 h post-reperfusion in every ischemic (surviving or dying) zone or at 5 h post-reperfusion in the ischemic core (Fig. 3a).
-
Cellular edema demonstrated by ADC was significantly more severe in the dying zone compared to the surviving zone, at 3 h, or from 3.5 to 8 h post-reperfusion (Fig. 3b).
-
Cellular edema demonstrated by ADC (< 60%) was observed at 1 h post-reperfusion in the surviving or dying zone and at 5 h post-reperfusion in the dying zone (Fig. 3b).
-
Cellular edema demonstrated by EDC (the severest level in the ADC analysis, < 60%) appeared from 1 to 3 h post-reperfusion in every ischemic (surviving or dying) zone and at 5 h post-reperfusion and thereafter in the dying zone (Fig. 4a).
-
Cellular edema demonstrated by EDC was significantly more severe in the dying zone compared to the surviving zone, at 2.5 and 5 h post-reperfusion, and thereafter (Fig. 4b).
-
A reduction in EDC below 50% was suggested to be an indicator of irreversible tissue damage (Fig. 4b), except in the ultra-acute phase < 1.5 h post-reperfusion.
Conclusion
Using our original mouse 3-VO model, it was demonstrated for the first time that vasogenic and cellular edema can appear simultaneously in the acute phase after focal ischemia. The MRI parameter, EDC, newly developed in the present study to detect the essence of cellular edema, revealed the threshold for distinguishing reversible and irreversible (at least necrotic or infarcted) damage of tissue. Persistent and severe (< 50%) reduction in EDC, detected 5.25 h after the onset of ischemic stroke, was indicated to be an earliest appearance on MRI of irreversible tissue damage or tissue destruction; cerebral infarction.
In the comparisons between the EDC ratios in the surviving and the dying/dead zone (Fig. 4b), there were significant differences at 5 h post-reperfusion and thereafter, indicating that an EDC reduction below 50%, excepting around the 1 h post-reperfusion period, is an early indicator of cerebral infarction.
References
Astrup J, Siesjo BK, Symon L (1981) Thresholds in cerebral ischemia—the ischemic penumbra. Stroke 12(6):723–725
Astrip J, Symon L, Branston NM, Lassen NA (1977) Cortical evoked potential and extracellular K+ and H+ at critical levels of brain ischemia. Stroke 8(1):51–57
von Kummer R, Dzialowski I (2017) Imaging of cerebral ischemic edema and neuronal death. Neuroradiology 59(6):545–553
Betz AL (1996) Alterations in cerebral endothelial cell function in ischemia. Adv Neurol 71:301–311
Quast MJ, Huang NC, Hillman GR, Kent TA (1993) The evolution of acute stroke recorded by multimodal magnetic resonance imaging. Magn Reson Imaging 11(4):465–471
Gill R, Sibson NR, Hatfield RH, Burdett NG, Carpenter TA, Hall LD, Pickard JD (1995) A comparison of the early development of ischaemic damage following permanent middle cerebral artery occlusion in rats as assessed using magnetic resonance imaging and histology. J Cereb Blood Flow Metab 15(1):1–11
Lansberg MG, Albers GW, Beaulieu C, Marks MP (2000) Comparison of diffusion-weighted MRI and CT in acute stroke. Neurology 54(8):1557–1561
Lansberg MG, Thijs VN, O’Brien MW, Ali JO, de Crespigny AJ, Tong DC, Moseley ME, Albers GW (2001) Evolution of apparent diffusion coefficient, diffusion-weighted, and T2-weighted signal intensity of acute stroke. AJNR Am J Neuroradiol 22(4):637–644
Lutsep HL, Albers GW, DeCrespigny A, Kamet GN, Marks MP, Moseley ME (1997) Clinical utility of diffusion-weighted magnetic resonance imaging in the assessment of ischemic stroke. Ann Neurol 41(5):574–580
Kato H, Kogure K, Ohtomo H, Izumiyama M, Tobita M, Matsui S, Yamamoto E, Kohno H, Ikebe Y, Watanabe T (1986) Characterization of experimental ischemic brain edema utilizing proton nuclear magnetic resonance imaging. J Cereb Blood Flow Metab 6(2):212–221
Naruse S, Aoki Y, Takei R, Horikawa Y, Uede S (1991) Effects of atrial natriuretic peptide on ischemic brain edema in rats evaluated by proton magnetic resonance method. Stroke 22(1):61–65
Moseley ME, Kucharczyk J, Mintorovitch J, Cohen Y, Kurhanewicz J, Derugin N, Asgari H, Norman D (1990) Diffusion-weighted MR imaging of acute stroke: correlation with T2-weighted and magnetic susceptibility-enhanced MR imaging in cats. AJNR Am J Neuroradiol 11(3):423–429
Roberts TP, Vexler Z, Derugin N, Moseley ME, Kucharczyk J (1993) High-speed MR imaging of ischemic brain injury following stenosis of the middle cerebral artery. J Cereb Blood Flow Metab 13(6):940–946
Lovblad KO, Laubach HJ, Baird AE, Crutin F, Schlaug G, Edelman RR, Warach S (1998) Clinical experience with diffusion-weighted MR in patients with acute stroke. AJNR Am J Neuroradiol 19(6):1061–1066
van Everdingen KJ, van Der Grond J, Kappelle LJ, Ramos LM, Mali WP (1998) Diffusion-weighted magnetic resonance imaging in acute stroke. Stroke 29(9):1783–1790
Tong DC, Yenari MA, Albers GW, O’Brien MW, Marks MP, Moseley ME (1998) Correlation of perfusion- and diffusion-weighted MRI with NIHSS score in acute (< 6.5 hour) ischemic stroke. Neurology 50(4):864–870
Hossmann KA, Fischer M, Bockhorst K, Hoehn-Berlage M (1994) NMR imaging of the apparent diffusion coefficient (ADC) for the evaluation of metabolic suppression and recovery after prolonged cerebral ischemia. J Cereb Blood Flow Metab 14(5):723–731
Busza AL, Allen KL, King MD, van Bruggen N, Williams SR, Gadian DG (1992) Diffusion-weighted imaging studies of cerebral ischemia in gerbils. Potential relevance to energy failure. Stroke 23(11):1602–1612
Mintorovitch J, Yang GY, Shimizu H, Kucharczyk J, Chan PH, Weinstein PR (1994) Diffusion-weighted magnetic resonance imaging of acute focal cerebral ischemia: comparison of signal intensity with changes in brain water and Na+, K(+)-ATPase activity. J Cereb Blood Flow Metab 14(2):332–336
Matsuoka Y, Hossmann KA (1982) Cortical impedance and extracellular volume changes following middle cerebral artery occlusion in cats. J Cereb Blood Flow Metab 2(4):466–474
Sykova E, Svoboda J, Polak J, Chvatal A (1994) Extracellular volume fraction and diffusion characteristics during progressive ischemia and terminal anoxia in the spinal cord of the rat. J Cereb Blood Flow Metab 14(2):301–311
Perez-Trepichio AD, Xue M, Ng TC, Majors AW, Furlan AJ, Awad IA, Jones SC (1995) Sensitivity of magnetic resonance diffusion-weighted imaging and regional relationship between the apparent diffusion coefficient and cerebral blood flow in rat focal cerebral ischemia. Stroke 26(4):667–674
Schlaug G, Siewert B, Benfield A, Edelman RR, Warach S (1997) Time course of the apparent diffusion coefficient (ADC) abnormality in human stroke. Neurology 49(1):113–119
Benveniste H, Hedlund LW, Johnson GA (1992) Mechanism of detection of acute cerebral ischemia in rats by diffusion-weighted magnetic resonance microscopy. Stroke 23(5):746–754
Le Bihan D, Breton E, Lallemand D, Grenier P, Cabanis E, Laval-Jeantet M (1986) MR imaging of intravoxel incoherent motions: application to diffusion and perfusion in neurologic disorders. Radiology 161(2):401–407
Yang D, Nakajo Y, Iihara K, Kataoka H, Nakagawara J, Zhao Q, Yanamoto H (2014) An integrated stroke model with a consistent penumbra for the assessment of neuroprotective interventions. Eur Neurol 71:4–18
Yanamoto H, Nagata I, Niitsu Y, Xue JH, Zhang Z, Kikuchi H (2003) Evaluation of MCAO stroke models in normotensive rats: standardized neocortical infarction by the 3VO technique. Exp Neurol 182(2):261–274
Yanamoto H, Nagata I, Hashimoto N, Kikuchi H (1998) Three-vessel occlusion using a micro-clip for the proximal left middle cerebral artery produces a reliable neocortical infarct in rats. Brain Res Brain Res Protoc 3(2):209–220
Back T (1998) Pathophysiology of the ischemic penumbra—revision of a concept. Cell Mol Neurobiol 18(6):621–638
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20(1):84–91
Koizumi J, Yoshida Y, Nakazawa T, Ooneda G (1986) Experimental studies of ischemic brain edema: a new experimental studies of ischemic brain edema: 1. A new experimental model of cerebral embolism in rats in which recirculation can be introduced in the ischemic area. Jpn J Stroke 8:1–8
Kim JH, Na DG, Chang KH, Song IC, Choi SH, Son KR, Kim KW, Sohn CH (2013) Serial MR analysis of early permanent and transient ischemia in rats: diffusion tensor imaging and high b value diffusion weighted imaging. Korean J Radiol 14(2):307–315
Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV (2001) Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation 103(13):1799–1805
Kitagawa K, Matsumoto M, Yang G, Mabuchi T, Yagita Y, Hori M, Yanagihara T (1998) Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery. J Cereb Blood Flow Metab 18(5):570–579
Lin X, Miao P, Wang J, Yuan F, Guan Y, Tang Y, He X, Wang Y, Yang GY (2013) Surgery-related thrombosis critically affects the brain infarct volume in mice following transient middle cerebral artery occlusion. PLoS One 8(9):e75561
Yamato K, Nakajo Y, Yamamoto-Imoto H, Kokame K, Miyata T, Kataoka H, Takahashi JC, Yanamoto Y (2017) A clinically relevant dose of activated protein C (APC) suppresses the development of experimental cerebral infarction. Soc Neurosci 2017
Yanamoto H, Miyamoto S, Nakajo Y, Nakano Y, Hori T, Naritomi H, Kikuchi H (2008) Repeated application of an electric field increases BDNF in the brain, enhances spatial learning, and induces infarct tolerance. Brain Res 1212:79–88
Yang D, Nakajo Y, Iihara K, Kataoka H, Yanamoto H (2013) Alogliptin, a dipeptidylpeptidase-4 inhibitor, for patients with diabetes mellitus type 2, induces tolerance to focal cerebral ischemia in non-diabetic, normal mice. Brain Res 1517:104–113
Nakajo Y, Yang D, Takahashi JC, Zhao Q, Kataoka H, Yanamoto H (2015) ERV enhances spatial learning and prevents the development of infarcts, accompanied by upregulated BDNF in the cortex. Brain Res 1610:110–123
Yamamoto H, Kokame K, Okuda T, Nakajo Y, Yanamoto H, Miyata T (2011) NDRG4 protein-deficient mice exhibit spatial learning deficits and vulnerabilities to cerebral ischemia. J Biol Chem 286(29):26158–26165
Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM (1986) Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 17(6):1304–1308
Hoehn M, Nicolay K, Franke C, van der Sanden B (2001) Application of magnetic resonance to animal models of cerebral ischemia. J Magn Reson Imaging 14(5):491–509
Minematsu K, Li L, Fisher M, Sotak CH, Davis MA, Fiandaca MS (1992) Diffusion-weighted magnetic resonance imaging: rapid and quantitative detection of focal brain ischemia. Neurology 42(1):235–240
Chen F, Suzuki Y, Nagai N, Jin L, Yu J, Wang H, Marchal G, Ni Y (2007) Rodent stroke induced by photochemical occlusion of proximal middle cerebral artery: Evolution monitored with MR imaging and histopathology. Eur J Radiol 63(1):68–75
Pham M, Helluy X, Kleinschnitz C, Kraft P, Bartsch AJ, Jakob P, Nieswandt B, Bendszus M, Stoll G (2011) Sustained reperfusion after blockade of glycoprotein-receptor-Ib in focal cerebral ischemia: an MRI study at 17.6 Tesla. PLoS ONE 6(4):e18386
Pham M, Helluy X, Braeuninger S, Jakob P, Stoll G, Kleinschnitz C, Bendszus M (2010) Outcome of experimental stroke in C57Bl/6 and Sv/129 mice assessed by multimodal ultra-high field MRI. Exp Transl Stroke Med 15:2–6
Yanamoto H, Nagata I, Niitsu Y, Zhang Z, Xue JH, Sakai N, Kikuchi H (2001) Prolonged mild hypothermia therapy protects the brain against permanent focal ischemia. Stroke 32(1):232–239
Griffin JH, Femandez JA, Liu D, Cheng T, Guo H, Zlokovic BV (2004) Activated protein C and ischemic stroke. Crit Care Med 32(5 Suppl):S247–S253
Hoehn-Berlage M, Eis M, Back T, Kohno K, Yamashita K (1995) Changes of relaxation times (T1, T2) and apparent diffusion coefficient after permanent middle cerebral artery occlusion in the rat: temporal evolution, regional extent, and comparison with histology. Magn Reson Med 34(6):824–834
Sakoh M, Ohnishi T, Ostergaard L, Gjedde A (2003) Prediction of tissue survival after stroke based on changes in the apparent diffusion of water (cytotoxic edema). Acta Neurochir Suppl 86:137–140
Gerriets T, Stolz E, Walberer M, Müller C, Kluge A, Kaps M, Fisher M, Bachmann G (2004) Middle cerebral artery occlusion during MR-imaging: investigation of the hyperacute phase of stroke using a new in-bore occlusion model in rats. Brain Res Brain Res Protoc 12(3):137–143
Hacke W, Kaste M, Bluhmki E, Brozman M, Dávalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D, ECASS Investigators (2008) Thrombolysis with Alteplase 3 to 4.5 hours after acute Ischemic stroke. N Engl J Med 359(13):1317–1329
Calder WA III (1983) Body size, mortality, and longevity. J Theor Biol 102(1):135–144
Calder WA III , (1981) Scaling of physiological processes in homeothermic animals. Annu Rev Physiol 43:301–322
Yanamoto H, Hashimoto N, Kassell NF, Lee KS (1996) Disruption of blood brain barrier triggered by reperfusion following transient focal ischemia in rats. Soc Neurosci 22(Part 3):2143
Dreier JP (2011) The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17(4):439–447
Yuzawa I, Sakadzic S, Srinivasan VJ, Shin HK, Eikermann-Haerter K, Boas DA, Ayata C (2012) Cortical spreading depression impairs oxygen delivery and metabolism in mice. J Cereb Blood Flow Metab 32(2):376–386
Hossmann KA (1996) Periinfarct depolarizations. Cerebrovasc Brain Metab Rev 8(3):195–208
Nedergaard M, Hansen AJ (1993) Characterization of cortical depolarizations evoked in focal cerebral ischemia. J Cereb Blood Flow Metab 13(4):568–574
Nedergaard M, Hansen AJ (1988) Spreading depression is not associated with neuronal injury in the normal brain. Brain Res 449(1–2):395–398
Dreier JP, Reiffurth C (2017) Exploitation of the spreading depolarization-induced cytotoxic edema for high-resolution, 3D mapping of its heterogeneous propagation paths. PNAS 114(9):2112–2114
Koroleva VI, Vykhodtseva NI, Elagin VA (1986) Spreading depression in the cortex and subcortical structures of the brain of the rat induced by exposure to focused ultrasound. Neirofiziologiia 18(1):55–61
Vinogradova LV, Koroleva VI, Bures J (1991) Re-entry waves of Leao’s spreading depression between neocortex and caudate nucleus. Brain Res 538(1):161–164
Schielke GP, Moises HC, Betz AL (1991) Blood to brain sodium transport and interstitial fluid potassium concentration during early focal ischemia in the rat. J Cereb Blood Flow Metab 11(3):466–471
Haas M, Forbush BIII (1998) The Na-K-Cl cotransporters. J Bioenerg Biomembr 30(2):161–172
O’Donnell ME, Tran L, Lam TI, Liu XB, Anderson SE (2004) Bumetanide inhibition of the blood-brain barrier Na-K-Cl cotransporter reduces edema formation in the rat middle cerebral artery occlusion model of stroke. J Cereb Blood Flow Metab 24(9):1046–1056
Simard JM, Chen M, Tarasov KV, Bhatta S, Ivanova S, Melnitchenko L, Tsymbalyuk N, West GA, Gerzanich V (2006) Newly expressed SUR1-regulated NC (Ca-ATP) channel mediates cerebral edema after ischemic stroke. Nat Med 12(4):433–440
Vella J, Zammit C, Di Giovanni G, Muscat R, Valentino M (2015) The central role of aquaporins in the pathophysiology of ischemic stroke. Front Cell Neurosci 9:108
Papadopoulos MC, Verkman AS (2013) Aquaporin water channels in the nervous system. Nat Rev Neurosci 14(4):265–277
Yanamoto H, Miyamoto S, Tohnai N, Nagata I, Xue JH, Nakano Y, Nakajo Y, Kikuchi H (2005) Induced spreading depression activates persistent neurogenesis in the subventricular zone, generating cells with markers for divided and early committed neurons in the caudate putamen and cortex. Stroke 36(7):1544–1550
Leao AAP (1944) Spreading depression of activity in the cerebral cortex. J Neurophysiol 7:359–390
Leao AAP, Morrison RS (1945) Propagation of spreading cortical depression. J Neurophysiol 8:33–45
Dreier JP, Fabricius M, Ayata C, Sakowitz OW, William Shuttleworth C, Dohmen C, Graf R, Vajkoczy P et al (2017) Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: review and recommendations of the COSBID research group. J Cereb Blood Flow Metab 37(5):1595–1625
Memezawa H, Minamisawa H, Smith ML, Siesjo BK (1992) Ischemic penumbra in a model of reversible middle cerebral artery occlusion in the rat. Exp Brain Res 89(1):67–78
Acknowledgements
We thank the valuable assistance of Nozomi Momosaki from the Laboratory of Neurology and Neurosurgery, National Cerebral and Cardiovascular Center. This work was supported by Japan Cardiovascular Research Foundation to HY (#J003).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Nakajo, Y., Zhao, Q., Enmi, Ji. et al. Early Detection of Cerebral Infarction After Focal Ischemia Using a New MRI Indicator. Mol Neurobiol 56, 658–670 (2019). https://doi.org/10.1007/s12035-018-1073-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1073-1