
Abstract
Cocaine is known to activate microglia both in vitro and in vivo. High expression of microglial Toll-like receptors (TLRs) and their downstream signal transducers play critical roles in determining microglial activation status. Emerging reports have also demonstrated that cocaine can enhance the strength of TLR signaling. Detailed molecular mechanisms underlying this phenomenon, however, remain elusive. In this study, we investigated the role(s) of miR-124 in regulating microglial TLR4 signaling in the context of cocaine. Herein, we found a dose- and time-dependent upregulation of KLF4 in cocaine-exposed BV-2 cells and rat primary microglial cells (rPMs). KLF4 also identified as a novel 3′-UTR target directly regulated by miR-124. In parallel, miR-124 regulated multiple TLR4 signaling molecules including TLR4, MyD88, TRAF6, and IRAK1. Repeated doses of cocaine (20 mg/kg; i.p.) administration in mice for 7 days further validated the in vitro key findings. Also, miR-124 overexpression significantly blocked the cocaine-mediated upregulation of pro-inflammatory cytokines. In contrast, miR-124 overexpression notably increased the expression of anti-inflammatory mediators in cocaine-exposed microglial cells. Intriguingly, stereotactic administration of lentivirus-miR-124 in the striatum significantly inhibited cocaine-mediated microglial activation and locomotor hyperactivity in vivo. In summary, these findings implicate the role of miR-124 in regulating TLR4 signaling, thereby indicating a new pathway responsible for cocaine-mediated microglial activation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Microglia, the brain resident macrophages, are the primary source of innate immune responses in the central nervous system (CNS). The activation status of microglia determines the intensity of inflammation in the CNS [1, 2]. Microglia express a variety of pattern-recognition receptors (PRR). Of these, the prototypic PRR, toll-like receptor 4 (TLR4), and its downstream signal transducers including myeloid differentiation primary-response protein 88 (MyD88), IL-1R-associated kinase 1 (IRAK1), TNF receptor-associated factor 6 (TRAF6), and NF-κB are well-established players in modulating microglial activation [3,4,5,6]. TLR4-induced NF-κB translocation causes transcriptional upregulation of a plethora of pro-inflammatory cytokines [7, 8]. Krüppel-like factor 4 (KLF4) is also a downstream transcriptional factor of the TLR4 pathway, and its expression is upregulated in microglial cells following lipopolysaccharide (LPS) stimulation [9]. Moderate activation of microglia is a necessary initial response to brain insult. Persistent activation of microglia, on the other hand, leading to a state of exacerbated neuroinflammation results in neuronal death [10,11,12]. Increased microglial activation has been shown in multiple neurodegenerative diseases such as Parkinson’s disease (PD), Alzheimer’s diseases, and amyotrophic lateral sclerosis [13,14,15] and drug addiction [16, 17].
Emerging evidence suggests a close link between drug addiction and the dysregulation of central immune pathways [18, 19]. Addictive drugs including cocaine have been consistently shown to activate microglia both in vitro and in vivo [17, 19,20,21,22]. Interestingly, inhibition of glial cell activation was shown to block cocaine- and methamphetamine-mediated behavioral changes in rodents, implying thereby that elevated neuroinflammation plays causative roles in the development of drug addiction [23, 24]. Previous reports have demonstrated TLR4 signaling [25] as well as reactive oxygen species (ROS), ER stress/autophagy axis to be involved in cocaine-mediated microglial activation [17].
MicroRNAs (miRs) are an important class of biological molecules with critical roles in many fundamental cellular processes such as proliferation, differentiation, and apoptosis [26,27,28]. The brain is one of the miR-enriched organs comprising of several miRs that are vital for its functioning including development, neurogenesis, and neuroinflammation [29]. Several brain-enriched miRs including miR-124 have been demonstrated to modulate microglial functions. Under normal conditions, miR-124 is highly expressed constitutively in microglia and contributes to its quiescence [30, 31]. Downregulation of miR-124 has been associated with multiple neuroinflammatory disorders such as PD, dementia, and multiple sclerosis [31,32,33,34]. Interestingly, the expression levels of miR-124 can be decreased by the classic TLR4 activator, LPS, and correspondingly, miR-124 overexpression has been shown to block LPS-induced microglial activation [35].
The current study was aimed at examining the modulation of miR-124 in microglia and to identify potential targets of miR-124 in the context of cocaine exposure. Our findings demonstrated that KLF4 and TLR4 signaling mediators are novel targets directly regulated by miR-124. Overexpression of miR-124 resulted in amelioration of cocaine-mediated microglial activation in vitro as evidenced by the downregulation of pro-inflammatory factors and upregulation of anti-inflammatory factors in cocaine-exposed microglia transfected with miR-124. Furthermore, intrastriatal administration of lentivirus-miR-124 injection in mice resulted in abrogation of cocaine-mediated microglial activation. Taken together, our findings implicate the role of miR-124 in regulating TLR4 signaling, thereby indicating a new pathway responsible for cocaine-mediated microglial activation.
Materials and Methods
Reagents
Cocaine hydrochloride (C5776) and 5-Azacytidine (5-AZA; A3656) were purchased from Sigma-Aldrich. TaqMan® microRNA assays for miR-124 (001182) quantification were purchased from Applied Biosystems, miR-124 mimic and inhibitor and negative control were purchased from Dharmacon, and lentivirus-miR-124 and control lentivirus were purchased from Biosettia (miR-LV084 and LV000). Antibodies to TLR4 (sc-10741), IRAK1 (sc-7883), TRAF6 (sc-7221), MyD88 (sc-11356), and KLF4 (sc-20691) were purchased from Santa Cruz Biotechnology. Goat anti-rabbit (sc-2004) and goat anti-mouse (sc-2005) secondary antibodies were also from Santa Cruz Biotechnology. Real-time quantitative polymerase chain reaction (qPCR) primers for TNF-α, IL-6, IL-1β, CCL2, TGFβ, IL-4, IL-10, and Arg1 were commercially available from Invitrogen.
BV-2 Cells and HEK293 Cell Culture
BV-2-immortalized cell line was obtained from Dr. Sanjay Maggirwar (University of Rochester Medical Center, Rochester, NY, USA) and was grown and routinely maintained in DMEM (Invitrogen, 11995-065) with 10% heat-inactivated fetal bovine serum (FBS, Invitrogen, 16000-044) at 37 °C and 5% CO2 and used up to 20 passages. HEK293 cells were purchased from ATCC (CRL-1573) and grown and routinely maintained in DMEM with 10% FBS at 37 °C and 5% CO2.
Rat Primary Microglial Cell Culture
Rat primary microglial cells were obtained from Sprague-Dawley newborn (1–3 days) pups and isolated according to the protocol described by Ni and Aschner [36]. Briefly, after digestion and dissociation of the dissected brain cortices in Hanks buffered salt solution (Invitrogen, 14025076) supplemented with 0.25% trypsin (Invitrogen, 25300-054), mixed glial cultures were prepared by resuspending the cell suspension in DMEM supplemented with 10% FBS with 100 U/ml penicillin and 0.1 mg/ml streptomycin. Cells were plated at 10 × 106 cells/flask density onto 75-cm2 cell culture flasks. The cell medium was replaced every 5 days, and after the first medium changed, macrophage colony-stimulating factor (0.25 ng/ml; Invitrogen, PHC9504) was added to the flasks to promote microglial proliferation. When confluent (7 to 10 days), mixed glial cultures were subjected to shaking at 37 °C at 220 g for 2 h to promote microglia detachment from the flasks. The cell medium, containing the released microglia cells, was collected from each flask and centrifuged at 1000g for 5 min to collect cells and plated onto cell culture plates for all subsequent experiments. The purity of microglial cultures was evaluated by immunocytochemistry using the antibody specific for Iba-1 (Wako Pure Chemical Industries, 019-19741) and used if >95% pure.
Western Blotting
Treated cells were lysed using the Mammalian Cell Lysis kit (Sigma, MCL1-1KT). In total, 10 μg of the proteins was electrophoresed on a 10% sodium dodecyl sulfate-polyacrylamide gel under reducing conditions followed by transfer to PVDF membranes (Millipore, IPVH00010). The blots were blocked with 5% nonfat dry milk (in 1× TTPS buffer). Western blots were then probed with antibodies specific for the indicated proteins. The protein amounts loaded were normalized according to the β-actin signal, using an anti-β-actin antibody (Sigma, A5441). The secondary antibodies were HRP conjugated to goat anti-mouse/rabbit IgG.
Quantitative Polymerase Chain Reaction
Total RNA was extracted using Quick-RNA™ MiniPrep Plus (Zymo Research, R1058) as per manufacturer’s protocol and quantified using Nanodrop. Reverse transcription reactions were performed using a Verso cDNA kit (Invitrogen, AB-1453/B), as per manufacturer’s instructions. qPCRs were performed by using SYBR Green ROX qPCR Mastermix (Qiagen, 330510). Reaction systems were set up as follows: 10 μl SYBR Green Mastermix, 0.5 μl forward primers, 0.5 μl reverse primers, and 9 μl distilled water. In total, 96-well plates were placed into a 7500 Fast Real-Time PCR system (Applied Biosystems, Grand Island, NY) for a program running. Actin levels were set as an internal control for calculation.
TaqMan® microRNA Assays for miR-124
TaqMan® microRNA assays for miR-124 were commercially available from Applied Biosystems. TaqMan® MicroRNA Reverse Transcription Kit (PN 4366596) was utilized according to indicated protocol. The reaction system (15 μl) was set as followed: 10 mM dNTPs (with dTTP) 1.5 μl, MultiScribe™ Reverse Transcriptase 50 U/μL 1.0 μl, 10✕ Reverse Transcription Buffer 1.5 μl, RNase Inhibitor, 20 U/μL 0.2 μl, nuclease-free water to 7 μl, total RNA 5 μl (~50 ng), and 3 μL of RT (5×) primer. The tube containing the reaction mixture was centrifuged and loaded onto the thermal cycler: 16 °C for 30 min, 42 °C for 30 min, and 85 °C for 5 min. The reverse transcription product was then diluted 1:10 for the following PCR reaction: TaqMan® PCR primer (20✕) 1 μl, RT reaction product 1.5 μl, TaqMan® 2✕ Universal PCR Master Mix, No AmpErase UNGa 10 μl (PN 4324018), and distilled water up to 20 μl. The PCR conditions were as follows: hold 95 °C for 10 min, then 40 cycles for 95 °C for 15 s and 60 °C for 1 min. All reactions were run in triplicate. The expression level of miR-124 was calculated by normalizing to U6 snRNA.
Luciferase Reporter Assays
KLF4 plasmids containing different 3′-UTR regions were provided by Dr. Michael Ruppert (pMIR-Report-Luc-KLF4, addgene). Briefly, the HEK293 cells were seeded into 96-well plates and cotransfected with KLF4 3′-UTR plasmids and miR-124 mimics/miR-scramble in a molar ratio 10:1. The luciferase activity was determined 24 h posttransfection, and the reporter assay was performed according to the manufacturer’s protocol (Promega). Renilla luciferase activity was normalized to firefly luciferase and expressed as a percentage of the control.
Ago2 Immunoprecipitation
BV-2 cells were plated onto 6-well plates and transfected with either miR-control or miR-124 mimics for 24 h. After treatment, cells were washed in the cold 1× PBS, scraped, and then lysed with a buffer containing 0.5% NP40, 150 mM KCL, 25 mM Tris-glycine pH 7.5, 2 mM EDTA, 0.5 mM DTT, and inhibitors of RNases, proteases, and phosphatases. In total, 10% of total lysate was removed and kept as the input samples and the remainder used for immunoprecipitation. In addition, 10 μg of anti-Ago2 (11A9, Sigma-Aldrich, SAB4200085) or anti-FLAG (M2, F1804, Sigma-Aldrich) antibodies were incubated overnight with protein A/G agarose beads (Thermo Scientific, 20423) at 4 °C. Precleared lysates were then incubated with the appropriate antibody-bound beads, and the immunoprecipitated proteins were washed and incubated with DNase I (Invitrogen, 18068015) followed by digestion with proteinase K (Zymo Research, D3001-2) for 15 min. RNA extraction was then performed using Quick-RNA™ MiniPrep Plus (Zymo Research, R1058) and quantified using Nanodrop. In total, Ago2-immunoprecipitated RNA samples were then used for determining the binding targets of miR-124, such as KLF4, MyD88, IRAK1, TRAF6, and TLR4 by qPCR.
miR-124 Oligo Transfection
MiR-124 mimics, inhibitors, and control were purchased from GE Health Corporation. BV-2 cells were seeded into 24-well plates (1 × 105/well) and transfected with 30 pmol of various oligos by Lipofectamine 2000 according to manufacturer’s protocol (Invitrogen). Twenty-four hours later, cellular homogenates were extracted to detect various proteins levels as indicated.
Chronic Cocaine Administration
Sprague-Dawley rats and C57BL/6N mice were purchased from Charles River Laboratories (Wilmington, MA, USA). All the animals were housed under conditions of constant temperature and humidity on a 12-h light and 12-h dark cycle, with lights on at 0700 hours. Food and water were available ad libitum. All animal procedures were performed according to the protocols approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center and the National Institutes of Health. For repeated cocaine administration, C57BL/6N mice were injected with cocaine (20 mg/kg, i.p.) or saline for 7 days. One hour after the last injection, mice were sacrificed for brain removal. The striatal protein homogenates were extracted and assessed for the levels of various proteins as indicated.
Stereotactic Injection of Lentivirus-miR-124
Male 8-week-old C57BL/6N mice were divided into four groups (n = 8) receiving the following treatments: (1) saline with miR-control lentivirus, (2) saline with miR-124 lentivirus, (3) cocaine with miR-control lentivirus, and (4) cocaine with miR-124 lentivirus. The lentiviruses (1.5 μl) were microinjected into the mouse brain striatum (at least 107 infectious units per ml) using the microinjection parameters (coordinates 2.8 mm behind the bregma, 1.7 mm lateral from the sagittal midline at a depth of 4.5 mm to skull surface). To increase the precision, a 10-ml Hamilton syringe with a 33G needle was utilized for intrastriatal injection as described.
Open-Field Test
Open-field consisted of the square arena (40 cm × 40 cm) and wall (35 cm high). Mice were carried to the test room in their home cages and were handled by the base of their tails at all times. Mice were placed at one of the four corners of the open field and allowed to explore the apparatus for 5 min, following which mice were returned to their home cages and received the final injection of cocaine/saline. The mice were then placed at the same corner of the arena, and a video camera was suspended directly over the arena for recording behavioral activities for 20 min using an automated video tracking system (EthoVision 3.1, Noldus Information Technology, Inc., Leesburg, VA). The parameters such as the total distance of running, latency time, and average speed were included and calculated using the Ethovision 3.1 software. After each test, the apparatus was thoroughly cleaned with cotton pad wetted with 70% ethanol. A single primary observer blinded to the experimental condition conducted the behavioral observations.
Statistical Analysis
All the data were expressed as mean ± SEM, and appropriate statistical significance was chosen based on the experimental design using GraphPad Prism version 5 (San Diego, CA, USA). The specific statistical analysis used is indicated in the text and each figure caption for all studies. Bonferroni post hoc test was used for one-way analysis of variance (ANOVA), and Tukey post hoc test was used for two-way ANOVA. Values were considered statistically significant when P < 0.05.
Results
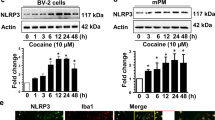
Cocaine-Upregulated KLF4 Levels In Vitro and In Vivo
KLF4, characterized as a zinc finger-containing transcriptional factor, belongs to the KLF protein family which includes 17 different members [37]. KLF4 is essential for embryonic development and regulates a variety of processes including cell proliferation, differentiation, apoptosis, migration, and invasion [34]. KLF4 is expressed in a variety of cell types including monocytes/macrophages. Previous reports have demonstrated that KLF4 is expressed in microglia and functions as a downstream factor of TLR signaling, plays critical roles in microglial activation, and ensuing neuroinflammation [38,39,40,41]. Multiple studies have shown that psychostimulants including cocaine can activate microglia both in vitro and in vivo [19,20,21,22]. To investigate whether cocaine-mediated microglial activation involved KLF4, BV-2 cells were exposed to varying doses of cocaine (1, 10, and 100 μM) for 24 h. Following cocaine exposure, total cellular proteins were isolated to determine levels of KLF4 expression by Western blotting. Our findings (Fig. 1a) showed that cocaine dose-dependently upregulated KLF4 protein levels (P < 0.05). Next, we isolated rPMs from the brain tissues of newborn rat pups and exposed them to cocaine, as was done for BV-2 cells. Consistent with the BV-2 cells, cocaine also upregulated KLF4 levels in rPMs (P < 0.05; Fig. 1b). Based on these experiments, we have chosen 10 μM cocaine as the dose for ensuing experiments. The rationale for choosing 10 μM cocaine is based on the fact that following intranasal cocaine administration plasma levels of cocaine range between 0.4 and 1.6 μM [42], while the plasma cocaine concentrations in tolerant abusers reach levels up to 13 μM [43]. Additionally, cocaine levels in postmortem brains of chronic cocaine users following acute intoxications have been reported higher than 100 μM [44]. Next, we performed time-course experiments to determine KLF4 protein levels in both BV-2 cells and rPMs exposed to 10 μM cocaine for varying time points. Our findings clearly showed that cocaine significantly (P < 0.05) increased KLF4 expression in a time-dependent manner. This upregulation was evident as early as 6 h post-treatment and persisted for 24 h in both BV-2 cells (Fig. 1c) and rPMs (Fig. 1d). We next sought to investigate whether cocaine also upregulated KLF4 levels in vivo. For this wild type, C57BL/6N mice were injected with cocaine (20 mg/kg, i.p.) for 7 consecutive days and the striatum was removed for assessment of KLF4 protein levels. Our findings demonstrated that KLF4 levels were significantly (P < 0.05) elevated in cocaine-treated mouse brains compared with saline-treated controls (Fig. 1e).

Cocaine increased KLF4 levels in vitro and in vivo. Representative Western blots showing the cocaine-mediated dose-dependent upregulation of KLF4 levels in both the BV-2 cells (a) and the rPMs (b). Representative Western blots showing the cocaine-mediated time-dependent upregulation of KLF4 levels in both the BV-2 cells (c) and the rPMs (d). Representative Western blots showing the increased KLF4 expression levels in the striatal brain region of mice chronically administered with cocaine (20 mg/kg, i.p.) for 7 days (e). β-Actin was used as a protein loading control for all the experiments. The data are presented as mean ± SEM from six independent experiments. One-way ANOVA followed by Bonferroni post hoc test was used to determine the statistical significance. *P < 0.05 vs control
KLF4 is a Novel 3′-UTR Target of miR-124
We next explored the mechanisms underlying cocaine-mediated upregulation of KLF4. miRs have been demonstrated to play critical roles in regulating microglial inflammatory responses [29, 45,46,47]. Our previous work showed that cocaine downregulated the expression of miR-124 levels via promoter DNA methylation in microglia [48]. Herein, we replicated these findings and again demonstrated that exposure of both BV-2 cells and rPMs to cocaine downregulated miR-124 levels in a time-dependent manner (Supplementary Fig. S1). We next sought to examine a possible functional linkage between miR-124 and KLF4 in the context of cocaine. Bioinformatics analyses (miRanda and TargetScan) suggested there are three binding sites located in the 3′-UTR of KLF4 mRNA (nucleotide position: 456-475, 772-791, and 814-833), indicating KLF4 as a direct target regulated by miR-124 (Fig. 2a). To investigate the effects of miR-124 on KLF4 levels, BV-2 cells were transfected with miR-124 mimics/scrambles followed by exposing the transfected cells to cocaine (10 μM) for 24 h. As shown in Fig. 2b, miR-124 overexpression significantly (P < 0.05) inhibited endogenous KLF4 levels while also attenuating cocaine-mediated upregulation of KLF4 in BV-2 cells. Cocaine-mediated upregulation of KLF4 levels and the inhibitory effects of miR-124 on KLF4 expression were also validated in rPMs (Fig. 2c). Reciprocally, BV-2 cells and rPMs transfected with miR-124 inhibitors demonstrated a significant (P < 0.05) upregulation of KLF4 expression levels (Fig. 2d, e). To confirm that miR-124 could directly bind to the KLF4 3′-UTR, we next cotransfected the recombinant KLF4 3′-UTR (fragment -1 or -2) luciferase reporter plasmids (schematic shown in Fig. 2f) with miR-124 mimics and miR-124 inhibitor, respectively, into HEK-293 cells, followed by an assessment of luciferase activity. Our findings demonstrated a decrease in luciferase activity in cells cotransfected with 3′-UTR-2 (containing the binding sites) and miR-124 mimics compared with cells cotransfected with 3′-UTR-1 and miR-124 mimics (Fig. 2f). In contrast, cells cotransfected with miR-124 mimics and KLF4-3′-UTR-1 had a negligible effect on luciferase activity. However, cells cotransfected with KLF4 3′-UTR plasmids and miR-124 inhibitors had no effects on luciferase activity. The binding of miR-124 with its target, KLF4, was further confirmed using Argonaute 2 immunoprecipitation assay (Ago2 IP). In principle, Ago2 is an indispensable component of the RNA-induced silencing complex (RISC) that is directed to target mRNAs by the miRNA guide strand. This complex is then immunoprecipitated, and the mRNA levels were quantified by qPCR. Based on this, we performed Ago2 IP in BV-2 cells that were transfected with either miR-124 mimics or miR-control for 24 h. As shown in Fig. 2h, Ago2 IP confirmed KLF4 as a direct target of miR-124 because KLF4 mRNA showed increased Ago2 binding in a miR-124-dependent manner. Taken together, these findings demonstrated KLF4 as a direct 3′-UTR target regulated by miR-124.

KLF4 is a novel target regulated by miR-124. a Putative miR-124 binding sites in KLF4 gene. The potential complementary residues are shown in red color. b Representative Western blots showing the overexpression of miR-124 significantly blocked the upregulation of KLF4 in BV-2 cells transfected with miR-124 mimics/control followed with cocaine (10 μM) exposure for 24 h. c Representative Western blots showing the overexpression of miR-124 significantly blocked the upregulation of KLF4 in rPMs transfected with miR-124 mimics/control followed with cocaine (10 μM) exposure for 24 h. d Representative Western blots showing the miR-124 inhibitor significantly increased the KLF4 in BV-2 cells transfected with miR-124 inhibitor/control followed with cocaine (10 μM) exposure for 24 h. e Representative Western blots showing the miR-124 inhibitor significantly increased the KLF4 in rPMs transfected with miR-124 inhibitor/control followed with cocaine (10 μM) exposure for 24 h. f Schematic diagram of KLF4-3′-UTR-1 and KLF4-3′-UTR-2 plasmids. KLF4-3′-UTR-1 plasmid has no miR-124 binding sites, while KLF4-3′-UTR-2 plasmid has three miR-124 binding sites. g Relative luciferase activity of 3′-UTR-1 and 3′-UTR-2 constructs of KLF4 cotransfected with miR-control, miR-124 mimics, and miR-124 inhibitor in HEK293 cells. Overexpression of miR-124 mimics/inhibitor had no effect on the luciferase activity of KLF4-3′-UTR-1 plasmids, while miR-124 mimics significantly inhibited KLF4-3′-UTR-2 luciferase activity. h Ago2 IP confirmed the enrichment of the miR-124 target mRNA, KLF4 in Ago2 IP as compared with total RNA isolated from BV-2 cells transfected with miR-124 mimics/control. β-Actin was used as a loading control for all the experiments. The data are presented as mean ± SEM from six independent experiments. One-way ANOVA followed by Bonferroni post hoc test was used to determine the statistical significance for multiple groups, and a Student’s t test was used to determine the statistical significance for between 2 groups. *P < 0.05 vs. control; # P < 0.05 vs. cocaine
Overexpression of miR-124 Inhibited Cocaine-Mediated TLR4 Upregulation
A striking feature of miR regulation is that a single miR can modulate several hundred mRNAs simultaneously to modify cellular activity. In addition to KLF4, we also assessed other molecules which are potential direct targets of miR-124 and also known to play a role in cocaine-mediated microglial activation. Recently, the interaction and activation of TLR4 following cocaine exposure were shown to be critical for cocaine-mediated neuroinflammation in vivo [25, 49]. In the current study, we sought to examine whether cocaine also regulated expression levels of TLR4 protein in microglial cells. BV-2 cells were exposed to cocaine at different doses (1, 10, and 100 μM for 24 h) for the assessment of TLR4 protein levels. Our findings demonstrated that cocaine exposure to BV-2 cells resulted in increased expression of TLR4 protein levels in a dose-dependent manner with highest expression at cocaine concentrations of 10 and 100 μM (P < 0.05; Fig. 3a). Next, we determined the time-dependent expression of TLR4 levels, in which BV-2 cells were exposed to 10 μM cocaine for different time periods (0–24 h). As shown in Fig. 3b, exposure of BV-2 cells to 10 μM cocaine significantly (P < 0.05) increased TLR4 protein levels on 6 h onwards while negligible effects at the earlier time point (3 h). Furthermore, upregulation of TLR4 expression was also observed in cocaine-administered mouse brains compared to saline-administered controls. Similar to the cocaine-mediated upregulation of KLF4 expression, we also observed upregulation of TLR4 in the presence of cocaine (Fig. 3c). Based on the bioinformatics analyses (miRanda and TargetScan) that TLR4 has two binding sites for miR-124 in its 3′-UTR region (Fig. 3d), we next sought to confirm that TLR4 is indeed a target of miR-124. BV-2 cells were transfected with miR-124 mimics followed with cocaine exposure for 24 h. Protein homogenates were assessed for detection of TLR4 levels, and as shown in Fig. 3E, overexpression of miR-124 significantly (P < 0.05) inhibited endogenous TLR4 levels while also abrogating cocaine-mediated upregulation of TLR4 levels. In contrast and as expected, transfection of BV-2 cells with miR-124 inhibitors resulted in increased expression of TLR4 (Fig. 3f). The regulation of TLR4 levels by miR-124 was further validated in rPMs wherein overexpression of miR-124 followed by cocaine exposure resulted in decreased levels of endogenous TLR4 (Fig. 3g). On the other hand, transfection of rPMs with miR-124 inhibitors followed by cocaine exposure resulted in statistically increased (P < 0.05) expression of TLR4 (Fig. 3g). Ago2 IP assay further confirmed the binding of miR-124 with TLR4 in BV-2 cells transfected with miR-124 mimics compared with miR-control (Fig. 3h). Taken together, these results provide evidence that cocaine exposure enhanced TLR4 levels both in vitro and in vivo and that overexpression of miR-124 blocked these effects.

Overexpression of miR-124 inhibited the cocaine-mediated TLR4 upregulation. a Representative Western blots showing the cocaine-mediated dose-dependent upregulation of TLR4 levels in BV-2 cells. b Representative Western blots showing the cocaine-mediated time-dependent upregulation of TLR4 levels in BV-2 cells. c Representative Western blots showing the increased TLR4 expression levels in the striatal brain region of mice chronically administered with cocaine (20 mg/kg, i.p.) for 7 days. d Putative miR-124 binding sites in TLR4 gene. The potential complementary residues are shown in red color. e Representative Western blots showing the overexpression of miR-124 significantly blocked the upregulation of TLR4 in BV-2 cells transfected with miR-124 mimics/control followed with cocaine (10 μM) exposure for 24 h. f Representative Western blots showing the miR-124 inhibitors significantly increased the TLR4 in rPMs transfected with miR-124 inhibitor/control for 24 h. g Representative Western blots showing the overexpression of miR-124 significantly blocked the upregulation of TLR4 in rPMs transfected with miR-124 mimics, while inhibition of miR-124 increased the TLR4 upregulation in rPMs transfected with the miR-124 inhibitor for 24 h. h Ago2 IP confirmed the enrichment of the miR-124 target mRNA, TLR4 in Ago2 IP as compared with total RNA isolated from BV-2 cells transfected with miR-124 mimics/control. β-Actin was used as a loading control for all the experiments. The data are presented as mean ± SEM from six independent experiments. One-way ANOVA followed by Bonferroni post hoc test was used to determine the statistical significance for multiple groups, and a Student’s t test was used to determine the statistical significance for between 2 groups. *P < 0.05 vs. control; # P < 0.05 vs. cocaine
Overexpression of miR-124 also Inhibited the Levels of MyD88, IRAK1, and TRAF6
Previous reports demonstrated that miR-124 negatively regulated TLR4 signaling by modulating the levels of MyD88 and TRAF6 in both alveolar macrophage and epithelial cells [50, 51]. In addition to these two TLR4 signaling transducers, we found that IRAK1 has one miR-124 binding site in its 3′-UTR, indicating IRAK1 could be yet another target regulated by miR-124 (Fig. 4a). The next step was to explore whether cocaine exposure could also lead to increased expression levels of these three signal transducers (MyD88, IRAK1, and TRAF6) and, if so, whether miR-124 overexpression could block these effects. BV-2 cells were transfected with either miR-124 mimics or scrambled siRNA followed by cocaine exposure for 24 h and protein homogenates prepared for targeted proteins detection. Our results clearly showed that cocaine significantly (P < 0.05) increased the levels of MyD88, IRAK1, and TRAF6 and that miR-124 overexpression abrogated cocaine-mediated upregulation of these mediators (Fig. 4b, d, and f). In contrast and as expected, transfection of BV-2 cells with miR-124 inhibitor resulted in increased expression of MyD88 (Fig. 4c), IRAK1 (Fig. 4e), and TRAF6 (Fig. 4g) compared with cells transfected with miR-control. These findings were also validated in rPMs transfected with miR-124 mimics or miR-124 inhibitors (Fig. 4h, i). Ago2 IP assay further confirmed the binding of miR-124 with MyD88, IRAk1, and TRAF6 in BV-2 cells transfected with miR-124 mimics compared with cells transfected with miR-control (Fig. 4j). Taken together, our results suggest that in addition to KLF4 and TLR4, miR-124 also modulates the levels of other signaling transducers such as MyD88, IRAK1, and TRAF6 in microglial cells.

Overexpression of miR-124 blocked the cocaine-mediated upregulation of MyD88, IRAK1, and TRAF6. Putative miR-124 binding sites in MyD88, IRAK1, and TRAF6 genes. The potential complementary residues are shown in red color (a). Representative Western blots showing the overexpression of miR-124 significantly blocked the upregulation of MyD88 (b), IRAK1 (d), and TRAF6 (f) in BV-2 cells transfected with miR-124 mimics/control followed with cocaine (10 μM) exposure for 24 h. Representative Western blots showing the inhibition of miR-124 significantly increased the upregulation of MyD88 (c), IRAK1 (e), and TRAF6 (g) in BV-2 cells transfected with miR-124 inhibitor/control followed with cocaine (10 μM) exposure for 24 h. Representative Western blots showing the overexpression of miR-124 significantly blocked the upregulation of MyD88, IRAK1, and TRAF6 in rPMs transfected with miR-124 mimics, while inhibition of miR-124 increased the upregulation of MyD88, IRAK1, and TRAF6 in rPMs transfected with the miR-124 inhibitor for 24 h (h, i). Ago2 IP confirmed the enrichment of the miR-124 target mRNAs, MyD88, IRAK1, and TRAF6 in Ago2 IP as compared with total RNA isolated from BV-2 cells transfected with miR-124 mimics/control (j). β-Actin was used as a loading control for all the experiments. The data are presented as mean ± SEM from 6 independent experiments. One-way ANOVA followed by Bonferroni post hoc test was used to determine the statistical significance for multiple groups, and a Student’s t test was used to determine the statistical significance for between 2 groups. *P < 0.05 vs. control; # P < 0.05 vs. cocaine
Overexpression of miR-124 Inhibits Cocaine-Mediated Microglial Activation In Vitro
The findings presented above demonstrate that miR-124 modulates the expression levels of multiple mediators in TLR4 signaling. Since miR-124 is known to maintain microglia in a quiescent state and also promote the transition of microglia from a pro-inflammatory to an anti-inflammatory phenotype [35, 52], we next sought to investigate the effect of miR-124 overexpression on cocaine-mediated microglial activation. BV-2 cells were transfected with miR-124 mimics or scrambled siRNA followed by exposure to cocaine and assessed for expression levels of M1 (pro-inflammatory) and M2 (anti-inflammatory) markers by qPCR. The representative M1 markers included TNF-α, CCL2, and NOS2 while the M2 markers included TGFβ, IL-4, and IL-10. As shown in Fig. 5a–c, cocaine exposure of BV-2 cells resulted in a significant (P < 0.05) increase in mRNA levels of the M1 markers (TNF-α 2.68 ± 0.17, CCL2 2.10 ± 0.09, NOS2 3.63 ± 0.20-fold respectively; P < 0.05) and overexpression of miR-124 abrogated cocaine-mediated effects on these M1 markers. Concomitantly, there was a substantial increase in expression of M2 anti-inflammatory markers in miR-124 overexpressing microglia (TGFβ 1.56 ± 0.03, IL-4 3.82 ± 0.14, IL-10 3.32 ± 0.07-fold respectively; P < 0.05), as shown in Fig. 5d–f. Our results thus imply that miR-124 overexpression blocked cocaine-mediated microglial activation by promoting microglia from a pro-inflammatory to an anti-inflammatory phenotype. In addition to M1 and M2 markers, we also demonstrated that the levels of two other miR-124 targets c/EBP (Fig. 5g) and ARG1 (Fig. 5h) were also decreased in cells overexpressing miR-124, a finding that is in agreement with previous reports [31].

Overexpression of miR-124 inhibited cocaine-mediated microglial activation in vitro. qPCR analysis showing the overexpression of miR-124 significantly inhibited the increased mRNA expressions of various proinflammatory mediators such as TNF-α (a), CCL2 (b), and NOS2 (c), in BV-2 cells transfected with miR-124 mimics/control followed with cocaine (10 μM) exposure for 24 h. In contrast, qPCR analysis showing the overexpression of miR-124 significantly increased mRNA expressions of various anti-inflammatory mediators such as TGFβ (d), IL-4 (e), and IL-10 (f) in BV-2 cells transfected with miR-124 mimics/control followed with cocaine (10 μM) exposure for 24 h. qPCR analysis showing the overexpression of miR-124 significantly inhibited the mRNA expressions of c/EBP and ARG1 in BV-2 cells transfected with miR-124 mimics/control followed with cocaine (10 μM) exposure for 24 h (g, h). β-Actin was used as an internal housekeeping gene for all the experiments. The data are presented as mean ± SEM from 6 independent experiments. One-way ANOVA followed by Bonferroni post hoc test was used to determine the statistical significance for multiple groups, and a Student’s t test was used to determine the statistical significance for between 2 groups. *P < 0.05 vs. control; # P < 0.05 vs. cocaine
Overexpression of Lentivirus-miR-124 Inhibits Cocaine-Mediated Microglial Activation In Vivo
Based on our findings that miR-124 overexpression inhibited cocaine-mediated microglial inflammatory responses in vitro, we next sought to validate these observations in vivo. For this wild-type C57BL/6N, mice (n = 8) were stereotactically injected with RFP-lentivirus (LV) containing miR-124 or miR-control into the striatum. Two weeks later, mice were administered with cocaine (20 mg/kg; i.p.) for 7 consecutive days (schematic shown in Fig. 6a). One hour after the last injection, mice were sacrificed and striatal tissues processed for assessment of RFP expression to examine the efficacy of miR-124 expression. Interestingly, mice that received LV injection revealed strong RFP expression colocalized with microglia in the striatum indicating successful virus transfection and expression in vivo (Fig. 6b). As expected, mice injected with LV-miR-124 exhibited increased miR-124 levels in the striatum compared to mice receiving control virus injection (Fig. 6c). Cocaine administration significantly (P < 0.05) downregulated miR-124 levels in the mice brains compared with mice receiving LV-miR-control (P < 0.05, Fig. 6c). We next wanted to assess the effect of miR-124 overexpression on KLF4 and TLR4 levels in the striatum in the presence of cocaine. Cocaine administration significantly increased KLF4 and TLR4 levels in the LV-miR-control group as expected. Interestingly, there was no significant changes in KLF4 and TLR4 levels in LV-miR-124 injected mice, indicating thereby that miR-124 overexpression blocked cocaine-mediated upregulation of KLF4 and TLR4 (Fig. 6d–f). We also observed that there is no difference in KLF4 levels between the two saline groups following miR-124 overexpression. One possible explanation for this discrepancy could be that the constitutive expression of miR-124 is already high in the saline group (maintaining a basal level of KLF4) and that further upregulation of miR-124 has little effect in modulating the KLF4 levels. Our findings also demonstrated that cocaine-mediated upregulation of microglial activation (increased Iba-1 levels) was abrogated in mice with LV-miR-124 injection (Fig. 6d, g). Additionally, to assess the role of miR-124 on cocaine-mediated locomotor activity, mice overexpressing LV-miR-124 or miR-control were administered cocaine for 7 days following which the mice were monitored for locomotor activity using the open-field test. Figure 6h shows the total distance traveled for 20 min following the last saline or cocaine injection. Regarding the distance traveled, there was no statistical difference observed between the saline-administered LV-miR-control and LV-RFP-miR-124 groups. Interestingly, however, cocaine injection had a notable stimulating effect on locomotor activity in both LV-miR-control and LV-RFP-124-injected mice, with a significant downregulation of locomotor activity in LV-miR-124 mice compared with the LV-miR-control group (P > 0.05). Taken together, these findings demonstrated that miR-124 overexpression blocked cocaine-mediated microglial activation in vivo, thereby lending support to the idea that miR-124 maintains microglia in a quiescent state by inhibiting neuroimmune signaling induced by cocaine.

Overexpression of miR-124 inhibited cocaine-mediated microglial activation in vivo. a Schematic showing the stereotactic injection of lentivirus (LV)-miR-124 followed by chronic cocaine administration (20 mg/kg; i.p. for 7 days) in mice. b Representative fluorescent microscopic image showing the overexpression of miR-124 colocalized with microglia in the striatum region of mice stereotactically injected with LV-miR-124. c qPCR analysis showing the expression levels of miR-124 in the striatal region of mice stereotactically injected with LV-miR-124 followed by cocaine administration. d–g Representative Western blots and quantifications showing the expression levels of KLF4, TLR4, and Iba-1 in the striatal region of mice stereotactically injected with LV-miR-124 followed by cocaine administration. β-Actin was used as a protein loading control for all the experiments. The data are presented as mean ± SEM from 8 independent experiments. Two-way ANOVA followed by Tukey post hoc test was used to determine the statistical significance for multiple groups, and a Student’s t test was used to determine the statistical significance for between 2 groups. *P < 0.05 vs. saline; # P < 0.05 vs. cocaine
Discussion
Although cocaine is well known to activate microglia leading to increased neuroinflammation, the detailed molecular mechanism(s) underlying this phenomenon remain poorly understood. It is now being recognized that development of addiction is associated with a neuroinflammatory phenotype, specifically with the implication of activation of microglia. MiR-124 is a brain-enriched miR that is highly expressed in microglia. This specific miR has been well established as a master regulator of microglial quiescence. Downregulation of miR-124 has been suggested in multiple neuroinflammatory diseases including PD, amyotrophic lateral sclerosis, with miR-124 overexpression shown to mitigate the pathogenesis associated with these brain diseases. Since cocaine addiction is linked to microglial activation and the fact that miR-124 is downregulated in response to cocaine exposure both in vitro and in vivo, we infer that miR-124 downregulation could be critical in the development of drug addiction. Following our previous studies showing that cocaine downregulated microglial miR-124 [48], herein we demonstrate that miR-124 can modulate the strength of TLR4 signaling by regulating multiple molecules including TLR4, MyD88, IRAK1, TRAF6, and KLF4 and that microglial overexpression of miR-124 inhibited cocaine-mediated activation both in vitro and in vivo. Since elevated neuroinflammatory levels are believed to contribute to the development of drug addiction, our results suggest that miR-124 can be envisioned as a potential therapeutic agent for the treatment of drug addiction via its inhibition of microglial activation.
Over the last several decades, much attention has been focused on the effects of abused drugs, such as cocaine, on signal transmission between neurons and the ensuing neuronal excitability in the context of addiction. Recent emerging evidence has also shed light on the cross-talk between neurons and glia (neuroimmune signaling), as a contributor to the development of drug addiction. Abused drugs including cocaine, amphetamine, and alcohol have long been recognized to activate microglia/astrocytes in multiple in vitro and in vivo model systems. Findings from our lab have demonstrated the ability of cocaine to activate microglia and rPM, leading in turn to the production and release of multiple pro-inflammatory factors including TNF-α, IL-6, and CCL2 [17, 19, 53]. Various other reports have also demonstrated increased neuroinflammation following exposure of rodents to cocaine and other abused drugs. For example, amphetamine administration (4 mg/kg at 2-h intervals, four times) was shown to activate microglia in mice [54]. In another study acute, a high dose (30 mg/kg) of methamphetamine exposure increased the mRNA levels of pro-inflammatory cytokines IL-6 and TNF-α in the striatum and hippocampus of mice [55]. Another study demonstrated that even lower doses of methamphetamine could activate microglia [56]. Furthermore, the relationship between psychostimulant exposure and ensuing neuroinflammation has also been confirmed by analyzing annotated datasets which are available in the GEO public database [57,58,59]. Clinical evidence has also revealed a close linkage between neuroinflammation and drug addiction. For example, it has also been shown that there is a statistically significant increase in activated macrophages/microglia in chronic cocaine abusers [60]. In another study comparing cognitive function with peripheral markers of immune activation, significant correlations were observed in methamphetamine users, but not in matched controls [61]. Another study using positron emission tomography suggested a diffuse persistent increase in activated microglia in all brain regions of human methamphetamine abusers compared to control subjects [20]. Studies by Watkin’s group have elegantly demonstrated that cocaine can directly interact with TLR4 to enhance microglial innate immune responses including increased secretion of IL-1β, leading in turn to upregulation of synaptic dopamine concentrations in the striatum. In this study using the TLR4 KO mice, the authors demonstrated that there was a decrease in cocaine-induced behavioral changes in both conditioned place preference (CPP) and self-administration assays, implying thereby that microglial activation via TLR4 was critical for cocaine addiction [25]. Intriguingly, inhibitors of glial activation have recently been suggested as potential treatment strategies for psychostimulant abuse [62]. Taken together, glial-neuronal cross-talk induced by drug abuse further facilitates the inflammatory factors that are critical targets for the development of psychostimulant abuse medications.
Herein, we have provided a novel molecular mechanism responsible for cocaine-mediated microglia activation (neuroinflammation). Previously, we demonstrated that cocaine exposure in microglia leads to downregulation of microglial miR-124 levels via an increase in the methylation status of the miR-124 promoter. Downregulation of miR-124, in turn, resulted in activation of the TLR4-mediated signaling via elevation of multiple miR-124-regulated mediators such as KLF4, TLR4, MyD88, TRAK1, and TRAF6. Since both microglial activation and elevated neuroimmune signaling have been suggested to activate the dopaminergic neuronal activity in the brain striatum, miR-124 could be envisioned to play critical roles in cocaine-associated reward circuitry and possibly, also in addiction to cocaine.
It is widely recognized that small noncoding RNAs, such as microRNAs, play critical roles in modulating glial activation and other cellular processes. As an example, miR-124, a brain-enriched miRNA, plays key roles in neurogenesis, synapse plasticity, and glia-neuronal interactions to maintain brain homeostasis. MiR-124 was first identified as a neuroprotective factor promoting neurogenesis during brain development and maintenance of synaptic homeostasis in the adult brain [63, 64]. There have been reports indicating miR-124 as one of the miRs that are highly expressed in resting microglia, and its expression levels were found to correlate with microglia quiescence [30, 31] closely. In another study, it was shown that overexpression of miR-124 blocked LPS-induced microglial activation in vitro [35]. Decreased miR-124 levels have been reported in various neuroinflammatory disorders including PD, dementia, and multiple sclerosis [31,32,33,34]. Drug addiction is recognized as a neuroinflammation-related disease with several abused drugs having demonstrated the ability to modulate miR-124 levels. It has been shown that chronic administration of cocaine decreased miR-124 levels in the nucleus accumbens (NAc) and that downregulation of miR-124 was critical for cocaine-induced synaptic plasticity and behavioral changes [65]. These findings are in agreement with our results that cocaine exposure leads to downregulation of miR-124, culminating ultimately into microglial activation. The role of miR-124 in drug addiction has also been reported by overexpression studies wherein lentivrus-miR-124 transduction in the NAc resulted in attenuation of cocaine-induced CPP [66]. Interestingly, reduced levels of miR-124 have also been found in the ventral tegmental area of methamphetamine self-administering rats by microarray analysis [67]. There are also reports demonstrating decreased miR-124 levels in ethanol-withdrawn rats, which were likely attributed to histone acetylation [68]. All these studies allude to the fact that miR-124 downregulation is critical for reward-related behavior changes induced by abused drugs, but detailed mechanisms remain unexplored. Our behavioral findings are also consistent with the published reports and lend further credence to the fact that cocaine-mediated downregulation of microglial miR-124 was involved in increased neuroimmune signaling mediated by TLR4 signaling, leading in turn to addictive behavior [25, 69]. We also acknowledge that BV-2 cells and rat primary microglia may not recapitulate the function of microglia in vivo. However, the novelty of our findings are: (1) KLF4, an important pro-inflammatory transcription factor, that was identified in this study as a novel miR-124 target; (2) miR-124 can modulate multiple molecules in TLR4 signaling including TLR4, MyD88, IRAK1, and TRAF6; (3) overexpression of miR-124 can inhibit cocaine-mediated microglial activation in vitro and in vivo.
In summary, we have demonstrated that miR-124 regulated microglial TLR4 signaling and overexpression of miR-124 inhibited cocaine-mediated microglial activation, and this could have an impact on the ensuing cross-talk between glia and neurons (Fig. 7). Overexpression of miR-124 has already been shown to prevent microglial activation through inhibition of reactive oxygen species production [70]. Taken together, miR-124 can be envisioned as a potential target for modulating drug abuse-mediated neuroinflammation and for ameliorating drug addiction.

Schematic diagram representing the miR-124-mediated regulation of TLR4 signaling in the context of cocaine
References
Salter MW, Beggs S (2014) Sublime microglia: expanding roles for the guardians of the CNS. Cell 158(1):15–24. doi:10.1016/j.cell.2014.06.008
Ransohoff RM, Cardona AE (2010) The myeloid cells of the central nervous system parenchyma. Nature 468(7321):253–262. doi:10.1038/nature09615
Heiman A, Pallottie A, Heary RF, Elkabes S (2014) Toll-like receptors in central nervous system injury and disease: a focus on the spinal cord. Brain Behav Immun 42:232–245. doi:10.1016/j.bbi.2014.06.203
Pedras-Vasconcelos J, Puig M, Verthelyi D (2009) TLRs as therapeutic targets in CNS inflammation and infection. Front Biosci (Elite Ed) 1:476–487
O'Neill LA, Golenbock D, Bowie AG (2013) The history of Toll-like receptors—redefining innate immunity. Nat Rev Immunol 13(6):453–460. doi:10.1038/nri3446
Fitzgerald KA, Rowe DC, Barnes BJ, Caffrey DR, Visintin A, Latz E, Monks B, Pitha PM et al (2003) LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J Exp Med 198(7):1043–1055. doi:10.1084/jem.20031023
Papageorgiou IE, Lewen A, Galow LV, Cesetti T, Scheffel J, Regen T, Hanisch UK, Kann O (2016) TLR4-activated microglia require IFN-gamma to induce severe neuronal dysfunction and death in situ. Proc Natl Acad Sci U S A 113(1):212–217. doi:10.1073/pnas.1513853113
Rivest S (2003) Molecular insights on the cerebral innate immune system. Brain Behav Immun 17(1):13–19
Feinberg MW, Cao Z, Wara AK, Lebedeva MA, Senbanerjee S, Jain MK (2005) Kruppel-like factor 4 is a mediator of proinflammatory signaling in macrophages. J Biol Chem 280(46):38247–38258. doi:10.1074/jbc.M509378200
Pena-Altamira E, Prati F, Massenzio F, Virgili M, Contestabile A, Bolognesi ML, Monti B (2016) Changing paradigm to target microglia in neurodegenerative diseases: from anti-inflammatory strategy to active immunomodulation. Expert Opin Ther Targets 20(5):627–640. doi:10.1517/14728222.2016.1121237
Walker DG, Lue LF (2015) Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther 7(1):56. doi:10.1186/s13195-015-0139-9
Houdek HM, Larson J, Watt JA, Rosenberger TA (2014) Bacterial lipopolysaccharide induces a dose-dependent activation of neuroglia and loss of basal forebrain cholinergic cells in the rat brain. Inflamm Cell Signal 1(1). doi:10.14800/ics.47
Moehle MS, West AB (2015) M1 and M2 immune activation in Parkinson's disease: foe and ally? Neuroscience 302:59–73. doi:10.1016/j.neuroscience.2014.11.018
Calsolaro V, Edison P (2016) Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement 12(6):719–732. doi:10.1016/j.jalz.2016.02.010
Brites D, Vaz AR (2014) Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front Cell Neurosci 8:117. doi:10.3389/fncel.2014.00117
Lacagnina MJ, Rivera PD, Bilbo SD (2017) Glial and neuroimmune mechanisms as critical modulators of drug use and abuse. Neuropsychopharmacology 42(1):156–177. doi:10.1038/npp.2016.121
Guo ML, Liao K, Periyasamy P, Yang L, Cai Y, Callen SE, Buch S (2015) Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 11(7):995–1009. doi:10.1080/15548627.2015.1052205
Cui C, Shurtleff D, Harris RA (2014) Neuroimmune mechanisms of alcohol and drug addiction. Int Rev Neurobiol 118:1–12. doi:10.1016/B978-0-12-801284-0.00001-4
Liao K, Guo M, Niu F, Yang L, Callen SE, Buch S (2016) Cocaine-mediated induction of microglial activation involves the ER stress-TLR2 axis. J Neuroinflammation 13(1):33. doi:10.1186/s12974-016-0501-2
Sekine Y, Ouchi Y, Sugihara G, Takei N, Yoshikawa E, Nakamura K, Iwata Y, Tsuchiya KJ et al (2008) Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci 28(22):5756–5761. doi:10.1523/JNEUROSCI.1179-08.2008
Grace PM, Strand KA, Galer EL, Urban DJ, Wang X, Baratta MV, Fabisiak TJ, Anderson ND et al (2016) Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc Natl Acad Sci U S A 113(24):E3441–E3450. doi:10.1073/pnas.1602070113
Crews FT, Sarkar DK, Qin L, Zou J, Boyadjieva N, Vetreno RP (2015) Neuroimmune function and the consequences of alcohol exposure. Alcohol Res 37(2):331–341 344-351
Snider SE, Hendrick ES, Beardsley PM (2013) Glial cell modulators attenuate methamphetamine self-administration in the rat. Eur J Pharmacol 701(1–3):124–130. doi:10.1016/j.ejphar.2013.01.016
Chen H, Uz T, Manev H (2009) Minocycline affects cocaine sensitization in mice. Neurosci Lett 452(3):258–261. doi:10.1016/j.neulet.2009.01.078
Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA, Pomrenze MB, Galer EL et al (2015) DAT isn't all that: cocaine reward and reinforcement require toll-like receptor 4 signaling. Mol Psychiatry 20(12):1525–1537. doi:10.1038/mp.2014.177
Carthew RW, Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136(4):642–655. doi:10.1016/j.cell.2009.01.035
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136(2):215–233. doi:10.1016/j.cell.2009.01.002
Hobert O (2008) Gene regulation by transcription factors and microRNAs. Science 319(5871):1785–1786. doi:10.1126/science.1151651
Cardoso AL, Guedes JR, de Lima MC (2016) Role of microRNAs in the regulation of innate immune cells under neuroinflammatory conditions. Curr Opin Pharmacol 26:1–9. doi:10.1016/j.coph.2015.09.001
Svahn AJ, Giacomotto J, Graeber MB, Rinkwitz S, Becker TS (2016) miR-124 contributes to the functional maturity of microglia. Dev Neurobiol 76(5):507–518. doi:10.1002/dneu.22328
Ponomarev ED, Veremeyko T, Barteneva N, Krichevsky AM, Weiner HL (2011) MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med 17(1):64–70. doi:10.1038/nm.2266
Gascon E, Lynch K, Ruan H, Almeida S, Verheyden JM, Seeley WW, Dickson DW, Petrucelli L et al (2014) Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat Med 20(12):1444–1451. doi:10.1038/nm.3717
Kanagaraj N, Beiping H, Dheen ST, Tay SS (2014) Downregulation of miR-124 in MPTP-treated mouse model of Parkinson's disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience 272:167–179. doi:10.1016/j.neuroscience.2014.04.039
Wang H, Ye Y, Zhu Z, Mo L, Lin C, Wang Q, Wang H, Gong X et al (2016) MiR-124 regulates apoptosis and autophagy process in MPTP model of Parkinson's disease by targeting to Bim. Brain Pathol 26(2):167–176. doi:10.1111/bpa.12267
Veremeyko T, Siddiqui S, Sotnikov I, Yung A, Ponomarev ED (2013) IL-4/IL-13-dependent and independent expression of miR-124 and its contribution to M2 phenotype of monocytic cells in normal conditions and during allergic inflammation. PLoS One 8(12):e81774. doi:10.1371/journal.pone.0081774
Ni M, Aschner M (2010) Neonatal rat primary microglia: isolation, culturing, and selected applications. Curr Protoc Toxicol Chapter 12:Unit 12 17. doi:10.1002/0471140856.tx1217s43
Black AR, Black JD, Azizkhan-Clifford J (2001) Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol 188(2):143–160. doi:10.1002/jcp.1111
Kaushik DK, Thounaojam MC, Kumawat KL, Gupta M, Basu A (2013) Interleukin-1beta orchestrates underlying inflammatory responses in microglia via Kruppel-like factor 4. J Neurochem 127(2):233–244. doi:10.1111/jnc.12382
Kaushik DK, Gupta M, Das S, Basu A (2010) Kruppel-like factor 4, a novel transcription factor regulates microglial activation and subsequent neuroinflammation. J Neuroinflammation 7:68. doi:10.1186/1742-2094-7-68
Kaushik DK, Mukhopadhyay R, Kumawat KL, Gupta M, Basu A (2012) Therapeutic targeting of Kruppel-like factor 4 abrogates microglial activation. J Neuroinflammation 9:57. doi:10.1186/1742-2094-9-57
Rosenzweig JM, Glenn JD, Calabresi PA, Whartenby KA (2013) KLF4 modulates expression of IL-6 in dendritic cells via both promoter activation and epigenetic modification. J Biol Chem 288(33):23868–23874. doi:10.1074/jbc.M113.479576
Van Dyke C, Barash PG, Jatlow P, Byck R (1976) Cocaine: plasma concentrations after intranasal application in man. Science 191(4229):859–861
Stephens BG, Jentzen JM, Karch S, Mash DC, Wetli CV (2004) Criteria for the interpretation of cocaine levels in human biological samples and their relation to the cause of death. Am J Forensic Med Pathol 25(1):1–10
Kalasinsky KS, Bosy TZ, Schmunk GA, Ang L, Adams V, Gore SB, Smialek J, Furukawa Y et al (2000) Regional distribution of cocaine in postmortem brain of chronic human cocaine users. J Forensic Sci 45(5):1041–1048
Karthikeyan A, Patnala R, Jadhav SP, Eng-Ang L, Dheen ST (2016) MicroRNAs: key players in microglia and astrocyte mediated inflammation in CNS pathologies. Curr Med Chem 23(30):3528–3546
Thome AD, Harms AS, Volpicelli-Daley LA, Standaert DG (2016) microRNA-155 regulates alpha-synuclein-induced inflammatory responses in models of Parkinson disease. J Neurosci 36(8):2383–2390. doi:10.1523/JNEUROSCI.3900-15.2016
Brites D, Fernandes A (2015) Neuroinflammation and depression: microglia activation, extracellular microvesicles and microRNA dysregulation. Front Cell Neurosci 9:476. doi:10.3389/fncel.2015.00476
Guo ML, Periyasamy P, Liao K, Kook YH, Niu F, Callen SE, Buch S (2016) Cocaine-mediated downregulation of microglial miR-124 expression involves promoter DNA methylation. Epigenetics 11(11):819–830. doi:10.1080/15592294.2016.1232233
Lewitus GM, Konefal SC, Greenhalgh AD, Pribiag H, Augereau K, Stellwagen D (2016) Microglial TNF-alpha suppresses cocaine-induced plasticity and behavioral sensitization. Neuron 90(3):483–491. doi:10.1016/j.neuron.2016.03.030
Ma C, Li Y, Li M, Deng G, Wu X, Zeng J, Hao X, Wang X et al (2014) microRNA-124 negatively regulates TLR signaling in alveolar macrophages in response to mycobacterial infection. Mol Immunol 62(1):150–158. doi:10.1016/j.molimm.2014.06.014
Ma C, Li Y, Zeng J, Wu X, Liu X, Wang Y (2014) Mycobacterium bovis BCG triggered MyD88 induces miR-124 feedback negatively regulates immune response in alveolar epithelial cells. PLoS One 9(4):e92419. doi:10.1371/journal.pone.0092419
Sun Y, Li Q, Gui H, Xu DP, Yang YL, Su DF, Liu X (2013) MicroRNA-124 mediates the cholinergic anti-inflammatory action through inhibiting the production of pro-inflammatory cytokines. Cell Res 23(11):1270–1283. doi:10.1038/cr.2013.116
Yao H, Yang Y, Kim KJ, Bethel-Brown C, Gong N, Funa K, Gendelman HE, Su TP et al (2010) Molecular mechanisms involving sigma receptor-mediated induction of MCP-1: implication for increased monocyte transmigration. Blood 115(23):4951–4962. doi:10.1182/blood-2010-01-266221
Asanuma M, Miyazaki I, Higashi Y, Tsuji T, Ogawa N (2004) Specific gene expression and possible involvement of inflammation in methamphetamine-induced neurotoxicity. Ann N Y Acad Sci 1025:69–75. doi:10.1196/annals.1316.009
Goncalves J, Martins T, Ferreira R, Milhazes N, Borges F, Ribeiro CF, Malva JO, Macedo TR et al (2008) Methamphetamine-induced early increase of IL-6 and TNF-alpha mRNA expression in the mouse brain. Ann N Y Acad Sci 1139:103–111. doi:10.1196/annals.1432.043
Wisor JP, Schmidt MA, Clegern WC (2011) Cerebral microglia mediate sleep/wake and neuroinflammatory effects of methamphetamine. Brain Behav Immun 25(4):767–776. doi:10.1016/j.bbi.2011.02.002
Piechota M, Korostynski M, Solecki W, Gieryk A, Slezak M, Bilecki W, Ziolkowska B, Kostrzewa E et al (2010) The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Biol 11(5):R48. doi:10.1186/gb-2010-11-5-r48
Renthal W, Maze I, Krishnan V, Covington HE 3rd, Xiao G, Kumar A, Russo SJ, Graham A et al (2007) Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56(3):517–529. doi:10.1016/j.neuron.2007.09.032
Ahmed SH, Lutjens R, van der Stap LD, Lekic D, Romano-Spica V, Morales M, Koob GF, Repunte-Canonigo V et al (2005) Gene expression evidence for remodeling of lateral hypothalamic circuitry in cocaine addiction. Proc Natl Acad Sci U S A 102(32):11533–11538. doi:10.1073/pnas.0504438102
Little KY, Ramssen E, Welchko R, Volberg V, Roland CJ, Cassin B (2009) Decreased brain dopamine cell numbers in human cocaine users. Psychiatry Res 168(3):173–180. doi:10.1016/j.psychres.2008.10.034
Loftis JM, Choi D, Hoffman W, Huckans MS (2011) Methamphetamine causes persistent immune dysregulation: a cross-species, translational report. Neurotox Res 20(1):59–68. doi:10.1007/s12640-010-9223-x
Beardsley PM, Hauser KF (2014) Glial modulators as potential treatments of psychostimulant abuse. Adv Pharmacol 69:1–69. doi:10.1016/B978-0-12-420118-7.00001-9
Deo M, Yu JY, Chung KH, Tippens M, Turner DL (2006) Detection of mammalian microRNA expression by in situ hybridization with RNA oligonucleotides. Dev Dyn 235(9):2538–2548. doi:10.1002/dvdy.20847
Krichevsky AM, Sonntag KC, Isacson O, Kosik KS (2006) Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells 24(4):857–864. doi:10.1634/stemcells.2005-0441
Chandrasekar V, Dreyer JL (2009) microRNAs miR-124, let-7d and miR-181a regulate cocaine-induced plasticity. Mol Cell Neurosci 42(4):350–362. doi:10.1016/j.mcn.2009.08.009
Chandrasekar V, Dreyer JL (2011) Regulation of MiR-124, Let-7d, and MiR-181a in the accumbens affects the expression, extinction, and reinstatement of cocaine-induced conditioned place preference. Neuropsychopharmacology 36(6):1149–1164. doi:10.1038/npp.2010.250
Bosch PJ, Benton MC, Macartney-Coxson D, Kivell BM (2015) mRNA and microRNA analysis reveals modulation of biochemical pathways related to addiction in the ventral tegmental area of methamphetamine self-administering rats. BMC Neurosci 16:43. doi:10.1186/s12868-015-0186-y
Mizuo K, Katada R, Okazaki S, Tateda K, Watanabe S, Matsumoto H (2012) Epigenetic regulation of MIR-124 under ethanol dependence and withdrawal. Nihon Arukoru Yakubutsu Igakkai Zasshi 47(3):155–163
Bachtell R, Hutchinson MR, Wang X, Rice KC, Maier SF, Watkins LR (2015) Targeting the Toll of drug abuse: the translational potential of Toll-like receptor 4. CNS Neurol Disord Drug Targets 14(6):692–699
Louw AM, Kolar MK, Novikova LN, Kingham PJ, Wiberg M, Kjems J, Novikov LN (2016) Chitosan polyplex mediated delivery of miRNA-124 reduces activation of microglial cells in vitro and in rat models of spinal cord injury. Nanomedicine 12(3):643–653. doi:10.1016/j.nano.2015.10.011
Acknowledgements
This work was supported by NIH grants: DA043138, DA033150, DA035203.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Figure 1
(DOCX 151 kb)
Rights and permissions
About this article

Cite this article
Periyasamy, P., Liao, K., Kook, Y.H. et al. Cocaine-Mediated Downregulation of miR-124 Activates Microglia by Targeting KLF4 and TLR4 Signaling. Mol Neurobiol 55, 3196–3210 (2018). https://doi.org/10.1007/s12035-017-0584-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0584-5