
Abstract
The molecular mechanisms responsible for the loss of dopaminergic neurons in Parkinson’s disease (PD) remain obscure. Loss of function of E3 ubiquitin ligases is associated with mitochondria dysfunction, dysfunction of protein degradation, and α-synuclein aggregation, which are major contributors to neurodegeneration in PD. Recent research has thus focused on E3 ubiquitin ligase glycoprotein 78 (GP78); however, the role of GP78 in PD pathogenesis remains unclear. Notably, cyclin-dependent kinase 5 (CDK5) controls multiple cellular events in postmitotic neurons, and CDK5 activity has been implicated in the pathogenesis of PD. Thus, we addressed the relationship between CDK5 and GP78 in MPTP-based PD models. We found that GP78 expression is decreased in MPTP-based cellular and animal PD models, and CDK5 directly phosphorylated GP78 at Ser516, which promoted the ubiquitination and degradation of GP78. Importantly, overexpression of GP78 or interference of GP78 Ser516 phosphorylation protected neurons against MPP+-induced cell death. Thus, our research reveals that the CDK5-GP78 pathway is involved in the pathogenesis of PD and could be a novel candidate drug target for the treatment of PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is characterized by the progressive loss of dopamine neurons in the substantia nigra pars compacta (SNpc) and the presence of Lewy bodies [1, 2]. Mutations in E3 ubiquitin ligases are a major cause of familial PD [3, 4]. These mutations impair E3 ligase activity and are associated with mitochondria dysfunction, alpha-synuclein aggregation, and dysfunction of protein degradation [5,6,7]. Studies on PINK1 and Parkin showed that mitochondrial dysfunction is a major contributor of neurodegeneration in PD [8,9,10,11]. However, molecular mechanisms responsible for loss of dopaminergic neurons in PD remain obscure. Recent research has thus focused on other E3 ubiquitin ligases, such as glycoprotein 78 (GP78) and Siah-1 [12, 13].
GP78 plays a critical role in endoplasmic reticulum-associated degradation (ERAD) [14, 15]. Before the discovery of its E3 ubiquitin ligase activity, GP78 was identified as a receptor for autocrine motility factor (AMF) [16]. The C-terminus of GP78 contains a catalytic RING finger, CUE, and ube2g2 E2 enzyme and p97 binding domains [17, 18]. GP78 ubiquitylates several substrates, including HMG CoA reductase, ApoB lipoprotein, SOD1, Ataxin3, and mutant cystic fibrosis transmembrane conductance regulator [19,20,21,22]. Recent evidence indicates that GP78 promotes the degradation of mitochondrial fission proteins and induces mitophagy [23, 24]. GP78 downregulation is found in many neurodegenerative diseases; however, the role of GP78 in PD pathogenesis remains unclear.
Phosphorylation is an important regulation mechanism for E3 ubiquitin ligases [25, 26]. Cyclin-dependent kinase 5 (CDK5) controls multiple cellular events in postmitotic neurons, and its activity has been implicated in the pathogenesis of several neurodegenerative disorders such as Alzheimer’s disease (AD), Huntington’s disease (HD), and PD [27,28,29,30]. CDK5 phosphorylation by Parkin decreases its E3 ubiquitin ligase activity and results in accumulation of toxic Parkin substrates in PD [31]. Here, we addressed the relationship between CDK5 and GP78 and investigated the mechanism of GP78 downregulation in a PD model. Results indicate that GP78 may be relevant to PD pathogenesis.
Materials and Methods
Reagents and Antibodies
Roscovitine, NH4Cl, MG132, adenosine triphosphate (ATP), 1-methyl-4-phenylpyridinium (MPP+), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), and lipopolysaccharide (LPS) were obtained from Sigma-Aldrich. The antibodies against GP78 and tyrosine hydroxylase were purchased from Abcam, and the antibodies against HA, GST, flag, and myc-tag were purchased from Cell Signaling Technology. The anti-CDK5, anti-p35, and anti-ubiquitin antibodies were purchased from Santa Cruz Biotechnology. The actin antibody was purchased from Sigma-Aldrich. All of the secondary antibodies used for immunoblotting or immunofluorescence were procured from Jackson ImmunoResearch. The antibodies were used according to the instructions recommended by the manufacturers.
Plasmids and Recombinant Proteins
The plasmids encoding full-length GP78, PCDNA-HA-CDK5, and myc-P35 plasmids were purchased from Addgene. Conventional molecular biological techniques were used to generate the following expression constructs with N-terminal GST or flag tags: GP78, GP78 S516D, △C GP78, △C GP78 S516D, and △C GP78 S516A. Mutations were performed using the Fast Mutagenesis System (TransGen Biotech). The GST-tagged fusion proteins were induced and purified from BL21 (DE3)-competent cells (TransGen Biotech) according to the instructions recommended by the manufacturers.
Cell Culture and Transfection
Neurons were cultured from Sprague-Dawley rats at embryonic 16–18 days on 6-wells or 24-wells plates that were precoated with poly-d-lysine (Sigma-Aldrich). The primary neurons were maintained in Neurobasal medium (Invitrogen) containing 2% B27 supplement (Invitrogen), 0.5 mM l-glutamine, and 25 μM glutamate, penicillin (100 g/ml), and streptomycin (100 g/ml). After 24 h of plating, cell division inhibitor Ara-C was added to the medium at a final concentration of 10 μM to remove glial cells. All the treatments or transfections were performed after plating 7 days in vitro. HEK293 cells were cultured in DMEM with 10% FBS and SH-SY5Y cells were cultured in DMEM/F12 with 10% FBS. HEK293 and SH-SY5Y cells were transfected with the indicated plasmids using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions.
Immunoprecipitation and Immunoblot Analysis
Primary cortical neurons and HEK293 cells were lysed in RIPA buffer with protease inhibitors. Lysates were incubated for 6 h at 4 °C with corresponding antibody and then were collected with protein G plus/protein A-agarose at 4 °C for 2 h. After being washed three times, the immune complexes were boiled for 5 min with SDS sample buffer. The immunocomplexes were then subjected to SDS-PAGE, transferred to a PVDF membrane, and immunoblotted with the indicated antibodies.
Immunofluorescence Microscopy
For primary neurons or cell lines, they were cultured on coverslips and fixed with 4% paraformaldehyde, then permeabilized with 0.3% Triton X-100 in PBS, blocked in 5% normal goat serum, and stained with anti-GP78 or anti-TH (tyrosine hydroxylase) antibody, and subsequently incubated with the corresponding fluorescent secondary antibodies, containing DyLight 488 and DyLight 594 (Jackson ImmunoResearch Inc.). For preparing mice brain tissue, mice were anesthetized with an overdose of chloral hydrate and sacrificed. All the brains were fixed in 4% paraformaldehyde for 24 h at 4 °C, and dehydration was performed with a 30% sucrose solution for 48 h. Then, the brain tissue was sliced into 30-μm sections using a freezing microtome (Leica CM1850) and processed for immunostaining. The nucleus of all samples was stained with DAPI (Sigma-Aldrich).
CDK5/p35 In Vitro Kinase Assay
The in vitro CDK/p35 kinase assay was performed according to the instructions recommended by the manufacturers (Millipore). Briefly, 2 μg of purified △C GP78 and △C GP78 S516A GST-fusion proteins were incubated with active CDK5/p35 in CDK kinase buffer (8 mM MOPS/NaOH, pH 7.0, 200 nM EDTA),and 20 μM ATP. The reaction was stopped after incubation at 30 °C for 30 min and boiled for 5 min with SDS sample buffer. The phosphorylation of substrates following SDS-PAGE analysis was detected by anti-Phos S/TP antibody and phos-tag SDS-PAGE.
In Vivo Ubiquitination Assay
HEK293 cells s were co-transfected with HA-ubiquitin and flag-GP78 or flag-GP78-S516D. After 24 h overexpression, HEK293 cells were treated with 20 μM MG132 for 6 h. Primary cortical neurons were treated with 5 μM MG132 for 24 h in the absence or presence of MPP+. HEK293 cells and neurons were lysed in RIPA buffer, and immunoprecipitation was performed with corresponding antibody and protein G plus/protein A-agarose. The samples were then subjected to SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblotted with the anti-ubiquitin antibody.
RT-PCR Analysis
The total RNA was extracted from neurons using TRIzol reagent (Invitrogen). Total RNA (1 μg) of each sample was reverse-transcribed using EasyScript First-Strand cDNA Synthesis SuperMix (TransGen) in a 20-μl volume. For PCR, the amplification was carried out in a total volume of 25 μl containing 0.5 μl of each primer, 1 μl of cDNA, 12.5 μl Taq Master Mix, and H2O to 25 μl, according to the manufacturer’s instructions. GAPDH was amplified as a reference standard for rat. Primers used were as follows: for rat GAPDH forward: 5′-AACAACCTGTTGCTGTAGCC-3′, reverse: 5′-ACCCACTCCTCCACCTTTGA-3′; for rat GP78 forward: 5′-TGCATTGAGCTGGGAGTTGC-3′, reverse: 5′-ATTTCGATGGGTCTCGGATTG-3′. PCR cycling conditions were 5 min 94 °C followed by 30 cycles of 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 60 s and a final extension at 72 °C for 10 min.
Animals and MPTP Injections
Sprague-Dawley rats and C57BL/6 mice were used in this study. MPTP was administered to 8-week-old male C57BL/6 mice (26–30 g) as previously reported. Briefly, the mice received a single intraperitoneal injection of 20 mg/kg MPTP once per day for five consecutive days and 0.9% saline was used as the vehicle. The ventral midbrains were extracted on indicated days, and samples were processed for analysis.
Statistical Analysis
All of the data were presented as the mean ± standard error of the mean (SEM). The statistical analyses were performed with two-tailed Student’s t tests. Analyses were carried out by GraphPad Prism5 software. P value of less than 0.05 was considered to be statistically significant,*P <0.05.
Results
GP78 Expression Is Decreased in Cellular and Animal Models of PD
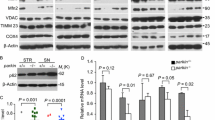
GP78 was expressed in tyrosine hydroxylase positive (TH+) dopaminergic neurons of the SNpc (Fig. 1a). To determine GP78 involvement in the pathogenesis of PD, we measured GP78 expressions in two PD models. GP78 expression was downregulated in the SNpc of MPTP-treated PD mice (Fig. 1b, c). Neurons exposed to MPP+ exhibited reduced GP78 expression (Fig. 1d, e) with no change at the messenger RNA (mRNA) level (Fig. 1f). These results suggest that regulation of GP78 downregulation is posttranslational.

GP78 is degraded in MPTP mouse and MPP+ cell models of PD. a, b Mouse brain slices were fixed and co-stained with anti-GP78 (green) and anti-TH (red). DNA was stained with DAPI (blue). Mice were injected with MPTP (20 mg/kg body weight) or saline (control) once a day for 5 days. c, d Brain lysates were prepared from SNpcGP78 that was determined by Western blot using an anti-GP78 antibody (n = 3). Treatment with 100 Μ MPP+ decreased GP78 levels. e Immunocytochemistry with anti-GP78 (red) and DAPI (blue). The scale bar represents 100 and 50 μm, respectively. f GP78 mRNA levels are unaffected by MPP+. Data are shown as the mean ± SEM of triplicate experiments. **P < 0.01, ***P < 0.001
Upregulation of CDK5 Activity Increases GP78 Degradation
Increased CDK5 activity was implicated in the pathogenesis of PD. Thus, we investigated if GP78 degradation was associated with CDK5 activity. CDK5 overexpression and its activator p35 prompted the degradation of flag-tagged GP78 co-expressed in HEK293 cells (Fig. 2a, b) and endogenous GP78 in neuroblastoma SH-SY5Y cells (Fig. 2c). Moreover, the highly selective CDK5 inhibitor roscovitine inhibited GP78 degradation in a cellular MPP+-induced PD model (Fig. 2d). These findings indicate that the upregulation of CDK5 activity directly promotes GP78 degradation.

CDK5 prompts GP78 degradation. a HEK293 cells were transfected with flag-GP78 with or without HA-CDK5 and myc-p35. Flag-GP78 was detected by Western blot using an anti-flag antibody. b Immunocytochemistry with anti-flag (green) and DAPI (blue). The scale bar represents 20 μm. c SH-SY5Y cells were transfected with HA-CDK5 and myc-p35. GP78 was determined by Western blot using anti-GP78. d MPP+-exposed cells were treated with 10 μM roscovitine. *P < 0.05, **P < 0.01
CDK5 Directly Phosphorylates GP78 at Ser516 In Vitro and In Vivo
We performed co-immunoprecipitation experiments to determine direct binding of GP78 with CDK5 and p35 in primary neurons. GP78 directly interacted with p35 but not CDK5 (Fig. 3a), indicating that GP78 associates with a CDK5/p35 complex in vivo. CDK5 increased GP78 phosphorylation in HEK293 cells co-transfected with full-length GP78 in the presence of HA-CDK5 and myc-p35 (Fig. 3b). To verify a kinase-substrate relationship, we analyzed the protein sequence of GP78 with two bioinformatics tools: GPS 2.1 and ScanSite. A candidate site was found at position 516 of GP78. This serine residue (Ser516)was highly conserved across sequences of all available species in the examined databases and identified in mass spectrometry assays of myr-RPALNSPVERP (amino acid sequence 511–521 of GP78) (Fig. 3c).We designed a Δc-GP78 (amino acid sequence 450–643 of GP78) and a mutant of Δc-GP78 in which the Ser516 was mutated to alanine (Ser516A). The CDK5/p35 complex phosphorylated Δc-GP78 (WT) but not the Ser516A mutant in vitro (Fig. 3d, e). The Ser516A mutant had lower phosphorylation levels in vivo when compared with the WT (Fig. 3f). Thus, GP78 is a direct substrate of CDK5/p35 complex in vitro and in cultured cells.

GP78 is a substrate of CDK5. a Co-immunoprecipitation of GP78 with CDK5 and p35. Primary neurons lysates were immunoprecipitated with an anti-GP78 antibody, followed by immunoblotting with anti-CDK5 and anti-p35. Data are shown as the mean ± SEM of triplicate experiments. b HEK293 cells were transfected with full-length GP78 with or without HA-CDK5 and myc-p35, then immunoprecipitated with anti-GP78 and immunoblotted with anti-Phos S/TP. c Mass spectrometry assays. CDK5/p35 phosphorylates myr-RPALNSPVERP in a kinase assay. d CDK5/p35 phosphorylates GP78 in a kinase assay. Purified GST-Δc-GP78 (WT) or GST-Δc-GP78-Ser516A fusion proteins were mixed with active CDK5/p35. Phosphorylation signals were analyzed using anti-phosS/TP. e Phos-tag SDS-PAGE. Western blotting using an antibody against GST showed changes in the GST-△c-GP78 (WT) and GST-△c-GP78-Ser516A phosphorylation profile. f HEK293 cells were co-transfected with flagΔc-GP78 (WT) or flag-Δc-GP78-Ser516A, HA-CDK5, and myc-p35. HEK293 cells lysates were immunoprecipitated with an anti-flag antibody, then immunoblotted with anti-phosS/TP antibody
GP78 Phosphorylation at Ser516 by CDK5 Triggers GP78 Degradation via the Ubiquitin-Proteasome Pathway
To elucidate the molecular mechanism of phosphorylation-dependent degradation of GP78, we used NH4Cl (autophagy inhibitor) and MG132 (ubiquitin-proteasome pathway inhibitor) to block MPP+-induced degradation of GP78 in primary cultured neurons. MG132 effectively protected GP78 against MPP+-induced degradation, whereas NH4Cl had little effect (Fig. 4a). The phosphorylation and ubiquitination of GP78 were both significantly increased in MPP+-treated primary cortical neurons (Fig. 4b). To determine the effect of phosphorylation on GP78 ubiquitination, we designed a mutant phosphomimetic form of GP78 by mutating Ser516 to aspartate (S516D). We transfected WT or S516D GP78 into HEK293 cells and added MG132 to block proteasome degradation. The GP78-S516D mutant showed higher ubiquitination when compared with the GP78-WT (Fig. 4c). Similar results were found for Δc-GP78 and Δc-GP78-S516D (Fig. 4d). We also studied the effect of phosphorylation at Ser516 on the endogenous degradation of GP78. We transfected WT or S516D GP78 into HEK293 cells and added cycloheximide to block protein synthesis. Phosphomimetic S516D degradation was much faster than WT GP78 (Fig. 4e, f). Thus, phosphorylation at Ser516 of GP78 by CDK5 triggers the ubiquitination of GP78 and its subsequent degradation via the ubiquitin-proteasome pathway.

GP78 phosphorylation at Ser516 prompts its ubiquitination and degradation. a MPP+-exposed cells were treated with 10 nM NH4Cl and 5 μM MG132 for 24 h. Cell lysates were analyzed by immunoblotting with anti-GP78. b Primary cortical neurons treated with 100 μM MPP+ and 5 μM MG132 for 24 h. Cell lysates were immunoprecipitated with anti-GP78 then immunoblotted with anti-phosS/TP and anti-ub. c HEK293 cells were co-transfected with HA-ub and flag-GP78 (WT) or flag-GP78-Ser516D (Ser516D). After 24 h, cells were treated with 20 μM MG132 for 6 h. Ubiquitination of WT or Ser516D was measured by IP-immunoblotting. d HEK293 cells were transfected with co-transfected with HA-ub and flagΔc-GP78 or flag-Δc-GP78-Ser516D. e HEK293 cells were transfected with flag-GP78-Ser516D or flag-GP78. After 24 h, cycloheximide (CHX) was added to inhibit protein synthesis. The level of flag-GP78-Ser516D and flag-GP78 was determined by immunoblotting with anti-flag. f Data quantification in e. The degradation curve of GP78 protein. Data are shown as mean ± SEM of triplicate experiments
GP78 Overexpression or Reduced GP78 Ser516 Phosphorylation Protected Neurons Against MPP+-Induced Cell Death
GP78 was downregulated in an MPTP-induced mouse model and MPP+-induced cell model of PD. We generated a GP78 adeno-associated virus (AAV) that overexpressed GP78 in primary neurons (Fig. 5a). GP78 overexpression protected neurons from MPP+-induced neuronal cell death (Fig. 5b). Next, we generated a synthetic myristoylated (myr) peptide (RPALNSPVERP, amino acid sequence 511–521 of GP78) that mimics the endogenous substrate region and, thus, interferes with the phosphorylation of GP78 at Ser516 via homologous competitive inhibition. RPALNSPVERP had little toxic effect on normal neurons (Fig. 5c) but effectively protected neurons against MPP+-induced neuronal cell death (Fig. 5d).

GP78 ameliorates MPP+-mediated neuronal cell death. a GP78 was overexpressed using GP78 AAV. b Primary cortical neurons were treated with GP78 AAV and AAV vector. After 12 h, 200 μΜ MPP+ was added for 36 h. Survival was detected by MTT. c Primary cortical neurons were treated with 0.1, 1, or 10 μΜ myr-RPALNSPVERP (peptide group) or a scrambled control peptide (scrambled group) for 36 h. d Primary cortical neurons were treated with 200 μΜ MPP+ and myr-RPALNSPVERP (0.1 or 1 μΜ) or scrambled control peptide for 36 h. *P < 0.05, ***P < 0.001
Discussion
Previously published studies have demonstrated that E3 ubiquitin ligase wildly contributes to the abnormal accumulation of misfolded proteins in many neurodegenerative diseases, such as Parkin for α-synuclein in PD. The findings observed in this study mirror those of the previous studies that have examined the effect of E3 ubiquitin ligase GP78 in PD. Above all, our data show that GP78, which expresses in the dopaminergic neurons of the SNpc, was significantly decreased in the classical PD models such as MPP+-induced primary cortical neurons and MPTP-treated mice. Moreover, the crucial proline-directed serine-threonine kinase CDK5 was found to robustly accelerate the degradation of GP78 on the condition that GP78 co-overexpressed with CDK5 and molecular chaperone p35 in HEK293. In contrast, the degradation of GP78 was obviously blocked by employing the selective chemical inhibitor (roscovitine) of CDK5. Subsequently, we performed a series of experiments to suggest that CDK5 could directly phosphorylate GP78 at Serine 516 in vitro and in vivo. Notably, the phosphorylation of GP78 by CDK5 promotes its degradation through the ubiquitin-proteasome pathway. We also found that AAV-mediated exogenous overexpression of GP78 effectively ameliorated the MPP+ insults in primary cortical neurons. Similarly, the myr-GP78 peptide, which competitively inhibits the phosphorylation of GP78 by CDK5, could also protect neurons against MPP+-induced neuronal death.
Growing evidence indicates that protein phosphorylation is an important regulator of E3 ubiquitin ligases [32]. Our previous study shows that CDK5 directly phosphorylates Cx43, and promotes its proteasome-dependent degradation in neurons [33]. MEKK1 phosphorylates Notch1-IC, which, in consequence, is recognized by Fbw7 E3 ligase and facilitates its degradation through the ubiquitin-proteasomal pathway in human breast cancer [34]. One of the more significant findings to emerge from this study is that CDK5 mediates the decrease of GP78 protein by going through phosphorylation-dependent ubiquitination and degradation pathway. In accordance with the present results, previous studies have demonstrated that phosphorylation-dependent ubiquitination and degradation pathway regulates tau-mediated Aβ toxicity in AD [35], colony formation in prostate cancer [36], and mitochondrial apoptosis in PD [37]. The PINK1/Parkin pathway, as the mostly classical phosphorylation-dependent ubiquitination and degradation pathway (PINK1 as kinase for the substrate phosphorylation and Parkin as E3 ligase for the substrate ubiquitination) during the progression of PD, is a central factor in regulating many signaling pathways controlling neuron metabolism, apoptosis, and death. Interestingly, the results of this study indicate that CDK5 is able to phosphorylate GP78 at site S516, and promotes its ubiquitination and degradation in the process of dopaminergic neuron death.
During the past decades, there has been an increasing amount of literature on the critical role of CDK5 in chronic neurodegenerative disease like AD. CDK5 is involved in multiple roles during neuronal death and has been established as a key mediator of tau hyperphosphorylation and neurofibrillary pathology, which are the two major characteristics of AD [38]. Notably, silencing of CDK5 by RNA inference (RNAi) could reduce the phosphorylation of tau and decrease the number of neurofibrillary tangles in the hippocampus [39]. More importantly, the Ser40, Thr121, and Ser163 triple phosphorylation of Cdh1 by CDK5 was necessary and sufficient for the E3 ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C) activation, cyclin B1 protein stabilization, and neuronal apoptotic death after the excitotoxicity of NMDAR stimulation, which commonly occurs in AD [40]. In conclusion, understanding the exact mechanism of CDK5 in the cellular biological process, especially for the regulation of E3 ubiquitin ligase, was crucial to ameliorate several neurodegenerative diseases.
However, more research about the mechanism of degradation of GP78 in neuronal death needs to be undertaken to clearly understand the association between GP78 and PD. GP78 is an E3 ubiquitin ligase that targets proteins for proteasome degradation through the ERAD pathway to protect cells from ER stress. Although our results have shown that overexpression of GP78 could rescue neuronal death, the downstream mechanism about cell survival is still unknown. A large and growing body of literature has investigated that the target proteins of GP78 are BOK (BCL-2 ovarian killer), SOD1 (superoxide dismutase-1), and Mfn1 (mitofusin-1, a mediator of mitochondrial fusion) [22, 23, 41]. In this study, we have also suggested that E3 ligase GP78 itself could be a substrate of ubiquitination by certain E3 ubiquitin ligases. There are lines of evidence proving that TRIM25 (tripartite motif-containing protein 25) has been identified as a chief contributing factor for the ubiquitination and degradation of GP78 [42]. In a future study, we plan to focus on whether the GP78-Mfn1 pathway and upstream E3 ubiquitin ligase TRIM25 are involved in dopaminergic neuron loss of PD.
Taken together, this finding has important implications for elucidating a novel molecular mechanism of the neuronal death and providing some therapeutic strategies with respect to clinically treat PD. Overexpressing GP78 or interfering the CDK5-GP78 pathway in midbrain dopaminergic neurons may open up new avenues for uncovering neuroprotective pathways.
References
Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302(5646):819–822. doi:10.1126/science.1087753
Moore DJ, West AB, Dawson VL, Dawson TM (2005) Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28:57–87. doi:10.1146/annurev.neuro.28.061604.135718
Cookson MR (2005) The biochemistry of Parkinson’s disease. Annu Rev Biochem 74:29–52. doi:10.1146/annurev.biochem.74.082803.133400
Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K et al (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25(3):302–305. doi:10.1038/77060
Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K et al (2001) Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 50(3):293–300
Khan NL, Graham E, Critchley P, Schrag AE, Wood NW, Lees AJ, Bhatia KP, Quinn N (2003) Parkin disease: a phenotypic study of a large case series. Brain 126(Pt 6):1279–1292
Ren Y, Zhao J, Feng J (2003) Parkin binds to alpha/beta tubulin and increases their ubiquitination and degradation. J Neurosci 23(8):3316–3324
Nardin A, Schrepfer E, Ziviani E (2016) Counteracting PINK/Parkin deficiency in the activation of mitophagy: a potential therapeutic intervention for Parkinson’s disease. Curr Neuropharmacol 14(3):250–259
Jones R (2010) The roles of PINK1 and Parkin in Parkinson’s disease. PLoS Biol 8(1):e1000299. doi:10.1371/journal.pbio.1000299
Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H et al (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open Biol 2(5):120080. doi:10.1098/rsob.120080
Zhuang N, Li L, Chen S, Wang T (2016) PINK1-dependent phosphorylation of PINK1 and Parkin is essential for mitochondrial quality control. Cell Death Dis 7(12):e2501. doi:10.1038/cddis.2016.396
Lee JT, Wheeler TC, Li L, Chin LS (2008) Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum Mol Genet 17(6):906–917. doi:10.1093/hmg/ddm363
Cai ZL, Xu J, Xue SR, Liu YY, Zhang YJ, Zhang XZ, Wang X, Wu FP et al (2015) The E3 ubiquitin ligase seven in absentia homolog 1 may be a potential new therapeutic target for Parkinson’s disease. Neural Regen Res 10(8):1286–1291. doi:10.4103/1673-5374.162763
Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM (2001) The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci U S A 98(25):14422–14427. doi:10.1073/pnas.251401598
Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ et al (2011) Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol 14(1):93–105. doi:10.1038/ncb2383
Nabi IR, Watanabe H, Raz A (1990) Identification of B16-F1 melanoma autocrine motility-like factor receptor. Cancer Res 50(2):409–414
Chen B, Mariano J, Tsai YC, Chan AH, Cohen M, Weissman AM (2006) The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its cue domain, RING finger, and an E2-binding site. Proc Natl Acad Sci U S A 103(2):341–346. doi:10.1073/pnas.0506618103
Li W, Tu D, Li L, Wollert T, Ghirlando R, Brunger AT, Ye Y (2009) Mechanistic insights into active site-associated polyubiquitination by the ubiquitin-conjugating enzyme Ube2g2. Proc Natl Acad Sci U S A 106(10):3722–3727. doi:10.1073/pnas.0808564106
Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S (2004) AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem 279(44):45676–45684. doi:10.1074/jbc.M409034200
Kostova Z, Tsai YC, Weissman AM (2007) Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Semin Cell Dev Biol 18(6):770–779. doi:10.1016/j.semcdb.2007.09.002
Tsai YC, Mendoza A, Mariano JM, Zhou M, Kostova Z, Chen B, Veenstra T, Hewitt SM et al (2007) The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat Med 13(12):1504–1509. doi:10.1038/nm1686
Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E, Wang G (2009) Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum Mol Genet 18(22):4268–4281. doi:10.1093/hmg/ddp380
Fu M, St-Pierre P, Shankar J, Wang PT, Joshi B, Nabi IR (2013) Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell 24(8):1153–1162. doi:10.1091/mbc.E12-08-0607
Jacobs JL, Zhu J, Sarkar SN, Coyne CB (2014) Regulation of mitochondrial antiviral signaling (MAVS) expression and signaling by the mitochondria-associated endoplasmic reticulum membrane (MAM) protein Gp78. J Biol Chem 289(3):1604–1616. doi:10.1074/jbc.M113.520254
Avraham E, Szargel R, Eyal A, Rott R, Engelender S (2005) Glycogen synthase kinase 3beta modulates synphilin-1 ubiquitylation and cellular inclusion formation by SIAH: implications for proteasomal function and Lewy body formation. J Biol Chem 280(52):42877–42886. doi:10.1074/jbc.M505608200
Kazlauskaite A, Martinez-Torres RJ, Wilkie S, Kumar A, Peltier J, Gonzalez A, Johnson C, Zhang J et al (2015) Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep 16(8):939–954. doi:10.15252/embr.201540352
Tian B, Yang Q, Mao Z (2009) Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat Cell Biol 11(2):211–218. doi:10.1038/ncb1829
Jessberger S, Gage FH, Eisch AJ, Lagace DC (2009) Making a neuron: Cdk5 in embryonic and adult neurogenesis. Trends Neurosci 32(11):575–582. doi:10.1016/j.tins.2009.07.002
Cheung ZH, Ip NY (2012) Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol 22(3):169–175. doi:10.1016/j.tcb.2011.11.003
Zhang P, Shao XY, Qi GJ, Chen Q, Bu LL, Chen LJ, Shi J, Ming J et al (2016) Cdk5-dependent activation of neuronal Inflammasomes in Parkinson’s disease. Mov Disord 31(3):366–376. doi:10.1002/mds.26488
Avraham E, Rott R, Liani E, Szargel R, Engelender S (2007) Phosphorylation of Parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. J Biol Chem 282(17):12842–12850. doi:10.1074/jbc.M608243200
Lahav-Baratz S, Sudakin V, Ruderman JV, Hershko A (1995) Reversible phosphorylation controls the activity of cyclosome-associated cyclin-ubiquitin ligase. Proc Natl Acad Sci U S A 92(20):9303–9307
Qi GJ, Chen Q, Chen LJ, Shu Y, Bu LL, Shao XY, Zhang P, Jiao FJ et al (2016) Phosphorylation of Connexin 43 by Cdk5 modulates neuronal migration during embryonic brain development. Mol Neurobiol 53(5):2969–2982. doi:10.1007/s12035-015-9190-6
Ahn JS, Ann EJ, Kim MY, Yoon JH, Lee HJ, Jo EH, Lee K, Lee JS et al (2016) Autophagy negatively regulates tumor cell proliferation through phosphorylation dependent degradation of the Notch1 intracellular domain. Oncotarget. doi:10.18632/oncotarget.12986
Lee S, Wang JW, Yu W, Lu B (2012) Phospho-dependent ubiquitination and degradation of PAR-1 regulates synaptic morphology and tau-mediated Abeta toxicity in Drosophila. Nat Commun 3:1312. doi:10.1038/ncomms2278
Kuntzel H, Pieniazek NJ, Pieniazek D, Leister DE (1975) Lipophilic proteins encoded by mitochondrial and nuclear genes in Neurospora crassa. Eur J Biochem 54(2):567–575
Soustre L, Diard F, Larroude C, Delorme G (1972) Primary pulmonary forms of histiocytosis in adults. J Radiol Electrol Med Nucl 53(11):837–838
Liu SL, Wang C, Jiang T, Tan L, Xing A, Yu JT (2016) The role of Cdk5 in Alzheimer’s disease. Mol Neurobiol 53(7):4328–4342. doi:10.1007/s12035-015-9369-x
Piedrahita D, Hernandez I, Lopez-Tobon A, Fedorov D, Obara B, Manjunath BS, Boudreau RL, Davidson B et al (2010) Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s mice. J Neurosci 30(42):13966–13976. doi:10.1523/JNEUROSCI.3637-10.2010
Maestre C, Delgado-Esteban M, Gomez-Sanchez JC, Bolanos JP, Almeida A (2008) Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J 27(20):2736–2745. doi:10.1038/emboj.2008.195
Llambi F, Wang YM, Victor B, Yang M, Schneider DM, Gingras S, Parsons MJ, Zheng JH et al (2016) BOK is a non-canonical BCL-2 family effector of apoptosis regulated by ER-associated degradation. Cell 165(2):421–433. doi:10.1016/j.cell.2016.02.026
Wang Y, Ha SW, Zhang T, Kho DH, Raz A, Xie Y (2014) Polyubiquitylation of AMF requires cooperation between the gp78 and TRIM25 ubiquitin ligases. Oncotarget 5(8):2044–2051. doi:10.18632/oncotarget.1478
Acknowledgments
This work was supported financially by the National Natural Science Foundation of China (Nos. 31371384 and 31571044 to B.T. and No. 31600821 to P.Z.), Program for New Century Excellent Talents in University (No. NCET-10-0415 to B.T.), and China Postdoctoral Scientific Foundation (No. 2015M582226 to P.Z.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal experiments were approved by the Institutional Animal Care and Use Committees of Huazhong University of Science and Technology.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Wang, Q., Jiao, F., Zhang, P. et al. CDK5-Mediated Phosphorylation-Dependent Ubiquitination and Degradation of E3 Ubiquitin Ligases GP78 Accelerates Neuronal Death in Parkinson’s Disease. Mol Neurobiol 55, 3709–3717 (2018). https://doi.org/10.1007/s12035-017-0579-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0579-2