
Abstract
Recent studies have demonstrated the utility and superiority of long non-coding RNAs (lncRNAs) as novel biomarkers for cancer diagnosis, prognosis, and therapy. In the present study, the prognostic value of lncRNAs in glioblastoma multiforme was systematically investigated by performing a genome-wide analysis of lncRNA expression profiles in 419 glioblastoma patients from The Cancer Genome Atlas (TCGA) project. Using survival analysis and Cox regression model, we identified a set of six lncRNAs (AC005013.5, UBE2R2-AS1, ENTPD1-AS1, RP11-89C21.2, AC073115.6, and XLOC_004803) demonstrating an ability to stratify patients into high- and low-risk groups with significantly different survival (median 0.899 vs. 1.611 years, p = 3.87e−09, log-rank test) in the training cohort. The six-lncRNA signature was successfully validated on independent test cohort of 219 patients with glioblastoma, and it revealed superior performance for risk stratification with respect to existing lncRNA-related signatures. Multivariate Cox and stratification analysis indicated that the six-lncRNA signature was an independent prognostic factor after adjusting for other clinical covariates. Further in silico functional analysis suggested that the six-lncRNA signature may be involved in the immune-related biological processes and pathways which are very well known in the context of glioblastoma tumorigenesis. The identified lncRNA signature had important clinical implication for improving outcome prediction and guiding the tailored therapy for glioblastoma patients with further prospective validation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme is the most aggressive of brain tumors and the most common glioma histology accounting for more than 40% of all gliomas [1]. Once diagnosed, the maximal surgical resection followed by adjuvant radiotherapy (RT) or chemotherapy is the standard treatment for glioblastoma patients [2, 3]. Despite traditional clinical risk factors (including tumor size, location, age, Karnofsky performance score (KPS), cytologic and histologic composition) are currently used to predict prognosis and guide treatment and management [4], but still received a poor prognosis with a more than 90% 5-year mortality [5]. Recent advances in omics study have greatly improved our understanding of the underlying molecular mechanisms of glioblastoma and revealed extensive inter-individual molecular heterogeneity, highlighting the clinical application of molecular biomarkers in diagnosis and prognosis to improve survival of glioblastoma patients.
During the past several years, particular attention has been paid recently to a newly discovered class of non-coding RNA (ncRNA), termed long non-coding RNAs (lncRNAs) which was defined arbitrarily as transcripts ranging in length from 200 nt to ~100 kilobases (kb) lacking protein-coding potential [6]. There is increasing evidence underscoring the importance of lncRNAs as new and enigmatic players in the complex genome regulatory network [7,8,9,10,11]. Recent transcriptomic analyses have identified a large number of differentially expressed lncRNAs in tumors compared to normal tissues or in cancer subtypes [12, 13]. Some of dysregulated lncRNAs have revealed oncogenic and tumor suppressive roles in cancer development, progression, and metastasis, such as well-known HOTAIR [14], PVT1 [15], H19 [16], and MALAT1 [17] that are reported to act as oncogenes, while MEG3 [18] and GAS5 [19] were reported to function as tumor suppressors. Given the fundamental role and intrinsic advantages of lncRNAs, they have been proposed to as promising diagnostic and prognostic biomarkers in cancers [20,21,22,23,24,25]. It has been observed that the perturbation of lncRNA expression appears to be critically involved in central nervous system pathologies including glioblastoma tumor, such as HOTAIR [26], MALAT1 [27], and HIF1A-AS2 [28]. Although there have been a few attempts to identify lncRNA biomarkers for prognosis prediction in glioblastoma [29], the clinical implication of lncRNA signature in glioblastoma patients has not been investigated sizably.
In this study, we performed a genome-wide analysis of lncRNA expression profiles in 419 glioblastoma patients from The Cancer Genome Atlas (TCGA) project to systematically investigate the prognostic value of lncRNAs. By using survival analysis and Cox regression model, we identified a biologically relevant six-lncRNA signature with the ability to predict the prognosis of patients with glioblastoma in the training cohort and validated its prognostic value in the testing cohort. More importantly, this six-lncRNA signature not only was an independent prognostic factor but also disclosed a better predictive performance than existing lncRNA-related signatures in predicting survival of patients with glioblastoma.
Material and Methods
Patient Data Sets
Clinical information of patients with glioblastoma was taken from The Cancer Genome Atlas (TCGA) project (https://cancergenome.nih.gov/) and molecular features and subtype information of patients with glioblastoma were derived from Ceccarelli’s study [30]. A total of 419 TCGA glioblastoma patients with lncRNA expression profiles and clinical follow-up information were utilized in our study. TCGA glioblastoma patients were partitioned into a training cohort for identifying prognostic lncRNA signature and building prognostic risk model, and a testing cohort for validating its prognostic value. The training cohort consisted of TCGA batches 1–7 with a total of 200 patients, while the testing cohort contained batch batches 8, 10, 16, 20, 26, and 38 with 219 patients. The detailed clinical features of all glioblastoma cohorts were listed in Table 1.
Acquisition and Processing of lncRNA Expression Profiles of Glioblastoma Patients
Genome-wide lncRNA and mRNA expression profiles of glioblastoma patients were fetched by repurposing the probe sets of the Affymetrix Human Exon 1.0 ST Array according to Du’s study [29]. Briefly, the probe sets corresponding lncRNA genes were achieved by re-mapping probe sets of Affymetrix Human Exon 1.0 ST to protein-coding transcripts, pseudogene, and lncRNAs. A total of 10,207 lncRNAs and 18,292 mRNAs with at least 4 probes uniquely mapped to them were obtained. The raw expression level of lncRNAs and mRNAs was calculated by summarizing the background-corrected intensity of all corresponding probes. Finally, the expression value of lncRNA was normalized using the quantile-normalized method and an empirical Bayes method for removing the heterogeneity of different biological samples and different batches in systematic measurement.
Statistical Analysis
To discover the potential lncRNA gene factors affecting the prognosis of glioblastoma patients, univariate Cox proportional hazard regression analysis is applied to determine the association between lncRNA expression and overall survival in the training cohort. The raw p values were adjusted by Benjamini and Hochberg multiple comparison methods to control the false discovery rate (FDR). If the corrected p value was less than 0.05, the corresponding lncRNAs were statistically significant and considered as the candidate prognostic lncRNAs of glioblastoma. Multivariate Cox proportional hazards regression was carried out among the pool of candidate prognostic lncRNAs, and lncRNAs with corresponding p values less than 0.1 were identified as optimal prognostic lncRNAs impacting the survival of glioblastoma patients.
An individual’s risk score model for each patient was built for predicting prognosis of glioblastoma patients by including expression level of each optimal prognostic lncRNA, weighted by their estimated regression coefficients of multivariate Cox regression model as follows:
Here, lncRNA i is the identifier of the ith selected lncRNAs. The risk score model was a measure of prognostic risk for each glioblastoma patient. Using the median risk score of the training cohort as the cutoff, patients with glioblastoma could be sorted into two groups, a high-risk group and a low-risk group. A high-risk score indicates poor survival for glioblastoma patients.
On each cohort, we validated the reliability and validity of the risk score formula. Kaplan-Meier analysis was utilized to compare the survival rate of the different patient groups. The survival difference between the low-risk and high-risk group was assessed by the log-rank test. Time-dependent ROC analysis for overall survival was used to display the performance of lncRNA risk model. Univariate and multivariate analyses with Cox proportional hazards regression for overall survival were performed on the individual clinical risk factors with and without the six-lncRNA signature in each cohort. Hazard ratios (HR) and 95% confidence intervals (CI) were estimated. All analyses were conducted using the R/Bio-Conductor.
Functional Enrichment Analysis
To identify co-expressed lncRNA-mRNA pairs, Pearson correlation coefficients were computed using the paired lncRNA and mRNA expression profiles. In order to investigate the biological roles of the six-lncRNA signature in glioblastoma, functional enrichment analysis of co-expressed protein-coding genes (PCGs) with prognostic lncRNAs were conducted for gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway using DAVID Bioinformatics Resources (version 6.8) [31, 32]. Significant functional categories were identified limited to GO terms in the “Biological Process” (GOTERM-BP-FAT) and KEGG pathway categories using functional annotation chart options with the human whole genome as background when adjusted p value by Benjamini <0.1 and enrichment score >1.5. Significant GO terms with similar function were visualized as interaction networks using the Enrichment Map plugin in Cytoscape [33].
Results
Identification of the Prognostic lncRNA Genes from the Training Cohort
The 419 glioblastoma patients from the TCGA project were assigned to the training sample cohort (n = 200, batch 1 to batch 7) and the testing sample cohort (n = 219, batch 8, 10, 16, 20, 26, and batch 38) by the batch. To single out the prognostic lncRNAs, expression data of each lncRNA were subjected to univariate Cox proportional hazards regression analysis in the training cohort. A total of 11 lncRNAs were found to be significantly associated with the glioblastoma patients’ overall survival (adjusted p value < 0.05) and were entered into the candidate pool for further selection. To determine the optimal prognostic lncRNAs, we adopted multivariate Cox proportional hazards regression among the pool of candidate prognostic lncRNAs to evaluate their independent prognostic values. Based on the Cox model, 6 of 11 candidate lncRNAs were found to retain their prognostic significance and thus were selected as independent remarkable prognostic factors (Table 2). Among six prognostic lncRNAs, four lncRNAs (AC005013.5, UBE2R2-AS1, ENTPD1-AS1, and RP11-89C21.2) with negative coefficient of univariate regression analysis may be protective factors owing to the close association between their high expression and longer patients’ survival, whereas the remaining two lncRNAs (AC073115.6 and XLOC_004803) tended to be prognostic risky factors and their high expression were associated with shorter survival.
The Six-lncRNA Prognostic Risk Model and Predictability Assessment in the Training Cohort
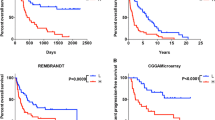
Given the significant and independent correlation between expression of six prognostic lncRNAs and overall survival, six prognostic lncRNAs was combined to form a six-lncRNA signature to predict patient’s outcome. To predict patient’s prognosis using lncRNA expression, an individual’s risk score model was developed using the regression coefficients of multivariate Cox regression model to weight the expression level of each lncRNA in the six-lncRNA signature as follows: risk score = (−0.5923 × expression value of AC005013.5) + (0.3662 × expression value of AC073115.6) + (−0.3275 × expression value of UBE2R2-AS1) + (0.2891 × expression value of ENTPD1-AS1) + (−0.3909 × expression value of RP11-89C21.2) + (0.2657 × expression value of XLOC_004803). Especially, the coefficients in the risk score model represented the relative contributions of six lncRNAs in the multiple Cox analysis. According to the risk score model, the six-lncRNA prognostic risk score was computed for each patient in the training cohort. All patients of training cohort were classified into a high-risk group (n = 100) and a low-risk group (n = 100) according to the median risk score. Overall survival was significantly different between the predicted two risk groups (p = 3.87e−09, log-rank test) (Fig. 1a). The median survival time of the high-risk group and the low-risk group was 0.899 and 1.611 years, respectively. In addition, the 3- and 5-year survival rates of the high-risk group were 5.13 and 1.03%, respectively, whereas the corresponding rates in the low-risk group were 23.6 and 14.4%. In the univariate Cox regression model of overall survival, as compared with the low-risk group, the high-risk group reveal a 2.44-fold increased risk of death (95% CI1.799–3.308, p = 9.52e−09) (Table 3).

Identification of a six-lncRNA signature significantly associated with survival of patients with glioblastoma. a Kaplan-Meier curve analysis of overall survival of glioblastoma patients in high- and low-risk groups. b Time-dependent ROC curves analysis. c Risk score distribution, survival status, and lncRNA expression patterns for patients in high- and low-risk groups by the six-lncRNA signature
Figure 1b showed the predictive performance of six-lncRNA prognostic risk model measured using time-dependent receiver operating characteristic (ROC) curves for 5-year survival. The area under the ROC (AUC) for the six-lncRNA prognostic model was 0.902 at 5 years of overall survival (Fig. 1b). We ranked the risk scores of patients in the training cohort and analyzed their distribution in Fig. 1c. The survival status of glioblastoma patients in the training cohort was marked on the dot plot (Fig. 1c). The heatmap revealed expression patterns of prognostic lncRNAs between two different prognostic patient groups. For patients with low-risk scores, the expression of four protective lncRNAs was up-regulated and the two risky lncRNAs were expressed at a low level. On the contrary, expression of prognostic lncRNAs displayed the opposite patterns among the patients with high-risk scores.
Analytic Validation of the Six-lncRNA Prognostic Risk Model
To test the robustness of the six-lncRNA signature, the prognostic value of the six-lncRNA signature was further validated using the testing cohort and entire TCGA patient cohort. With the six-lncRNA signature, the prognostic risk score was calculated for patients in the testing cohort based on the expression value of six prognostic lncRNAs. Each patient in the testing cohort was marked as a high-risk case or a low-risk case by comparing this patient’s risk score with the cutoff derived from the training cohort. There were 110 high-risk patients and 109 low-risk patients among the testing cohort. As shown in Fig. 2a, Kaplan-Meier survival curves based on the six-lncRNA signature were significantly different in the predicted two risk groups (p = 1.87e−02, log-rank test), and the median survival time of patients in the high-risk group and the low-risk group were 1.17 and 1.28 years, respectively (Fig. 2a). Similarly, the survival rate for low-risk patients was higher than that of the high-risk group throughout the follow-up time. The survivals at 3 years in the high-risk and low-risk group were 5.54 and 13.49%. The survival rate of high-risk group was zero at 5 years, and that of the low-risk group was 6.99%. In univariate analysis, the hazard ratios of high-risk group versus low-risk group for overall survival were 1.467 (95% CI 1.063–2.025, p = 1.96e−02) (Table 3). ROC analyses at 5 years for overall survival of testing patients were performed based on our six-lncRNA prognostic risk model. In the testing cohort, AUC for the six-lncRNA signature was 0.842. The distribution of risk score, survival status, and expression of six prognostic lncRNAs in testing samples were illustrated in Fig. 2c. Comparable to the training cohort, expression of risky lncRNAs was lower in the low-risk group, and protective lncRNAs were expressed at a higher level in the low- risk group.

Independent validation of the six-lncRNA signature in the independent testing cohort. a Kaplan-Meier curve analysis of overall survival of glioblastoma patients in high- and low-risk groups. b Time-dependent ROC curves analysis. c Risk score distribution, survival status, and lncRNA expression patterns for patients in high- and low-risk groups by the six-lncRNA signature
The performance of predicting prognosis for patients in the entire TCGA cohort by the six-lncRNA signature was similar to the above results (Fig. 3). Patients of the entire TCGA cohort were divided into the high-risk group (n = 210) and low-risk group (n = 209). The high-risk patients had shorter median survival than low-risk patients (0.959 vs. 1.375 years, p = 1.67e−09, log-rank test) (Fig. 3a). The survival rates at 3 and 5 years in high-risk group were lower than those in the low-risk group (5.5 vs. 19.2% at 3 years, 0.68 vs. 11.09% at 5 years). Patients with high-risk signature had a nearly twofold higher risk of death (95% CI 1.565–2.435, p = 3.02e−09) (Table 3). The same ROC analysis was undertaken for the entire TCGA cohort and parallel results were observed as above. The AUC of ROC analysis at 5 years for overall survival was 0.883 (Fig. 3b). The distribution of risk score, survival status, and lncRNA expression of glioblastoma patients in the entire TCGA cohort were presented in Fig. 3c, which were similar to those observed in the training cohort and testing cohort.

Performance evaluation of the six-lncRNA signature in the entire TCGA cohort. a Kaplan-Meier curve analysis of overall survival of glioblastoma patients in high- and low-risk groups. b Time-dependent ROC curves analysis. c Risk score distribution, survival status, and lncRNA expression patterns for patients in high- and low-risk groups by the six-lncRNA signature
Independence of Prognostic Value of the Six-lncRNA Signature from Other Clinical Variables and Molecular Features
We assessed whether the prognostic value of the six-lncRNA signature was independent of other clinical variables. Multivariate Cox regression analysis was performed using age, KPS score, gender, and our prognostic risk score model as covariables. Results from multivariate Cox analysis revealed that the six-lncRNA signature was significantly or marginally significantly associated with overall survival in each cohort when adjusted for age, gender, and KPS score (Table 3). However, we also observed that two clinical variables, age and gender, were also significant in the multivariate analysis. Therefore, stratification analysis was performed to determine the independence of the six-lncRNA signature according to age and gender. According to the age, glioblastoma patients could be stratified into a young patient group (age ≤ 50, n = 123) and an old patient group (age > 50, n = 296). Using the six-lncRNA signature, patients of each age group could be classified into either the high-risk or low-risk group. For each age group, there was significantly different overall survival between high-risk group and low-risk group (log-rank test p = 5.08e−04 for the young patient group and log-rank test p = 1.54e−05 for the old patient group) (Fig. 4a, b). Next, all glioblastoma patients were also stratified by gender. The six-lncRNA signature could classify 263 male patients into high-risk group (n = 130) and low-risk group (n = 133) with significantly different overall survival (p = 4.99e−05, log-rank test) (Fig. 4c). Similarly, even among 156 female patients, the six-lncRNA signature could be used to separate patients into the high-risk group (n = 80) and low-risk group (76), and there was the statistically significant difference in overall survival between the predicted two risk group (p = 4.93e−06, log-rank test) (Fig. 4d).

Stratification analysis by age and gender. Kaplan-Meier curve analysis of overall survival in high- and low-risk groups for young patients (a) and old patients (b). Kaplan-Meier curve analysis of overall survival in high- and low-risk groups for male patients (c) and female patients (d). Kaplan-Meier curve analysis of overall survival in high- and low-risk groups for patients with GCIMP−/IDHwt (e)
Furthermore, we investigated whether the predictive power of the six-lncRNA signature was independent of IDH mutation status and MGMT promoter methylation status using multivariate Cox regression analysis. The result of multivariate Cox regression analysis suggested that the six-lncRNA signature were statistically significantly associated with survival (p = 0.0214) when adjusted for IDH mutation status and MGMT promoter methylation status (Table 4), indicating that the predictive ability of the six-lncRNA signature is also independent of these two molecular features for the survival of patients with glioblastoma. However, we also found that IDH mutation status was statistically significantly associated with survival (p = 0.0265) in the multivariate analysis. Therefore, the 419 glioblastoma patients used in this study were divided into three groups: GCIMP/IDHmut patients (n = 27), GCIMP−/IDHwt patients (n = 282), and those with unknown IDH mutation status (n = 110). Then we tested whether the six-lncRNA signature could predict survival of patients with GCIMP−/IDHwt. The patients with GCIMP−/IDHwt were classified as high-risk (n = 151) or low-risk (n = 131) according to their six-lncRNA signature (median survival 0.992 vs. 1.315 years, p = 8.44e−06, log-rank test) (Fig. 4e), indicating that the six-lncRNA signature was able to identify a subgroup of GCIMP−/IDHwt patients who have favorable survival.
Finally, we compared expression level of lncRNAs in the signature across five glioblastoma subtypes (classical, the glioma-CpG island methylator phenotype (GCIMP), mesenchymal, neural, and proneural) and found no significant difference in the distribution of expression levels for all six lncRNAs across five glioblastoma subtypes (Fig. 5), implying that the six-lncRNA signature is not subtype-specific marker. These results thus indicated that the six-lncRNA signature is an independent prognostic factor related to overall survival for patients with glioblastoma.

Expression map of the six lncRNA signature across five glioblastoma subtypes. Kruskal-Wallis test was used to compare the expression levels for each lncRNAs across five glioblastoma subtypes
Comparison of the Six-lncRNA Signature with Existing lncRNA-Related Signatures
Recently, two lncRNA-related signatures were reported to predict prognosis of glioblastoma patients [34, 35]. Therefore, we compared the prognostic value of our six-lncRNA signature (hereinafter referred to as LncSig) to that of different lncRNA sets for predicting prognosis of glioblastoma patients: the lncRNAs derived from Zhang’s study [35] (hereinafter referred to as ZhangSig) and ceRNA signature derived from Cao’s study [34] (hereinafter referred to as ceRSig). Utilizing the same TCGA patient cohort, the LncSig and ZhangSig were successful and performed very well in predicting survival (Fig. 6a), with the notable exception of ceRSig which was not significant (p = 0.088, log-rank test) (Fig. 6c). Further comparison of Kaplan-Meier survival curves showed that patients with high-risk scores predicted by the LncSig had a worse prognosis than those with high-risk scores predicted by ZhangSig and ceRSig, and patients with low-risk scores predicted by the LncSig had a better prognosis than those with low-risk scores predicted by ZhangSig and ceRSig (Fig. 6b, d). We also performed time-dependent ROC analysis to compare the prognostic power between the LncSig and other two existing lncRNA-related signatures in the entire TCGA patient cohort. As shown in Fig. 6e, the AUC at 5 years of overall survival for the LncSig is 0.883, which was significantly higher than that of ZhangSig (AUC = 0.724) and ceRSig (AUC = 0.764) (Fig. 6e). These results demonstrated the better prognostic power of the six-lncRNA signature in predicting survival than two existing lncRNA-related signatures.

Comparison of sensitivity and specificity for survival prediction by the six-lncRNA signature and two existing lncRNA-related signatures. a Kaplan-Meier curve analysis of overall survival of glioblastoma patients in high- and low-risk groups predicted by ZhangSig. b Comparison of survival differences in high- and low-risk groups predicted by ZhangSig and the six-lncRNA signature. c Kaplan-Meier curve analysis of overall survival of glioblastoma patients in high- and low-risk groups predicted by ceRSig. d Comparison of survival differences in high- and low-risk groups predicted by ceRSig and the six-lncRNA signature. e The ROC analysis at 5 years of overall survival for the six-lncRNA signature, ZhangSig and ceRSig
In silico Functional Analysis of the Six-lncRNA Signature
In order to gain new insights into the function of the six-lncRNA signature, we performed in silico functional analysis to reveal potential biological roles of the six-lncRNA signature in glioblastoma. For this purpose, we examined the expression correlation between prognostic lncRNAs and mRNAs by calculating the Pearson correlation coefficient through paired lncRNA and mRNA expression profiles in 419 glioblastoma patients. A total of 931 protein-coding genes (PCGs) were expressed as highly correlated with at least one of lncRNAs contained in the six-lncRNA signature (Pearson correlation coefficient >0.5 and p < 0.01). We performed GO and KEGG enrichment analysis to uncover specific functional categories of 931 co-expressed PCGs. As a result, 931 co-expressed PCGs clustered most significantly in 21 GO functional categories (Fig. 7a) and three KEGG pathways (p values < 0.1 after Benjamini adjustment) (Fig. 7b, Supplementary file 1). This analysis revealed an overrepresentation of co-expressed PCGs with prognostic lncRNAs involved in immune-related biological processes and pathways such as immune response, leukocyte mediated immunity, B cell mediated immunity, innate immune response, leukocyte activation, Toll-like receptor signaling pathway, and NOD-like receptor signaling pathway. Further analysis revealed an enrichment of co-expressed PCGs with prognostic lncRNAs in neurological disease class from The Genetic Association Database (Supplementary file 2). These results suggested that the six-lncRNA signature might be an immune-related signature and variation in expression of prognostic lncRNAs might affect immune-related biological processes and pathways involved in glioblastoma biology.

In silico functional analysis of the six-lncRNA signature based on co-expressed protein-coding genes. a The functional enrichment map of significantly enriched GO terms. b Significantly enriched KEGG pathways
Discussion
Although aggressive and multimodal treatment (such as surgery, radiotherapy, and Temozolomide-based chemotherapy) has improved survival of glioblastoma patients, the treatment outcome of glioblastoma patients remains unsatisfactory. The prognosis and tumor response to therapy varied greatly between different individual patients with similar clinical risk factors, as glioblastoma is a heterogeneous disease characterized by variant morphologic and molecular altered heterogeneous [36], leading to an urgent need to identify additional molecular prognostic indicators beyond traditional clinical risk factors for glioblastoma. In the past decade, several molecular markers focusing on mRNAs or miRNAs have been proposed to predict the prognosis of glioblastoma patients. More recently, the perturbation of lncRNA expression has been widely observed in different cancer types, indicating the fundamental role of lncRNAs in cancer biology [37, 38]. Increasing evidence have suggested that lncRNAs were expressed in much more tissue- and cell type-specific manner than PCGs and miRNAs [39, 37], and their expression level has been shown to be more closely associated function compared with PCGs as lncRNA do not encode proteins [29], which implied that lncRNA expression may be a better indicator of the tumor status compared to PCGs and miRNAs [40]. It is well known that the perfect biomarkers need to be stable and easily detected in biofluid/liquid biopsy which allows noninvasive diagnosis. Therefore, some inherent nature of lncRNAs, such as long transcripts (often unstable) and multiple transcripts, made them as the best biomarkers in daily practice but still faced challenges [37]. Recently, tumor-specific lncRNAs that are protected from the RNases present in body fluids have been detected in exosomes, microvesicles, apoptotic bodies, and apoptotic microparticles [41,42,43,44], highlighting clinical implication of lncRNAs as fluid-based markers for the diagnostics and treatment of specific cancer. Given the significance and inherent advantages of lncRNAs, the roles of lncRNAs in cancer diagnosis and prognosis are steadily increasing as being studied [45, 23, 25, 46, 22, 47, 48, 24, 49]. Therefore, there is an urgent need to investigate the prognostic value of lncRNAs in glioblastoma patients.
In this study, we examined the association between lncRNA expression and prognosis in glioblastoma patients by performing a genome-wide analysis for 10,207 lncRNAs of 200 patients in the training cohort and identified 11 lncRNAs significantly associated with the glioblastoma patients’ overall survival. Using multivariate Cox and risk scoring methods, we developed a six-lncRNA signature which was able to classify glioblastoma patients into the high-risk group and low-risk group with significantly different overall survival. Because there is still possibilities of overtraining or false positives for the development of six-lncRNA signature, we further validated the prognostic value of the six-lncRNA signature using an independent cohort of 219 glioblastoma patients. Results with independent validation suggested that the six-lncRNA signature has good reproducibility and robustness in predicting prognosis for glioblastoma patients. The current traditional clinical risk factors have limited success in predicting survival of patients with glioblastoma because of molecular heterogeneity. The results of multivariate analysis showed that the six-lncRNA signature is independent of traditional clinical risk factors and molecular features. When the subgroup stratified analysis was conducted to test the independence of the signature, we found that the six-lncRNA signature could clearly distinguish patients at high-risk from those at low-risk within age and gender subgroups. Large-scale genomic analyses have identified five subtypes (classical, GCIMP, mesenchymal, neural, and proneural) with different biologic and clinical behaviors in glioblastoma [50]. Therefore, we also investigated the expression pattern of lncRNAs in the signature in specific subtypes and found that there are no different expression patterns for all six prognostic lncRNAs across five glioblastoma subtypes. These findings suggested that the six-lncRNA signature may potentially enable clinicians to determine and select patients at high-risk from those with identical clinical or molecular characteristics for rationalizing treatment decisions.
More recently, the prognostic value of lncRNA-related signature has been reported in two studies. Cao et al. proposed the lncRNA-based prognostic model for glioblastoma based on the “ceRNA” theory [34]. Besides, in the study of Zhang et al., a prognostic signature consisting of six lncRNAs was constructed using their expressions [35]. Although these two previously studies demonstrated the prognostic value of lncRNAs, they assessed the expression of a relatively small portion of currently known lncRNAs (393 lncRNAs in Cao’s study and 470 lncRNAs in Zhang’s study). Furthermore, there are no overlapping lncRNAs of our six-lncRNA signature with the abovementioned lncRNA-related signatures which could be explained by the molecular heterogeneity or methodology used. The lncRNA-related signature in the study by Cao et al. was identified using ceRNA network-derived methods and involved in RNA processing and cell cycle-related functions. Therefore, this approach was limited to those lncRNAs with ceRNA activity. The report by Zhang et al. identified a six-lncRNA signature only limited to univariate analysis of 470 lncRNAs. Moreover, six lncRNAs identified by Zhang et al. may be implicated in the pathogenesis of myocardial infarction, breast cell apoptosis, and Prader-Willi syndrome [35]. Comparing our six-lncRNA signature with that of the abovementioned two lncRNA-related signatures suggested that our six-lncRNA signature has better prognostic power in predicting survival than two existing lncRNA-related signatures when tested in the same TCGA patient cohort. In order to gain insights into the putative functional role of the six-lncRNA signature, we performed in silico functional analysis by conducting lncRNA-PCG-correlated analysis and enrichment analysis. Functional analysis showed that PCGs identified in the correlated analysis are significantly clustered in immune response, leukocyte-mediated immunity, B cell-mediated immunity, innate immune response, leukocyte activation, Toll-like receptor signaling pathway, NOD-like receptor signaling pathway, and neurological disease class. This finding suggested that the six-lncRNA signature may contribute to glioblastoma tumorigenesis by interacting with or regulating PCGs that are well known to be involved in the immune-related biological processes and pathways. Mounting evidence has supported a link between the immune system and glioblastoma prognosis and therapy [51, 52]. Moreover, some well-defined mRNA signatures in glioblastoma are related to the immune system [53,54,55]. Although functional implication of the six-lncRNA signature has been inferred by in silico functional analysis, the careful functional characterization of lncRNAs should be delineated using biological experimental analyses (such as RNA interference or in vitro cell-based assays).
Conclusions
In summary, we identified an immune-related lncRNA signature comprising six lncRNAs (AC005013.5, UBE2R2-AS1, ENTPD1-AS1, RP11-89C21.2, AC073115.6, and XLOC_004803), which can be used as an independent prognostic marker in stratifying risk subgroups in terms of survival for patients with glioblastoma. The six-lncRNA signature was successfully validated on independent cohort, comprising a total of 219 patients, and it had superior performance for risk stratification compared to existing glioblastoma lncRNA-related signatures. With further prospective validation, the six-lncRNA signature may improve outcome prediction and guide the tailored therapy for patients with glioblastoma.
References
Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA et al (2014) The epidemiology of glioma in adults: a “state of the science” review. Neuro-Oncology 16(7):896–913. doi:10.1093/neuonc/nou087
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL et al (2007) Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 21(21):2683–2710. doi:10.1101/gad.1596707
Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN (2007) Molecularly targeted therapy for malignant glioma. Cancer 110(1):13–24. doi:10.1002/cncr.22741
Walid MS (2008) Prognostic factors for long-term survival after glioblastoma. Perm J 12(4):45–48
Carlsson SK, Brothers SP, Wahlestedt C (2014) Emerging treatment strategies for glioblastoma multiforme. EMBO Mol Med 6(11):1359–1370. doi:10.15252/emmm.201302627
Ma L, Bajic VB, Zhang Z (2013) On the classification of long non-coding RNAs. RNA Biol 10(6):925–933. doi:10.4161/rna.24604
Kornienko AE, Guenzl PM, Barlow DP, Pauler FM (2013) Gene regulation by the act of long non-coding RNA transcription. BMC Biol 11:59. doi:10.1186/1741-7007-11-59
Mercer TR, Mattick JS (2013) Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol 20(3):300–307. doi:10.1038/nsmb.2480
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136(4):629–641. doi:10.1016/j.cell.2009.02.006
Zhou M, Wang X, Li J, Hao D, Wang Z, Shi H, Han L, Zhou H et al (2015) Prioritizing candidate disease-related long non-coding RNAs by walking on the heterogeneous lncRNA and disease network. Mol BioSyst 11(3):760–769. doi:10.1039/c4mb00511b
Sun J, Shi H, Wang Z, Zhang C, Liu L, Wang L, He W, Hao D et al (2014) Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol BioSyst 10(8):2074–2081. doi:10.1039/c3mb70608g
Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR et al (2015) The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 47(3):199–208. doi:10.1038/ng.3192
Zhou M, Zhao H, Xu W, Bao S, Cheng L, Sun J (2017) Discovery and validation of immune-associated long non-coding RNA biomarkers associated with clinically molecular subtype and prognosis in diffuse large B cell lymphoma. Mol Cancer 16(1):16. doi:10.1186/s12943-017-0580-4
Hajjari M, Salavaty A (2015) HOTAIR: an oncogenic long non-coding RNA in different cancers. Cancer Biol Med 12(1):1–9. doi:10.7497/j.issn.2095-3941.2015.0006
Johnsson P, Morris KV (2014) Expanding the functional role of long noncoding RNAs. Cell Res 24(11):1284–1285. doi:10.1038/cr.2014.104
Raveh E, Matouk IJ, Gilon M, Hochberg A (2015) The H19 long non-coding RNA in cancer initiation, progression and metastasis - a proposed unifying theory. Mol Cancer 14:184. doi:10.1186/s12943-015-0458-2
Gutschner T, Hammerle M, Diederichs S (2013) MALAT1—a paradigm for long noncoding RNA function in cancer. J Mol Med (Berl) 91(7):791–801. doi:10.1007/s00109-013-1028-y
Zhou Y, Zhang X, Klibanski A (2012) MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol 48(3):R45–R53. doi:10.1530/JME-12-0008
Pickard MR, Williams GT (2015) Molecular and cellular mechanisms of action of tumour suppressor GAS5 LncRNA. Genes (Basel) 6(3):484–499. doi:10.3390/genes6030484
Nana-Sinkam SP, Croce CM (2011) Non-coding RNAs in cancer initiation and progression and as novel biomarkers. Mol Oncol 5(6):483–491. doi:10.1016/j.molonc.2011.10.003
Zhou M, Guo M, He D, Wang X, Cui Y, Yang H, Hao D, Sun J (2015) A potential signature of eight long non-coding RNAs predicts survival in patients with non-small cell lung cancer. J Transl Med 13:231. doi:10.1186/s12967-015-0556-3
Zhou M, Zhao H, Wang Z, Cheng L, Yang L, Shi H, Yang H, Sun J (2015) Identification and validation of potential prognostic lncRNA biomarkers for predicting survival in patients with multiple myeloma. J Exp Clin Cancer Res 34:102. doi:10.1186/s13046-015-0219-5
Zhou M, Xu W, Yue X, Zhao H, Wang Z, Shi H, Cheng L, Sun J (2016) Relapse-related long non-coding RNA signature to improve prognosis prediction of lung adenocarcinoma. Oncotarget 7(20):29720–29738. doi:10.18632/oncotarget.8825
Zhou M, Sun Y, Sun Y, Xu W, Zhang Z, Zhao H, Zhong Z, Sun J (2016) Comprehensive analysis of lncRNA expression profiles reveals a novel lncRNA signature to discriminate nonequivalent outcomes in patients with ovarian cancer. Oncotarget 7(22):32433–32448. doi:10.18632/oncotarget.8653
Sun J, Cheng L, Shi H, Zhang Z, Zhao H, Wang Z, Zhou M (2016) A potential panel of six-long non-coding RNA signature to improve survival prediction of diffuse large-B-cell lymphoma. Sci Rep 6:27842. doi:10.1038/srep27842
Pastori C, Kapranov P, Penas C, Peschansky V, Volmar CH, Sarkaria JN, Bregy A, Komotar R et al (2015) The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc Natl Acad Sci U S A 112(27):8326–8331. doi:10.1073/pnas.1424220112
Vassallo I, Zinn P, Lai M, Rajakannu P, Hamou MF, Hegi ME (2016) WIF1 re-expression in glioblastoma inhibits migration through attenuation of non-canonical WNT signaling by downregulating the lncRNA MALAT1. Oncogene 35(1):12–21. doi:10.1038/onc.2015.61
Mineo M, Ricklefs F, Rooj AK, Lyons SM, Ivanov P, Ansari KI, Nakano I, Chiocca EA et al (2016) The long non-coding RNA HIF1A-AS2 facilitates the maintenance of mesenchymal glioblastoma stem-like cells in hypoxic niches. Cell Rep 15(11):2500–2509. doi:10.1016/j.celrep.2016.05.018
Du Z, Fei T, Verhaak RG, Su Z, Zhang Y, Brown M, Chen Y, Liu XS (2013) Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat Struct Mol Biol 20(7):908–913. doi:10.1038/nsmb.2591
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y et al (2016) Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164(3):550–563. doi:10.1016/j.cell.2015.12.028
Huang da W, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37(1):1–13. doi:10.1093/nar/gkn923
Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1):44–57. doi:10.1038/nprot.2008.211
Merico D, Isserlin R, Stueker O, Emili A, Bader GD (2010) Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5(11):e13984. doi:10.1371/journal.pone.0013984
Cao Y, Wang P, Ning S, Xiao W, Xiao B, Li X (2016) Identification of prognostic biomarkers in glioblastoma using a long non-coding RNA-mediated, competitive endogenous RNA network. Oncotarget 7(27):41737–41747. doi:10.18632/oncotarget.9569
Zhang XQ, Sun S, Lam KF, Kiang KM, Pu JK, Ho AS, Lui WM, Fung CF et al (2013) A long non-coding RNA signature in glioblastoma multiforme predicts survival. Neurobiol Dis 58:123–131. doi:10.1016/j.nbd.2013.05.011
Cancer Genome Atlas Research N (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455(7216):1061–1068. doi:10.1038/nature07385
Huarte M (2015) The emerging role of lncRNAs in cancer. Nat Med 21(11):1253–1261. doi:10.1038/nm.3981
Qi P, Du X (2013) The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod Pathol 26(2):155–165. doi:10.1038/modpathol.2012.160
Gibb EA, Brown CJ, Lam WL (2011) The functional role of long non-coding RNA in human carcinomas. Mol Cancer 10:38. doi:10.1186/1476-4598-10-38
Hauptman N, Glavac D (2013) Long non-coding RNA in cancer. Int J Mol Sci 14(3):4655–4669. doi:10.3390/ijms14034655
Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H et al (2007) Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 132(1):330–342. doi:10.1053/j.gastro.2006.08.026
Mohankumar S, Patel T (2016) Extracellular vesicle long noncoding RNA as potential biomarkers of liver cancer. Brief Funct Genomics 15(3):249–256. doi:10.1093/bfgp/elv058
Qi P, Zhou XY, Du X (2016) Circulating long non-coding RNAs in cancer: current status and future perspectives. Mol Cancer 15(1):39. doi:10.1186/s12943-016-0524-4
Wu Y, Wang YQ, Weng WW, Zhang QY, Yang XQ, Gan HL, Yang YS, Zhang PP et al (2016) A serum-circulating long noncoding RNA signature can discriminate between patients with clear cell renal cell carcinoma and healthy controls. Oncogene 5:e192. doi:10.1038/oncsis.2015.48
Zhu X, Tian X, Yu C, Shen C, Yan T, Hong J, Wang Z, Fang JY et al (2016) A long non-coding RNA signature to improve prognosis prediction of gastric cancer. Mol Cancer 15(1):60. doi:10.1186/s12943-016-0544-0
Hu Y, Chen HY, Yu CY, Xu J, Wang JL, Qian J, Zhang X, Fang JY (2014) A long non-coding RNA signature to improve prognosis prediction of colorectal cancer. Oncotarget 5(8):2230–2242 10.18632/oncotarget.1895
Zhou M, Wang X, Shi H, Cheng L, Wang Z, Zhao H, Yang L, Sun J (2016) Characterization of long non-coding RNA-associated ceRNA network to reveal potential prognostic lncRNA biomarkers in human ovarian cancer. Oncotarget 7(11):12598–12611. doi:10.18632/oncotarget.7181
Zhou M, Diao Z, Yue X, Chen Y, Zhao H, Cheng L, Sun J (2016) Construction and analysis of dysregulated lncRNA-associated ceRNA network identified novel lncRNA biomarkers for early diagnosis of human pancreatic cancer. Oncotarget 7(35):56383–56394. doi:10.18632/oncotarget.10891
Li J, Chen Z, Tian L, Zhou C, He MY, Gao Y, Wang S, Zhou F et al (2014) LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut 63(11):1700–1710. doi:10.1136/gutjnl-2013-305806
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L et al (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17(1):98–110. doi:10.1016/j.ccr.2009.12.020
Vauleon E, Tony A, Hamlat A, Etcheverry A, Chiforeanu DC, Menei P, Mosser J, Quillien V et al (2012) Immune genes are associated with human glioblastoma pathology and patient survival. BMC Med Genet 5:41. doi:10.1186/1755-8794-5-41
Zhou M, Wiemels JL, Bracci PM, Wrensch MR, McCoy LS, Rice T, Sison JD, Patoka JS et al (2010) Circulating levels of the innate and humoral immune regulators CD14 and CD23 are associated with adult glioma. Cancer Res 70(19):7534–7542. doi:10.1158/0008-5472.CAN-10-0815
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV et al (2014) Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344(6190):1396–1401. doi:10.1126/science.1254257
Chifman J, Pullikuth A, Chou JW, Bedognetti D, Miller LD (2016) Conservation of immune gene signatures in solid tumors and prognostic implications. BMC Cancer 16(1):911. doi:10.1186/s12885-016-2948-z
Arimappamagan A, Somasundaram K, Thennarasu K, Peddagangannagari S, Srinivasan H, Shailaja BC, Samuel C, Patric IR et al (2013) A fourteen gene GBM prognostic signature identifies association of immune response pathway and mesenchymal subtype with high risk group. PLoS One 8(4):e62042. doi:10.1371/journal.pone.0062042
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 61602134) and Postdoctoral scientific research developmental fund of Heilongjiang Province (Grant No. LBH-Q16172).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure of Potential Conflicts of Interest
The authors declare that they have no potential conflicts of interests.
Rights and permissions
About this article

Cite this article
Zhou, M., Zhang, Z., Zhao, H. et al. An Immune-Related Six-lncRNA Signature to Improve Prognosis Prediction of Glioblastoma Multiforme. Mol Neurobiol 55, 3684–3697 (2018). https://doi.org/10.1007/s12035-017-0572-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0572-9