
Abstract
E6ap is a known transcriptional coregulator for estrogen receptor alpha (Er, Erα) in the presence of estrogen. Protein kinase A (PKA) contains two regulatory subunits derived from four genes. Recent evidence demonstrates that PKA regulates E6ap activity. Data generated in our lab indicated estrogen dependent regulation of Pkar2a levels. Our project sets to investigate a possible feedback mechanism constituting of Erα and E6ap transcriptional regulation of Pkar2a expression. Western blot evaluated protein regulation correlations with E2 in mouse neuroblastoma lines. Bioinformatics detected estrogen response element (ERE) sequences. quantitative polymerase chain reaction (qPCR) validated the western blot results. ERE oligonucleotides were synthesized. Reporter gene transcriptional activity was evaluated via Luciferase assay output. Electromobility shift assay (EMSA) assessed direct binding between Erα relevant sequences. Chromatin immunoprecipitation (ChIP) and Re-ChIP were conducted in quantifying protein complex recruitment levels. Pkar2a protein expression directly correlated with E2, and four putative ERE sequences were identified. Pkar2a mRNA expression reverted to baseline with either E2 or E6ap absent. In the presence of E2, ERE-1 and ERE-4 possessed Luciferase reporter gene transcriptional capabilities. ERE-1 portrayed band shifts, representing direct binding to Erα with E2 supplementation. With E2, ERE-1 significantly enhanced Erα and E6ap recruitment levels to the Pkar2a promoter. Pkar2a is directly regulated by Erα and E6ap in the presence of estrogen stimulus. This work indicates a feedback mechanism in the interplay between PKA and E6ap, which may prove crucial for the role of both proteins in cancers and neurogenetic diseases like Angelman syndrome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
E6-associated protein (E6ap, Ube3a) is a protein that was discovered in the context of studying oncogenic types of the human papillomavirus (HPV). In analyzing the viral protein E6, E6ap was implicated as present in formation of the ubiquitination complex responsible for degrading p53 [1]. This E3-ubiquitin ligase function of E6ap has been described extensively through the literature [2]. A relatively novel role for E6ap as a steroid hormone receptor (SHR) coregulator was unraveled by Nawaz et al. [3, 4]. E6ap was shown to modulate the transcriptional gene expression of downstream targets of estrogen receptor, androgen receptor, and progesterone receptor amongst others [3, 5].
Estrogen receptors (Er, Erα) are a group of intracellular proteins characterized by their ability to bind 17β-estradiol hormone molecules (E2) and consequentially perform downstream alterations in the cell [6]. Upon E2 activation, Erα dimerizes with an identically activated receptor, undergoes translocation to the nucleus and regulates particular levels of gene expression [7]. It is generally regarded as a transcription factor contingent on its DNA-binding to the estrogen response element (ERE) sequence [6, 8]. Standardized to a consensus, variations in this critical sequence correspond to substantial changes in the binding and transcriptional capabilities of Erα [9].
Protein kinase A (PKA) are a ubiquitous enzymatic intracellular family of proteins responsible for the phosphorylation of residues on other proteins [10]. The addition of a phosphoryl group is localized to serine or threonine residues and may augment or suppress the activity of a particular target protein [11]. Phosphorylation may also direct the compartmental localization of certain targets [12]. Through such a broad functionality, PKA portrays a potent regulatory effect over numerous downstream molecules in appropriate cells [13]. cAMP is a second messenger traditionally known for having PKA as a primary intracellular receptor [14].
PKA is composed of two regulatory subunits (R1 and R2) and two catalytic subunits [15, 16]. Upon cAMP activation, released catalytic subunits are responsible for the direct phosphorylation of proteins [17]. The two regulatory subunits may originate from four genes coding for distinct regulatory isoforms R1A, R1B, R2A, and R2B [18]. These isoforms display variance in tissue and subcellular distribution as well as biological characteristics [19]. The relative expression of such subunits is contingent on the activity of specific transcriptional regulators present in different cell types. This expression subsequently impacts the functionality of PKA at its pertinent sites, and thus its influence upon various downstream targets. Such targets play prominent roles in several clinical disorders as neuroblastoma cancers and neurological diseases [20–24].
PKA and E6ap have been demonstrated to interact through PKA-dependent phosphorylation of E6ap. Yi et al. show that through PKA phosphorylation of E6ap residue T485, which falls in a region exterior to the catalytic domain, E6ap activity is indiscriminately inhibited [25]. Identifying PKA as an upstream modulator of E6ap is crucial, particularly due to further characterization of an autism-linked mutation perturbing the described control and constraining E6ap activity to a constitutive state [25].
PKA and estrogen receptors have notably portrayed interactions highlighted throughout the literature. The role of PKA in the characterization of Tamoxifen resistance, displayed by certain breast cancer patients, has been especially prominent. Er is susceptible to phosphorylation at a series of locations resulting in altered transcriptional activity [26]. Original analysis of PKA regulatory subunit Iα (PKARIα) mRNA from breast cancer patient tumors exhibited significantly elevated quantities in tumors subsequently responding to Tamoxifen hormonal therapy [27]. Further studies illuminated that PKA-dependent phosphorylation of Erα at the serine-305 hinge region residue was sufficient to induce Tamoxifen resistance [28]. Synthesizing from prior data, Michalides et al. concluded that PKA activation from PKARIα downregulation modified Tamoxifen to act as an Erα growth stimulator [28]. Recent evidence elucidated the PKA-anchoring protein AKAP13 as responsible for the Tamoxifen resistance developed through enabling the Erα serine-305 phosphorylation mechanism [29].
In summary, E6ap has been identified as a SHR coregulator functioning in conjunction with Er to promote transcription upon activation by E2. Furthermore, potentially downstream modulators as PKA have been shown to interact in feedback with these upstream transcriptional regulators, particularly in the context of neurological and oncological pathologies. However, the complete description of pathways including hormonal based feedback regulation of upstream elements generally remains unclear. In light of data generated in our lab indicating estrogen dependent regulation of Pkar2a levels by estrogen, we set our project to investigate a possible feedback mechanism constituting of Erα and E6ap transcriptional regulation of Pkar2a expression.
Materials and Methods
Cell Lines
The mouse neuroblastoma cell line Neuro2a (from American Type Culture Collection. ATCC) was routinely grown, unless otherwise indicated, in media with the following composition: 50% OptiMEM (Invitrogen), 45% DMEM + Glutamax (Invitrogen), 5% fetal bovine serum (FBS, Atlanta Biologicals), and 1× penicillin/streptomycin (Invitrogen). Human cervical cancer cell line, HeLa, was routinely grown, unless otherwise indicated, in media with the following composition: 90% DMEM + Glutamax (Invitrogen), 10% FBS (Atlanta Biologicals) and 1× penicillin/streptomycin (Invitrogen). All cell lines were incubated at 37 °C in the presence of 5% CO2.
Western Blot
Cell lysates were placed on ice for 30 min and cleared by centrifugation at 3000g for 15 min at 4 °C. The supernatants were collected and frozen at −80 °C until used for analysis. The protein concentrations of lysates were measured using the Bio-Rad protein assay kit.
Thirty micrograms of total protein from each sample was resolved on 10% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) and transferred onto nitrocellulose membranes (Protran, Schleicher & Schuell, Inc., Keene, NH). Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline (20 mM Tris base (pH 7.5) and 150 mM NaCl) containing 0.05% Tween 20 (TBS-T), then probed with the primary antibody. The primary antibodies were diluted in 5% nonfat dry milk in TBS-T as indicated and used for immunoblotting: Anti-Pkar2a (1:1000 dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and anti-Glyceraldehyde 3-phosphate dehydrogenase (anti-Gapdh) (1:2000 dilution; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). After washing in TBS-T, membranes were incubated with their appropriate horseradish peroxidase-conjugated secondary antibodies (Bio-Rad Laboratories, Inc.) and developed using an enhanced chemiluminescence detection system (Amersham Biosciences, Arlington Heights, IL) according to the instructions of the manufacturer and were exposed to X-ray film (Phoenix Research Products, Hayward, CA).
Densitometric Analysis
To quantify the bands obtained via Western blot analysis, we applied ImageJ software based analysis (http://rsb.info.nih.gov/ij/). The area of the specific signal was corrected for the corresponding signal from the area of the loading control. Normalized optical density (OD) values were used to plot matching diagrams.
siRNA Knockdown
E6ap siRNAs were procured from Dharmacon (Chicago, IL) as ON-TARGET plus Smart Pools. Non-targeting siRNAs (siScramble) were used as negative controls. 1 × 105 Neuro2a cells were plated in 6-well plates with 5% charcoal stripped fetal bovine serum (cFBS, Atlanta Biologicals) in phenol red free DMEM (Invitrogen) for 24 h. Cells were then forward transfected with 150 pmol of E6ap siRNA or control siScramble using Lipofectamine RNAiMax (Invitrogen) according to the manufacturerʼs protocols. Forty-eight hours after transfection, hormone treatments were begun using ethanol vehicle control, 10 nM 17β-estradiol (Sigma) or 10 μM Tamoxifen (Invitrogen). The 48-h time point was chosen as an appropriate duration after the system has ensured reaching its steady-state plateau. Earlier time points have been shown to demonstrate equivalent results up til the 72-h time point.
RNA Extraction and cDNA Synthesis
Neuro2a cells grown in 6-well plates were washed once with PBS and 350 μL of buffer RLT (Qiagen) was added to each well. Cells were then frozen at −80 °C for later processing. RNA was isolated from 6-well plates following the manufacturerʼs protocol for Qiagen RNeasy mini kit. Lysate was homogenized by either passing the lysate through a QIAShredder (Qiagen) or pipetting up and down with a 1 mL pipette tip (10×). 2 μg of total RNA was used to create cDNA using Fermentas Maxima First Strand cDNA synthesis kit according to the manufacturerʼs protocol.
Bioinformatics/Putative ERE Generation
12 kb promoter sequence (10 kb 5′ of the transcriptional start site (TSS) and 2 kb 3′ of TSS) of Pkar2a gene was scanned for putative ERE using two different databases: JASPAR [30] and Cluster-Buster [31]. Putative ERE was chosen if it was predicted by both databases and if it passed all stringent criteria outlined. Cluster-Buster criteria: Cluster score threshold = 7 and motif score threshold = 7. JASPAR criteria: minimum degree of confidence 80%. This identified four potential putative ERE sequences.
A scrambled (Scr) sequence was arbitrarily generated and crossed against the consensus (Cons) to ensure lack of overlap [32, 33]. The complete sequence design accounted for a SmaI restriction site in the multiple cloning site (MCS) of the pGL3-basic vector. Twenty base pairs on both the 5′ and 3′ ends of the SmaI site were duplicated to flank 2 ERE sequences followed by 2 spacer sequences each. Downstream of the second spacer was a TATA sequence and filler base pair to ensure an aligned reading frame. These composite 100 base pair sequences generating ERE-1, ERE-2, ERE-3, and ERE-4 in addition to the consensus and scrambled sequences were synthesized as forward and reverse oligonucleotide strands from Sigma-Aldrich (Table S1). All of the oligonucleotide strands were suspended in an appropriate quantity of nuclease-free water yielding a 100 μM solution. One microliter of the 100 μM forward and reverse solutions was added to 98 μL of nuclease-free water for a 100:1 dilution of each oligonucleotide couple. These new solutions underwent an annealing protocol (Sigma) using a thermal cycler (MJ Research) at 95 °C for 2 min with ramp cooling to 25 °C over 45 min producing double stranded sequences with proper overhangs.
Luciferase Reporter Assay
A 4818 bp pGL3-Basic plasmid (Promega) containing a MCS, ampicillin resistance gene, and Luciferase reporter gene was elected to serve as our assay vector. SmaI (New England Biolabs (NEB)) was utilized as a restriction enzyme and incubated for 15 min with pGL3-Basic to produce blunt ends in the MCS. The cleaved product was filtered through a 1% agarose gel and retrieved through dissection and resuspension. The consensus, scrambled and ERE-1 through ERE-4 oligonucleotide sequences were respectively cloned into cleaved pGL3-Basic vectors via the Gibson Assembly Kit and protocol (NEB). The cloned vectors were transformed with DH5α E. coli cells plated on ampicillin-selective agar dishes with overnight incubation. Ten colonies were selected from the transformation growth plates of each vector and inoculated into LB broth. The set of LB broth tubes were incubate overnight with vigorous shaking. The replicated cells were precipitated by centrifuge with discarding of the supernatant. The cloned vectors were purified following cell lysis and extraction using a Plasmid Miniprep Kit (Qiagen). Concentrations of these products were quantified through absorptive spectroscopy. HeLa cells were seeded in stripped serum media for 48 h. Cells were trypsinized and seeded in a 96 well-plate at 104 cells/well with stripped serum and antibiotics. After 24 h, the cells were transfected with 30 μL Opti-MEM, 1.2 μL Lipofectamine LTX, 600 ng ERE-Luceferase pGL3-Basic vector, 12 ng Erα and 6 ng Renilla Luciferase for standardization of signal. Plates were incubated at 37 °C for 6 h, and solution was changed to 100 μL stripped serum media with antibiotics. Half of the wells were stimulated with E2 at 100 nM for 20 h. Wells were washed and cells were suspended in lysis media with gentle shaking for 15 min. As per Promega Luciferase protocol, the well-plate was loaded into a dispenser/reader device. One hundred microliters of LAR II and Stop and Glo Reagent were sequentially dispensed, with each followed by a 5–10-s read time for Luciferase luminal output and Renilla luminal output, respectively.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay (EMSA) was conducted for the purpose of evaluating direct binding between Erα and the set of ERE sequences. A 10 well 6% polyacrylamide gel was prepared and pre-run for 1 h. Erα activation was accomplished by incubating 5 μL of Erα with 1 μL of 10–4 M E2 and 4 μL nuclease-free water for 80 min on ice. Binding reactions were generated according to Thermo Scientific protocols utilizing final concentrations of 1× binding-buffer, 50 ng/μL poly(dI·dC), 10% glycerol, 0.05% NP-40, 10 mM MgCl2, 1 mM EDTA, 250 mM Erα, 0.2 μg Anti-Erα Antibody, 10 fmol 5′-biotin labeled DNA, and 50 pmol unlabeled DNA competitor. Five microliters of 5× Loading Buffer was added, and 20 μL of total solution loaded into each well. A potential difference of 100 V was established across the gel, which ran for 1 h. A 9 × 7.5-cm Hybond N+ nylon membrane was cut, given a clipped corner for orientation, and soaked in 0.5× TBE for 10 min with filter paper. The gel and membrane were sandwiched in a clean electrophoretic transfer unit flanked by filter paper and sponges. Binding reactions were transferred to the membrane at 380 mA for 30 min. The transferred DNA was cross-linked to the membrane by passing the membrane under a hand-held 312 nm UV lamp at 0.5 cm for 10 min. The dry membrane was processed using LightShift™ Chemiluminescent EMSA Kit (Thermo Scientific). Membrane blocking was followed by supplementation of Stabilized Streptavidin-Horseradish Peroxidase Conjugate. The membrane was washed four times with Wash Buffer then incubated with substrate equilibration buffer for 5 min. A substrate working solution consisting of 6 mL Luminol/Enhancer Solution and 6 mL Stable Peroxide Solution was prepared. This solution covered the membrane surface for 5 min. A digital CCD camera was employed at an exposure time of 2 min in capturing the chemiluminescent bands.
Chromatin Immunoprecipitation and ReChIP Assays
Neuro2a cells were grown in cFBS in 100-mm plates. Seventy-two hours after media change to estrogen devoid media (5% cFBS) cells were treated with estrogen for 1 h. Cells were then double fixed with 2 mM EGS (Thermo Scientific) for 35 min then with 1% formaldehyde in phosphate-buffered saline (PBS) for 15 min, washed in ice cold PBS for 5 min, and then quenched with glycine stop buffer (125 mM Glycine in PBS) for 5 min. Cells were subsequently washed in ice cold PBS for 5 min. Cells were then scraped in PBS containing phenylmethylsulfonyl fluoride (PMSF), spun, and resuspended in lysis buffer (50 mM HEPES pH 8.0, 1 mM EDTA, 140 mM NaCl, 10% glycerol, 0.5% NP-40, and 0.25% Triton X-100) to sit on ice for 30 min. Lysate was then homogenized with a Type-B Dounce homogenizer using 15 strokes per sample. Nuclear fraction was then collected by pelleting and resuspended in 600 μL of sonication buffer (10 mM Tris pH 8.0, 1 mM EDTA, 140 mM NaCl, 1% SDS, 0.1% sodium deoxycholate, and 1% Triton X-100). Cells were then sonicated using a Misonix S-4000 sonicator 11 times for 10 s at 20% power with a 30 s interval (~55 J). Ten microliters of the sheared DNA was aliquoted out and considered input DNA. For chromatin immunoprecipitation (ChIP), 100 μL of sheared DNA was diluted 10-fold in buffer Y (16.7 mM Tris pH 8.1, 167 mM NaCl, 0.01% SDS, 1.1% Triton X-100, and 1.2 mM EDTA). Diluted sheared DNA was precleared with 2 μg of non-targeting IgG antibody and salmon sperm coated DNA Agarose Beads (Millipore) for 1 h at 4 °C. Beads were spun at 0.6 relative centrifugal force (RCF) for 1 min and supernatant was moved to a fresh tube. Two micrograms of specific antibody was then used for overnight IP at 4 °C. Salmon sperm coated DNA Agarose Beads were then added for 1.5-h incubation. Beads were spun at 0.6 RCF for 1 min. Beads were washed once in TSEI (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.1, 150 mM NaCl), once in TSEII (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.1, 500 mM NaCl), once in TSEIII (0.5 M LiCl, 1% NP-40, 1% Sodium Deoxycholate, 1 mM EDTA, 10 mM Tris pH 8.1, 150 mM NaCl) and then twice in TE (1 mM EDTA, 10 mM Tris pH 8.1) for 5 min each. DNA was then eluted twice using Elution Buffer (1% SDS and 0.1 M NaHCO3) for 15 min each elution, 75 μL of buffer each time. 10 μL of input DNA was diluted 1:10 with buffer Y. One hundred fifty microliters of elutions and input DNA were then reverse cross-linked by adding NaCl to a final concentration 200 mM and incubating for 5 h or overnight at 65 °C. Proteins were removed by bringing elutions to a final concentration of 10 mM EDTA and 40 mM Tris pH 6.8 with 1.25 units of Proteinase K for 1.5 h at 45 °C. DNA from elutions was then purified using a Qiagen PCR purification kit. DNA was used directly in qPCR reactions. Antibodies used targeted pRNA Pol II (Abcam 5095-100), E6ap (H182, Santa Cruz), and Erα (HC20). To identify other protein components in protein-DNA complexes, Re-ChIP assays were performed in which the cross-linked immunocomplex was eluted from the first ChIP with 10 mM dithiothreitol at 37 °C for 30 min, and then the product was diluted 50-fold in 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl (pH 8.1). Re-ChIP assay was performed with a different antibody.
Quantitative Real-Time PCR
For mRNA qPCR, 4% of the cDNA was used in 12.5-μL reactions for the real-time PCR reactions. Cycle threshold (CT) values were normalized to Calnexin mRNA levels. Total primer concentration was 200 μM for forward and reverse primers. iQ SYBR Green mastermix from Bio-Rad was used in 12.5 μL total reaction volumes. Real-time PCR was carried out on a LightCycler 480 (Roche). CT values were first normalized to input DNA and then compared to zero-minute time point. PCR cycles used were 3 min melting at 95 °C, 1 min extension at 60 °C, and 10 s melting at 95 °C for 40 cycles.
Input DNA from the ChIP protocol was diluted 1:50 and ChIP DNA was diluted 1:5 for the qPCR reactions. Ten-microliter reaction volumes were used. CT values of ChIP samples were normalized to input DNA CT values. Parameters for the qPCR were same as the mRNA qPCR. Primers employed are listed in Table S2.
Statistical Analysis
Determination of statistical significance between the various outcomes measured and respective controls occurred in experiments involving qPCR mRNA expressional levels, Luciferase assay luminal readings and ChIP recruitment levels. Quantification of such significances was conducted using the Student’s t test analysis. A two-tailed p value of 0.05 was applied as a standard of significance. The analyses were performed using R™ software (version 3.1.1; R Foundation for Statistical Computing, Vienna, Austria) and plots generated using Excel™ software (version 14.0; Microsoft Corporation, Redmond, WA).
Results
E2 Regulates Pkar2a Protein Expression in N2A Cells
Western blot analysis was undertaken to visualize functional protein changes in N2A cells within the context of estrogen-deprived environments. The qualitative intensity exhibited by a band was employed to correlate directly with expressional levels of the respective protein. The control protein Gapdh band was visualized to be relatively equivalent in the E2 deprived and E2 provided states. In contrast, the Pkar2a band demonstrated a distinct enhancement in intensity with the addition of E2 relative to the E2 absent state. Indeed, densitometric analysis of the bands quantitatively support the qualitative observation and present an approximate 3-fold increase in protein expression of Pkar2a after treating Neuro2a cells with E2 (Fig. 1).

a Western blot band results demonstrating qualitative amplification of Pkar2a protein expression upon supplementation of mouse neuroblastoma cells with E2. Glyceraldehyde 3-phosphate dehydrogenase (Gapdh) stable expression displayed as control reference. Molecular marker (MM) bands shown at far left. b Densitometric analysis of Pkar2a protein expression in Neuro2a cells treated with E2 or vehicle. Average normalized OD values were used to plot respective diagram. OD optical density, MM molecular marker
E2 and E6ap Regulate Pkar2a mRNA Expression
qPCR was utilized in measuring Pkar2a mRNA expressional levels in order to delineate factors responsible for generating an appropriate response to E2. Perturbation of such factors would be rationalized to inhibit a significant expressional alteration upon E2 supplementation relative to the devoid state. Control mRNA quantities were evaluated in the context of siScramble and the absence of E2. This baseline measure was normalized to a fold change of unity. Supplementation of E2 resulted in a 2.74-fold change with standard error (SE) of ±0.03. Further addition of Tamoxifen suppressed expression below baseline to a 0.78-fold change (SE ± 0.17). Repetition of these conditions in the context of E6ap silencing using siE6ap provided a 0.83-fold change (SE ± 0.18) in the absence of E2. With supplementation of E2, however, a lack of significant mRNA expression was observed with a 0.73-fold change (SE ± 0.15). Similarly, further addition of Tamoxifen yielded a 0.75-fold change (SE ± 0.03) (Fig. 2).

qPCR analysis of Pkar2a mRNA expression regulated by E2 in Neuro2a cells at 6 h after 10 nM E2 treatment following Scrambled silencing and E6ap silencing, respectively. Both were silenced in the context of control (−), E2 (E), and E2 with Tamoxifen (E + T). The bar designated with an asterisk portrays a significant Student’s t test analysis (p < 0.05)
Bioinformatics Yield four Putative EREs
Bioinformatics was employed in identifying sites potentially responsive to Er within the Pkar2a promoter in accounting for the observed E2 modulation. Scanning of the Pkar2a promoter sequence utilizing the two independent databases described (JASPAR and Cluster-Buster) generated four putative ERE sequences per the inputted filtration criteria. Only one of the four ERE sequences produced was predicted with high confidence (>85%) and was termed ERE-1. Base pair compositions of the identified sequences are listed as a subgroup of Table S1.
ERE-1 and ERE-4 Are Functional in the Presence of E2
Luciferase reporter gene assay provided a measure of the relative gene transcriptional activity present. In the context of E2 activated Erα, a successful ERE sequence would exhibit increased Luciferase gene expression and subsequent luminal output. Use of this reporter gene assay was thus to quantify, through a standard surrogate, the degree of downstream transcriptional activity conferred by each identified putative ERE. Luciferase output is referenced and normalized to Renilla luminal output to account for fluctuations in cell number and state for each well. The scrambled ERE demonstrated an average Luciferase luminal output to Renilla luminal output of 2.84 in the absence of E2, and 1.96 in the presence of E2 (p value = 0.021). Such average ratios for the consensus ERE were 9.53 and 87.39 respectively (p = 0.007). ERE-1 ratios were 8.54 and 60.32 (p = 0.033). ERE-2 ratios were 2.63 and 1.58 (p = 0.058). ERE-3 ratios were 0.94 and 0.72 (p = 0.296). ERE-4 ratios were 14.86 and 59.73 (p = 0.001). The scrambled ERE demonstrated a 95% confident limit (2 times the standard error) in the output ratios of 0.28 in the absence of E2, and 0.13 in the presence of E2. Such 95% confident limits for the consensus ERE were 0.76 and 11.18, respectively. ERE-1 confident limits were 0.71 and 15.77. ERE-2 confident limits were 0.07 and 0.44. ERE-3 confident limits were 0.03 and 0.26. ERE-4 confident limits were 1.41 and 4.11 (Fig. 3).

Putative Pkar2a ERE promoter motifs functionality in a Luciferase reporter gene assay. A synthetized oligonucleotide sequence containing two scrambled (Scr-ERE), consensus. Cons-ERE or putative Pkar2a ERE (ERE) motifs were inserted upstream of the Luciferase gene in the pGL3-basic vector. These Luciferase reporter constructs were cotransfected with a control reporter vector pRL-SV40 encoding for Renilla Luciferase into Hela cells. Luciferase activity was measured for samples treated with vehicle (−E2) or after treatment of E2 deprived cells with 10 nM E2 for 20 h. n = 3 experiments. Bars designated with asterisks portray a significant Student’s t test analysis (p < 0.05)
Erα Displays Direct Binding to ERE-1 in Presence of E2
EMSA allowed for DNA band visualization as a result of a chemiluminescent reaction coupled to the 5′-biotin attachment. This experiment is critical in demonstrating the physical binding necessary for a potential ERE to elicit Erα-dependent transcriptional activity. The scrambled ERE sequence displayed no shift in the presence of Erα relative to its baseline, independent of the presence or absence of E2. In the presence of E2, the consensus ERE sequence exhibited an increased intensity shift with addition of Erα, along with a clear super-shift upon further addition of anti-Erα antibody. In the absence of E2, the Erα shift was attenuated, and Erα with antibody super-shift completely eliminated. In both cases, competitive saturation of the solutions with excess unlabeled consensus ERE resulted in returning of the band to baseline. In the presence of E2, the ERE-1 sequence exhibited a shift with addition of Erα, as well as a super-shift with supplemental antibody. In the absence of E2, the Erα based shift was eliminated; however, a band corresponding to the super-shift position was retained in the Erα plus antibody lane. In both cases, competitive saturation with excess unlabeled ERE-1 produced a smeared returning of the labeled band to baseline. Saturation with excess unlabeled consensus ERE yielded a relatively cleaner returning of the labeled ERE-1 band to baseline. A lack of any significant shifts from baseline was demonstrated in the bands corresponding to ERE-2, ERE-3 and ERE-4. As such, the results were uniformly invariant between the states characterized by the presence or absence of E2 (Fig. 4).

a, b EMSA bands displaying shifts and super-shifts in consensus ERE (Cons) and c ERE-1 in the presence of E2 relative to the deprived state. These shifts are competitively suppressed by excess unlabeled ERE-1 “Unlab(e)” and unlabeled consensus “Unlab(c)”. Such shifts are not observed in the remaining (a, b, d–f) ERE sequences: Scr, ERE-2, ERE-3, and ERE-4
Recruitment of E6ap-Erα to ERE-1 Is Associated with Promotion of Pkar2a mRNA Expression
ChIP analysis is ultimately required to display appropriate formation of a specific complex accounting for the mechanisms suggested by the prior experimental outcomes. Quantification of protein recruitment to specific DNA sites analyzed by ChIP through qPCR demonstrated substantial variability. All outcomes were measured in fold recruitment with controls, defined by the absence of E2, uniformly normalized to unity. ChIP assessment of pRNA Pol II recruitment to the TSS was standardized as a positive control (SE ± 0.15) and displayed a 7.3-fold recruitment with addition of E2 (SE ± 0.6). ChIP/Re-ChIP results evaluating Erα-E6ap complex recruitment to each putative ERE sequence were similarly ascertained using qPCR. In the cases of ERE-1 (SE ± 0.04), ERE-2 (SE ± 0.04), ERE-3 (SE ± 0.2), and ERE-4 (SE ± 0.1), the fold recruitments exhibited upon E2 addition were 5.7 (SE ± 0.3), 1.0 (SE ± 0.3), 1.2 (SE ± 0.3), and 1.6 (SE ± 0.4), respectively (Fig. 5). Statistically significant changes in the degree of recruitment were observed in the positive control and ERE-1. These results demonstrate congruency and act in confirmation towards the outcomes suggested by the previously mentioned assays.

Plots of ChIP detected pRNA Pol II recruitment at a the Pkar2a TSS. ChIP/Re-ChIP detected E6ap-Erα complex recruitment at b ERE-1, c ERE-2, d ERE-3, and e ERE-4 on the promoter of Pkar2a. For each element evaluated, fold recruitment was normalized to unity in the absence of estradiol (−E2) and displayed as a relative multiple with the addition of estradiol (+E2). Bars designated with an asterisk portray significant outcomes (p < 0.05)
Discussion
The Pkar2a mRNA expression was determined to be contingent upon the presence of both E2 and E6ap. The lack of either generated a qPCR detected fold change statistically comparable to the negative control defined as lacking both. The dependency on E2 and the abrogation of increased expression by treatment with the Erα antagonist Tamoxifen implicate underlying transcriptional activity through Erα as a potential mechanism of expression. Similarly, the requirement for E6ap indicates its presence in the transcriptional complex formed, with emphasis on its role as a coregulator for Erα. Such findings are in accordance with the dual-functions comprising the cellular roles of E6ap [34].
In evaluating the transcriptional capabilities of Erα in the context of each ERE sequence, a reported gene assay was performed. Given that Renilla normalized output was significantly enhanced in the cases of ERE-1 and ERE-4 with the addition of E2, such sequences exhibited capacity to upregulate a downstream gene. This is relevant to the confirmed presence of ERE-1 and ERE-4 in the promoter region of Pkar2a. They may play the role of a nidus in explaining the E2 dependent transcription of Pkar2a mRNA through Erα.
Experimental detection of direct binding of Erα to ERE-1 was ascertained through EMSA. A distinct band corresponding to the Erα-ERE-1 site was visualized following the addition of E2 to the mixture. The antibody dependent super-shift band was similarly acquired in this case; however, it was also apparent in the E2 deficient state implying a propensity towards enhancing binding. Asides from the trivial consensus sequence, this direct binding to Erα is exclusively found for ERE-1, as opposed to the Scr and remaining ERE sequences. ChIP and Re-ChIP results reinforced the EMSA outcomes. Immunoprecipitation of a complex consisting of Erα and E6ap at the promoter region for Pkar2a using ERE-1 was significant with E2 supplementation. The ChIP positive control employed was based upon the amplified association of pRNA Pol II to the TSS with E2. These results effectively confirmed the capacity of the ERE-1 sequence to bind Erα in the presence of E2 and upregulate the transcription of Pkar2a mRNA in conjunction with E6ap in neuroblastoma cells (Fig. 6).

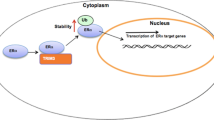
General schematic representation of E2-activated Erα regulation of Pkar2a gene transcription in the context of coregulation by E6ap in neuroblastoma cells. Complex formation of Erα and E6ap at the ERE promote recruitment of RNA polymerase and transcription factors to the transcription start site. ERE estrogen response element, TFs general transcription factors, TSS transcription start site
E6ap was traditionally studied exclusively as an E3-ubiquitin ligase protein. In light of its relatively recent recognition as a transcriptional coregulator to SHRs, its domain of functions has been substantially expanding [34]. Mapping such relationships governing its interactions with particular sets of genes may uncover roles for endocrine hormonal influence in regions outside of their currently established scope. Interestingly, recent evidence demonstrates PKA control of E6ap through phosphorylation, potentially revealing a feedback control loop pertinent to our findings [25]. Elucidating these molecular pathways may provide the benefit of exposing multiple opportunities for intervention in a series of interacting components. In the particular instance of Pkar2a regulation via E6ap, therapeutics developed for endocrine purposes may now portray novel applicability. This specific upstream mechanism of transcriptional modulation by E6ap is significant by virtue of the vast range of activities the PKA effector is responsible for, including metabolic and neuronal development [35, 36].
The gene encoding E6ap, UBE3A, is harbored on chromosome 15 in humans [37]. Certain genetic pathologies are documented to be location-specific in targeting chromosome 15. Angelman syndrome and Prader-Willi syndrome are two such disorders with roots in the epigenetic modifications of parental inheritance patterns [38–40]. In particular, Angelman syndrome is characterized by a loss of the genetic infrastructure required for encoding functional E6ap [37, 41, 42]. The clinical manifestations of the disorder may be partially attributed to a deficiency in the biochemical activities E6ap is known to partake in. Analogously, insufficiencies in our knowledge regarding the downstream molecular behaviors E6ap is responsible in regulating are prohibitive towards therapeutic development in these diseases [34]. Supplementation of such downstream elements E6ap aids in expressing may be beneficial to patients with respect to certain symptoms. Such an approach is commonly successful in biochemical pathologies involving the absence of a critical enzyme or molecule.
In conclusion, Pkar2a is directly regulated by Erα and E6ap in the presence of estrogen stimulus. ERE-1 is found to act as an appropriate site for E2-dependent Erα transcription of Pkar2a mRNA in Neuro2a cells. This activity is contingent upon the presence of E6ap, a known SHR coregulator. PKA is known to play a prominent role towards the clinical characteristics of neuroblastoma cancers in addition to neurological disorders [21–23]. In particular, Angelman syndrome, which is defined by loss of E6ap functionality, may thus attribute certain factors to perturbation of this critical pathway. Knowledge regarding this novel dependence of Pkar2a transcription to such molecules and receptors may aid future investigations in evaluating methods of intervention. Potential therapeutics may be focused at restoring or interfering with upstream or downstream components with intentions to compensate for inherent deficiencies in the other.
Abbreviations
- E6ap:
-
E6-associated protein
- Pkar2a:
-
Protein kinase A regulatory subunit R2A
- E2:
-
Estradiol
- Erα:
-
Estrogen receptor
- ERE:
-
Estrogen response element
- TSS:
-
Transcription start site
- Cons:
-
Consensus sequence
- Scr:
-
Scrambled sequence
References
Scheffner M, Huibregtse JM, Vierstra RD, Howley PM (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75(3):495–505
Rolfe M, Beer-Romero P, Glass S, Eckstein J, Berdo I, Theodoras A, Pagano M, Draetta G (1995) Reconstitution of p53-ubiquitinylation reactions from purified components: the role of human ubiquitin-conjugating enzyme UBC4 and E6-associated protein (E6AP). Proc Natl Acad Sci U S A 92(8):3264–3268
Nawaz Z, Lonard DM, Smith CL, Lev-Lehman E, Tsai SY, Tsai MJ, O’Malley BW (1999) The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol Cell Biol 19(2):1182–1189
Nawaz Z, O’Malley BW (2004) Urban renewal in the nucleus: is protein turnover by proteasomes absolutely required for nuclear receptor-regulated transcription? Mol Endocrinol 18(3):493–499. doi:10.1210/me.2003-0388
Gao X, Loggie BW, Nawaz Z (2002) The roles of sex steroid receptor coregulators in cancer. Mol Cancer 1:7
Heger Z, Zitka O, Krizkova S, Beklova M, Kizek R, Adam V (2013) Molecular biology of beta-estradiol-estrogen receptor complex binding to estrogen response element and the effect on cell proliferation. Neuro Endocrinol Lett 34(Suppl 2):123–129
McKenna NJ, Lanz RB, O’Malley BW (1999) Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev 20(3):321–344. doi:10.1210/edrv.20.3.0366
Mueller-Fahrnow A, Egner U (1999) Ligand-binding domain of estrogen receptors. Curr Opin Biotechnol 10(6):550–556
Klinge CM (2001) Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res 29(14):2905–2919
Roskoski R Jr (2015) A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res 100:1–23. doi:10.1016/j.phrs.2015.07.010
Johnson LN (2009) The regulation of protein phosphorylation. Biochem Soc Trans 37(Pt 4):627–641. doi:10.1042/BST0370627
Whitmarsh AJ, Davis RJ (2000) Regulation of transcription factor function by phosphorylation. Cell Mol Life Sci 57(8–9):1172–1183
Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP (2013) PKA: lessons learned after twenty years. Biochim Biophys Acta 1834(7):1271–1278. doi:10.1016/j.bbapap.2013.03.007
Kopperud R, Krakstad C, Selheim F, Doskeland SO (2003) cAMP effector mechanisms. Novel twists for an “old” signaling system. FEBS Lett 546(1):121–126
Otten AD, McKnight GS (1989) Overexpression of the type II regulatory subunit of the cAMP-dependent protein kinase eliminates the type I holoenzyme in mouse cells. J Biol Chem 264(34):20255–20260
Titani K, Sasagawa T, Ericsson LH, Kumar S, Smith SB, Krebs EG, Walsh KA (1984) Amino acid sequence of the regulatory subunit of bovine type I adenosine cyclic 3',5'-phosphate dependent protein kinase. Biochemistry 23(18):4193–4199
Mellon PL, Clegg CH, Correll LA, McKnight GS (1989) Regulation of transcription by cyclic AMP-dependent protein kinase. Proc Natl Acad Sci U S A 86(13):4887–4891
Skalhegg BS, Tasken K (2000) Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front Biosci 5:D678–D693
Lania AG, Mantovani G, Ferrero S, Pellegrini C, Bondioni S, Peverelli E, Braidotti P, Locatelli M et al (2004) Proliferation of transformed somatotroph cells related to low or absent expression of protein kinase a regulatory subunit 1A protein. Cancer Res 64(24):9193–9198. doi:10.1158/0008-5472.CAN-04-1847
Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, Van Houten B, Cherra SJ 3rd et al (2011) Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinsonʼs disease. Cell Death Differ 18(12):1914–1923. doi:10.1038/cdd.2011.74
Dagda RK, Pien I, Wang R, Zhu J, Wang KZ, Callio J, Banerjee TD, Dagda RY et al (2014) Beyond the mitochondrion: cytosolic PINK1 remodels dendrites through protein kinase A. J Neurochem 128(6):864–877. doi:10.1111/jnc.12494
Kim SN, Kim SG, Park SD, Cho-Chung YS, Hong SH (2000) Participation of type II protein kinase A in the retinoic acid-induced growth inhibition of SH-SY5Y human neuroblastoma cells. J Cell Physiol 182(3):421–428. doi:10.1002/(SICI)1097-4652(200003)182:3<421::AID-JCP13>3.0.CO;2-2
Kume T, Kawato Y, Osakada F, Izumi Y, Katsuki H, Nakagawa T, Kaneko S, Niidome T et al (2008) Dibutyryl cyclic AMP induces differentiation of human neuroblastoma SH-SY5Y cells into a noradrenergic phenotype. Neurosci Lett 443(3):199–203. doi:10.1016/j.neulet.2008.07.079
Sen RP, Delicado EG, Miras-Portugal MT (1999) Differential modulation of nucleoside transport types in neuroblastoma cells by protein kinase activation. Neuropharmacology 38(7):1009–1015
Yi JJ, Berrios J, Newbern JM, Snider WD, Philpot BD, Hahn KM, Zylka MJ (2015) An autism-linked mutation disables phosphorylation control of UBE3A. Cell 162(4):795–807. doi:10.1016/j.cell.2015.06.045
Le Goff P, Montano MM, Schodin DJ, Katzenellenbogen BS (1994) Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem 269(6):4458–4466
Miller WR, Hulme MJ, Bartlett JM, MacCallum J, Dixon JM (1997) Changes in messenger RNA expression of protein kinase A regulatory subunit ialpha in breast cancer patients treated with tamoxifen. Clin Cancer Res 3(12 Pt 1):2399–2404
Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, Floore A, Velds A et al (2004) Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer Cell 5(6):597–605. doi:10.1016/j.ccr.2004.05.016
Bentin Toaldo C, Alexi X, Beelen K, Kok M, Hauptmann M, Jansen M, Berns E, Neefjes J et al (2015) Protein kinase A-induced tamoxifen resistance is mediated by anchoring protein AKAP13. BMC Cancer 15:588. doi:10.1186/s12885-015-1591-4
Portales-Casamar E, Thongjuea S, Kwon AT, Arenillas D, Zhao X, Valen E, Yusuf D, Lenhard B et al (2010) JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res 38:D105–D110. doi:10.1093/nar/gkp950
Frith MC, Li MC, Weng Z (2003) Cluster-buster: finding dense clusters of motifs in DNA sequences. Nucleic Acids Res 31(13):3666–3668
Gruber CJ, Gruber DM, Gruber IM, Wieser F, Huber JC (2004) Anatomy of the estrogen response element. Trends Endocrinol Metab 15(2):73–78. doi:10.1016/j.tem.2004.01.008
Martinez E, Wahli W (1989) Cooperative binding of estrogen receptor to imperfect estrogen-responsive DNA elements correlates with their synergistic hormone-dependent enhancer activity. EMBO J 8(12):3781–3791
El Hokayem J, Nawaz Z (2014) E6AP in the brain: one protein, dual function, multiple diseases. Mol Neurobiol 49(2):827–839. doi:10.1007/s12035-013-8563-y
Dagda RK, Das Banerjee T (2015) Role of protein kinase A in regulating mitochondrial function and neuronal development: implications to neurodegenerative diseases. Rev Neurosci 26(3):359–370. doi:10.1515/revneuro-2014-0085
Dickson LM, Gandhi S, Layden BT, Cohen RN, Wicksteed B (2016) Protein kinase A induces UCP1 expression in specific adipose depots to increase energy expenditure and improve metabolic health. Am J Physiol Regul Integr Comp Physiol:ajpregu 00114:02016. doi:10.1152/ajpregu.00114.2016
Kishino T, Lalande M, Wagstaff J (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15(1):70–73. doi:10.1038/ng0197-70
Buiting K (2010) Prader-Willi syndrome and Angelman syndrome. Am J Med Genet C Semin Med Genet 154C(3):365–376. doi:10.1002/ajmg.c.30273
Butler MG, Palmer CG (1983) Parental origin of chromosome 15 deletion in Prader-Willi syndrome. Lancet 1(8336):1285–1286
Magenis RE, Brown MG, Lacy DA, Budden S, LaFranchi S (1987) Is Angelman syndrome an alternate result of del(15)(q11q13)? Am J Med Genet 28(4):829–838. doi:10.1002/ajmg.1320280407
Chamberlain SJ, Lalande M (2010) Angelman syndrome, a genomic imprinting disorder of the brain. J Neurosci 30(30):9958–9963. doi:10.1523/JNEUROSCI.1728-10.2010
Horsthemke B, Wawrzik M, Gross S, Lich C, Sauer B, Rost I, Krasemann E, Kosyakova N et al (2011) Parental origin and functional relevance of a de novo UBE3A variant. Eur J Med Genet 54(1):19–24. doi:10.1016/j.ejmg.2010.09.005
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by a postdoctoral fellowship from FAST (Foundation for Angelman Syndrome Therapeutics) granted to Dr. Jimmy El Hokayem.
Disclosure Statement
The authors have nothing to disclose.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Obeid, JP., Zeidan, Y.H., Zafar, N. et al. E6-Associated Protein Dependent Estrogen Receptor Regulation of Protein Kinase A Regulatory Subunit R2A Expression in Neuroblastoma. Mol Neurobiol 55, 1714–1724 (2018). https://doi.org/10.1007/s12035-017-0449-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0449-y