
Abstract
Parthanatos is a form of PARP-1-dependent programmed cell death. The induction of parthanatos is emerging as a new strategy to kill gliomas which are the most common type of primary malignant brain tumor. Oxidative stress is thought to be a critical factor triggering parthanatos, but its underlying mechanism is poorly understood. In this study, we used glioma cell lines and H2O2 to investigate the role of JNK in glioma cell parthanatos induced by oxidative stress. We found that exposure to H2O2 not only induced intracellular accumulation of ROS but also resulted in glioma cell death in a concentration- and incubation time-dependent manner, which was accompanied with cytoplasmic formation of PAR polymer, expressional upregulation of PARP-1, mitochondrial depolarization, and AIF translocation to nucleus. Pharmacological inhibition of PARP-1 with 3AB or genetic knockdown of its level with siRNA rescued glioma cell death, as well as suppressed cytoplasmic accumulation of PAR polymer and nuclear translocation of AIF, which were consistent with the definition of parthanatos. Moreover, the phosphorylated level of JNK increased markedly with the extension of H2O2 exposure time. Either attenuation of intracellular ROS with antioxidant NAC or inhibition of JNK phosphorylation with SP600125 or JNK siRNA could significantly prevent H2O2-induced parthanatos in glioma cells. Additionally, inhibition of JNK with SP600125 alleviated intracellular accumulation of ROS and attenuated mitochondrial generation of superoxide. Thus, we demonstrated that JNK activation contributes to glioma cell parthanatos caused by oxidative stress via increase of intracellular ROS generation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Malignant glioma is the most common type of primary malignant brain tumor with higher morbidity [1, 2]. Resistance to apoptosis is found to make glioma cells survive currently used chemotherapy or radiotherapy [2]. Poly(ADP-ribose) synthetase 1 (PARP-1), as a highly expressed nuclear protein that senses DNA-strand breaks and is involved in multiple facets of DNA repair and maintenance of genomic stability in glioma cells [3], was thought to be a factor responsible for the resistance of glioma cells to apoptosis after being mildly activated, and inhibition of PARP-1 activation has been a strategy to recover the sensitivities of glioma cells to chemotherapy or radiotherapy [4–7]. However, recent evidences have shown that hyper-activation of PARP-1 leads to cell death [8, 9], which is established as a new modality of programmed cell death and named as “parthanatos” to distinguish it from caspase-dependent apoptosis, necrosis, and other modalities of cell death [3].
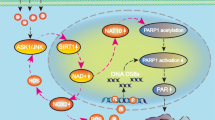
According to a cell death nomenclature committee, parthanatos is defined by two main criteria. One is that excessive poly(ADP-ribose) (PAR) synthesis should accompany cell death, and the other is that the cell death should be completely or partially prevented by PARP-1 deletion or inhibitor treatment [10]. Experimental studies reveal that parthanatos is not only involved in multiple pathological conditions such as diabetes, inflammation, cerebral hypoxia/ischemia, and head trauma [10–13] but also contributed to chemical-induced death in cancer cells such as glioma, esophageal squamous-cell carcinoma, and gastric cancer [8, 9, 14]. As a distinct cell death pathway, parthanatos is characterized by a series of choreographed biochemical events including hyper-activation of PARP-1, cytoplasmic accumulation of PAR polymer, mitochondrial depolarization, and nuclear translocation of apoptosis inducing factor (AIF). Finally, the nuclear AIF initiates chromatinolysis or condensation and results in cell death [3, 10].
Although the activation of PARP-1 is thought to be induced by chromosomal DNA strand nicks and breaks due to the attack of genotoxic agents [3], reactive oxygen species (ROS) was also found to participate in the regulation of parthanatos under the condition of genotoxic stress. Chiu et al. proved that inhibition of ROS attenuated PARP-1-dependent cell death in the mouse embryonic fibroblasts treated with MNNG, an alkylating agent that damages DNA directly [15]. Similarly, ROS was found in our previous study to account for the hyper-activation of PARP-1 in the glioma cells treated with deoxypodophyllotoxin [8]. However, it is still poorly understood which signal pathway is involved in PARP-1 hyper-activation in glioma cells under the condition of oxidative stress, despite that mitogen-activated protein kinase (MAPK) subfamily extracellular signal regulated kinase (ERK) pathway was reported to participate in the regulation of parthanatos caused by oxidative stress in human lymphocytes [16]. c-Jun-N-terminal protein kinase (JNK), which is another subfamily of MAPK, could also be activated by ROS and plays an important role in regulation of programmed glioma cell death [17]. We thus speculate that JNK might have a regulatory effect on parthanatos caused by oxidative stress. Given that H2O2 is a member of ROS that also include superoxide and hydroxyl radicals [18] and extensively used to induce oxidative stress in various types of cancer cells such as gastric carcinoma, cervical cancer, colorectal cancer, breast cancer, and glioma [19–26], we thus used human glioma cell lines and H2O2 in this study to investigate the role of JNK in glioma cell parthanatos during oxidative stress.
Materials and Methods
Reagents
H2O2, 3AB, N-acetyl-l-cysteine (NAC), U0126, SB203580, and SP600125 were purchased from Sigma (St. Louis, MO). Anti-PARP-1, anti-AIF, anti-JNK, anti-phosphor-JNK, and anti-Histone 3 antibodies were from Abcam (Cambridge, MA). Anti-PAR polymer antibody was from Calbiochem (Danvers, MA). Anti-β-actin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Scramble small interfering RNA (siRNA), PARP-1 siRNA, and JNK siRNA were purchased from GenePharma Company (Suzhou, China). Other reagents were purchased from Sigma.
Cell Lines and Culture
Human SHG-44, U251, and U87 glioma cells were obtained from the cell bank of Shanghai Biological Institute, Chinese Academy of Science (Shanghai, China). The cells were cultured in DMEM supplemented with 10 % fetal bovine serum, glutamine (2 mmol/L), penicillin (100 U/mL), and streptomycin (100 μg/mL) and maintained at 37 °C and 5 % CO2 in a humid environment. Cells at the mid-log phase were used in the experiments.
Cell Viability Assay
SHG-44 (1 × 104 cells/well), U251 (5 × 104 cells/well), and U87 (5 × 104 cells/well) glioma cells were seeded onto 96-well microplate and cultured for 24 h and then were treated with target compounds at indicated concentrations for indicated periods. The cellular viability was assessed using an MTT assay and was expressed as a ratio to the absorbance value at 570 nm of the control cells.
LDH Release Cell Death Assay
SHG-44 (1 × 104 cells/well), U251 (5 × 104 cells/well), and U87 (5 × 104 cells/well) glioma cells were seeded onto 96-well microplate and cultured for 24 h. Lactate dehydrogenase cytotoxicity assay kit (Beyotime Biotech, Nanjing, China) was used to assay cellular death rate. According to the manufacturer’s instructions, the absorbance value of each sample was read at 490 nm, and cell death ratio was calculated by using the following formula: cell death ratio % = (Asample − Acontrol / Amax − Acontrol) × 100. Asample is the sample absorbance value; Acontrol is the absorbance value of control group; and Amax is the absorbance value of positive group.
Assessment of Cell Death and Measurement of Mitochondrial Membrane Potential by Flow Cytometry
SHG-44 and U251 glioma cells were collected at indicated time after being treated with target compounds. Then, the Annexin V-FITC detection kit (Invitrogen, Grand Island, NY) was used for assessment of cell death modality as described by the manufacturer’s instruction. The collected cells were washed twice with PBS and resuspended in 400 μL 1× binding buffer (10 mM HEPES/NaOH, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4). Cells (100 μL) were transferred to a 5-mL culture tube containing 5 μL of Annexin V-FITC and 10 μL of propidium iodide and then incubated for 15 min at room temperature in the dark. After 1× binding buffer was added into each tube, the stained cells were analyzed by flow cytometry (FACScan, Becton Dickinson, San Jose, CA). The rate of cell death was analyzed using CELLquest software (Becton Dickinson). Data acquisition was conducted by collecting 20,000 cells per tube, and the numbers of viable and dead cells were determined for each experimental condition.
Mitochondrial membrane potential was determined by the retention of the dye Rhodamine 123 (Beyotime Biotech, Nanjing, China). The collected cells were washed twice with PBS, incubated with 10 μg/mL Rhodamine 123 at 37 °C for 30 min, and washed with PBS, and the fluorescence intensities of Rhodamine 123 were analyzed with flow cytometry.
Measurement of Intracellular ROS and Mitochondrial Superoxide
SHG-44 (5 × 104 cells/well) and U251 (5 × 104 cells/well) glioma cells were seeded onto 96-well microplate and cultured for 24 h and then treated with target compounds. The average level of intracellular H2O2 was evaluated by using redox-sensitive dye DCFH-DA (Beyotime Biotech, Nanjing, China). All the experimental cells were washed twice in PBS and stained in the dark for 30 min with 20 μmol/L DCFH-DA. After the cells were dissolved with 1 % Triton X-100, the fluorescence was measured at an excitation wavelength of 485 nm and an emission wavelength 530 nm using a fluorescence spectrometer (HTS 7000, Perkin Elmer, Boston, MA). The H2O2 levels were expressed as arbitrary unit per milligram protein, then as the percentage of control.
Mitochondrial superoxide was assayed by using MitoSOX red (Invitrogen company, Eugene, OR) as described by the manufacturer’s instruction. The cells were incubated 10 min with 2.0 ml MitoSOX reagent working solution at 5 μmol/L at 37 °C in dark and washed with PBS. The red fluorescence density was measured at an excitation wavelength of 510 nm and an emission wavelength at 580 nm and was expressed as a ratio to the fluorescence in control cells.
Other groups of SHG-44 and U251 glioma cells were seeded onto a culture dish in a diameter of 3 cm and cultured for 24 h. After being treated with H2O2, the cells were stained with DCFH-DA or MitoSOX red as described above and observed under fluorescence microscope (Olympus IX71, Tokyo, Japan).
Immunocytochemical Staining
SHG-44 glioma cells (4 × 105) were seeded onto a culture dish with a diameter of 3 cm and cultured for 24 h. After being treated with 250 μmol/L H2O2 for an indicated time, the cells were fixed in 4.0 % paraformaldehyde, washed with PBS, and incubated with 1% Triton X-100 for 10 min. After the nonspecific antibody binding sites were blocked by 10 % goat serum in PBS containing 0.3 % Triton X-100 and 0.5 % bovine serum albumin (BSA), the cells were incubated with primary antibodies against AIF (1:100) followed by incubation in Alexa-Fluor 488-conjugated goat anti-rabbit IgG (1:200) for 30 min at room temperature. Then, all the cells were incubated with Heochst33342 for 30 min. Finally, the cells were visualized, and images were acquired under laser scanning confocal microscope.
Transfection of siRNA
SHG-44 (5 × 105 cells/mL) and U251 (15 × 105 cells/mL) glioma cells were seeded onto a culture dish with a diameter of 10 cm. Transfection of siRNA was performed by using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions. The siRNA targeting PARP-1 was 5′-GAGCACUUCAUGAAAUUAUTT-3′, and the targeting JNK siRNA was 5′-GCCCAGUAAUAUAGUAGUATT-3′. After siRNA transfection overnight, the cells were incubated with H2O2 at indicated dose for subsequent experiments.
Gel Electrophoresis and Western Blotting
The collected SHG-44 and U251 glioma cells by centrifugation following harvest with a scraper were homogenized with a glass Pyrex microhomogenizer (20 strokes) in ice-cold Tris-buffered saline (TBS; 15 mmol/L, pH 7.6) containing 250 mmol/L sucrose, 1 mmol/L magnesium chloride, 2.5 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol, 1.25 mg/mL pepstatin A, 10 mg/mL leupeptin, 2.5 mg/mL aprotinin, 1.0 mmol/L PMSF, 0.1 mmol/L sodium orthovanadate, 50 mmol/L sodium fluoride, and 2.0 mmol/L tetrasodium pyrophosphate. Homogenates were centrifuged at 1000g for 10 min at 4 °C to obtain the pallets containing nucleus and the supernatants including cytoplasm. Protein was quantified by Bio-Rad protein assay kit (Bio-Rad, Hercules, CA), and equal protein amounts were electrophoresed on 10 % SDS gels and transferred to PVDF membranes (Millipore, Billerica, MA). Membranes were blocked with 3 % bovine serum albumin in TBS for 30 min at room temperature, then incubated overnight at 4 °C with anti-PAR polymer (1:500, Calbiochem, Danvers, MA), anti-AIF (1:1000, Abcam, Cambridge, MA), anti-PARP-1 (1:1000, Abcam), anti-phosphor-JNK (1:1000, Abcam), anti-JNK (1:1000, Abcam), anti-Histone-3 (1:2000, Abcam), and β-actin (1:1000, Santa Cruz Biotechnology). After being incubated 1 h at room temperature with horseradish peroxidase-conjugated IgG (1:1500, Cell Signaling Technology, Danvers, MA), the membranes were washed three times at room temperature with PBS (pH 7.4), and immunoreactive proteins were visualized by enhanced chemiluminescence (Amersham Biosciences, Piscataway NJ) and exposure to Kodak® X-omat LS film (Eastman Kodak Company, New Haven, CT). Densitometry was performed with Kodak® 1D image analysis software (Eastman Kodak Company).
Statistical Analysis
All data represent at least four independent experiments and are expressed as mean ± SD. Statistical comparisons were made using one-way ANOVA. P values of less than 0.05 were considered to represent statistical significance.
Results
H2O2-Induced Glioma Cell Death
MTT assay was used to examine H2O2-induced changes in the viabilities of glioma cells. As shown in Fig. 1, when compared with that of control group, the viabilities of SHG-44, U251, and U87 glioma cells reduced obviously after 24 h exposure to H2O2 at indicated concentrations (Fig. 1a–c). Correspondingly, the cell death ratio increased significantly as revealed by lactate dehydrogenase (LDH) release assay (Fig. 1e, f). Thus, these data indicated that exogenous H2O2 induced glioma cell death in a concentration-dependent manner, and 250 and 500 μmol/L H2O2 were used in the subsequent experiments.

H2O2 inhibited the viabilities of glioma cells and triggered cell death. a MTT assay showed that exogenous H2O2 inhibited the viabilities of SHG-44, U251, and U87 glioma cells in a concentration-dependent manner. b LDH release assay proved that H2O2 induced concentration-dependent cell death in SHG-44, U251, and U87 cells. Data were shown as mean ± SD from three independent experiments. *p < 0.01 versus 0 μmol/L H2O2 group
H2O2-Induced Parthanatos in Glioma Cells
In order to investigate whether parthanatos contributes to the glioma cell death caused by H2O2, the cytoplasmic level of PAR polymer was firstly examined by western blotting. As shown in Fig. 2, the cytoplasmic PAR polymer increased obviously in a concentration-dependent manner when SHG-44 and U251 glioma cells were exposed for 24 h to H2O2 at the dosages from 62.5 to 500 μmol/L or in an incubation time-dependent manner after the cells were incubated from 6 to 24 h with 500 μmol/L H2O2. Meanwhile, the expressional level of PARP-1 was upregulated markedly as well in either cytoplasms or nuclei of glioma cells. These results indicated that exposure to H2O2 not only resulted in expressional upregulation of PARP-1 but also led to its hyper-activation in glioma cells. Consistent with the finding from western blotting showing that nuclear AIF increased with the elevation of cytoplasmic PAR polymer, the images acquired by confocal microscopy revealed that AIF accumulated markedly in the nuclei of the SHG-44 cells exposed for 12 h to 250 μmol/L H2O2 (Fig. 2g). This result indicated that H2O2-induced mitochondrial damage might be associated with cytoplasmic PAR polymer, because AIF is a protein located normally within mitochondria. Furthermore, the mitochondrial membrane potential that controls AIF release was measured by using flow cytometry with Rhodamine 123 staining, showing that 24 h exposure to H2O2 induced concentration-dependent decline in mitochondrial membrane potential (Fig. 3h). Therefore, these results suggested exposure to H2O2 resulted in expressional upregulation and hyper-activation of PARP-1, and the PAR polymer which accumulated within the cytoplasm led to mitochondrial depolarization and AIF translocation into the nucleus.

H2O2 induced changes in parthanatos-related proteins. a, b H2O2 induced cytoplasmic formation of PAR polymer dependently on its concentration and exposure time in SHG-44 and U251 cells. c–f Cytoplasmic and nuclear of PARP-1 and nuclear AIF increased with the elevation of H2O2 concentration and the extension of incubation time in SHG-44 and U251 cells. g Observation with confocal microscopy proved that AIF accumulated in the nuclei of SHG-44 glioma cells at 24 h following exposure to 250 μmol/L H2O2

Pharmacological inhibition of PARP-1 with 3AB protected glioma cells against H2O2 toxicity. a, b MTT assay showed that pretreatment with 3AB at indicated concentrations reversed significantly H2O2-induced reduction in the viabilities of SHG-44 and U251 cells (*p < 0.01 versus the group treated with H2O2 alone). c, d Flow cytometry with Annexin V/PI double staining proved that 500 μmol/L 3AB rescued H2O2-induced cell death in SHG-44 and U251 cells. e–g Five hundred micromoles per liter of 3AB inhibited the increase of cytoplasmic PAR-polymer, suppressed the upregulation of PARP-1 in both cytoplasm and nucleus, and mitigated AIF translocation into nuclei in SHG-44 and U251 cells. h Flow cytometry with Rhodamine 123 staining demonstrated that 500 μmol/L 3AB prevented H2O2-induced decline of mitochondrial membrane potential at 24 h in SHG-44 cells. Data were shown as mean ± SD from three independent experiments
To verify the role of PARP-1 in H2O2-induced glioma cell death, PARP-1 was pharmacologically inhibited by its specific inhibitor 3AB. MTT assay proved that pretreatment 1 h with 3AB at indicated doses increased significantly the viabilities of the SHG-44 and U251 glioma cells exposed to H2O2 (Fig. 3a, b). Flow cytometry analysis with Annexin V and PI double staining demonstrated that the reduced percentage of living cells due to exposure to H2O2 at 250 and 500 μmol/L was, respectively, improved by 500 μmol/L 3AB from 74.82 and 39.36 to 88.58 and 55.11 % in SHG-44 cells and from 77.74 and 41.52 to 87.23 and 85.23 % in U251 cells (Fig. 3c, d). Moreover, the glioma cells with positive staining of Annnexin V (Annexin V+/PI−, Annexin+/PI+) were obviously mitigated. At molecular level, western blotting revealed that 500 μmol/L 3AB inhibited the expressional upregulation of PARP-1, suppressed the increase of cytoplasmic PAR polymer, and mitigated nuclear accumulation of AIF (Fig. 3e–g). Moreover, mitochondrial depolarization at 24 h caused by H2O2 at either 250 or 500 μmol/L was counteracted obviously by administration of 3AB (Fig. 3h).
Then, we knocked down PARP-1 with siRNA to further confirm its role in H2O2-induced glioma cell death. As shown in Fig. 4, western blotting analysis proved that, either in the absence of H2O2 or being treated 24 h with 250 μmol/L H2O2, the glioma cells transfected with PARP-1 siRNA had a lower level of PARP-1, cytoplasmic PAR polymer, and nuclear AIF, respectively, when compared with that in the scramble siRNA group (Fig. 4). This indicated that H2O2-induced elevation in both the cytoplasmic level of PAR polymer and the nuclear level of AIF were regulated by PARP-1. Moreover, MTT assay and flow cytometry analysis proved that H2O2-induced reduction in the cellular viabilities and the living glioma cells (Annexin V−/PI−) was reversed when PARP-1 was knocked down by siRNA (Fig. 4d–f). Particularly, the cells with positive staining of Annexin V were suppressed obviously after being transfected with PARP-1 siRNA.

Knockdown of PARP-1 with small interfering RNA prevented the toxic effect of H2O2 on glioma cells. a, b Western blotting analysis showed that ablation of PARP-1 with siRNA attenuated the upregulation of PARP-1 in cytoplasm and nucleus and mitigated the increase of nuclear AIF in SHG-44 and U251 cells. c Knockdown of PARP-1 with siRNA inhibited cytoplasmic formation of PAR polymer in the SHG-44 and U251 cells that were exposed for 24 h to 250 μmol/L H2O2. d MTT assay showed that the reduced cellular viabilities due to 250 μmol/L H2O2 was partially reversed in both SHG-44 and U251 cells transfected with PARP-1 siRNA. e, f Flow cytometry with Annexin V/PI double staining proved that H2O2-induced cell death in SHG-44 and U251 cells was inhibited when PARP-1 was knocked down by siRNA. Data were shown as mean ± SD from three independent experiments
Therefore, these data suggested that the glioma cell death induced by H2O2 was PARP-1-dependent, which was in line with the criterion used for determination of parthanatos [10].
Intracellular ROS Contributed to Glioma Cell Parthanatos
Intracellular accumulation of ROS is a prominent feature of oxidative stress, and we thus measured exogenous H2O2-induced changes in the intracellular level of ROS by using DCFH-DA. Observation under fluorescence microscope revealed that the glioma cells exposed for 12 h to H2O2 at 500 μmol/L had much brighter green fluorescence than the cells in control group (Fig. 5a), indicating that H2O2 induced marked accumulation of intracellular ROS. Statistical analysis of the fluorescence density showed that the intracellular ROS increased continuously in glioma cells when the incubation time with H2O2 was extended from 1 to 12 h. Moreover, the intracellular ROS level in the 500 μmol/L group was significantly higher than that in the 250 μmol/L group at the incubation time of 6 and 12 h (Fig. 5b, c). This indicated that exogenous H2O2 induced oxidative stress in glioma cells.

H2O2-induced overproduction of ROS could be inhibited by antioxidant NAC. a Fluorescence microscopy with DCFH-DA staining revealed that the green fluorescence in the glioma cells exposed for 12 h to 500 μmol/L H2O2 was obviously brighter than that in control group. b, c Statistical analysis of the fluorescence density showed that exogenous H2O2 induced intracellular accumulation of ROS in a concentration- and incubation time-dependent manner (*p < 0.01 versus control group and 1 h group; # p < 0.05 versus 6 h). d, e Intracellular ROS level induced by 12 h exposure to exogenous H2O2 was significantly mitigated by pretreatment with 2 mmol/L NAC (*p < 0.01 versus the group treated with H2O2 alone)
In contrast, pretreatment with antioxidant NAC at 2 mmol/L not only counteracted the increase of intracellular ROS in the glioma cells exposed for 12 h to H2O2 (Fig. 5d, e) but also significantly reversed H2O2-induced reduction in the cellular viabilities of glioma cells at 24 h (Fig. 6a, b). Flow cytometry analysis demonstrated as well that the living cells were improved significantly, when glioma cells were treated with NAC prior to 24 h exposure to H2O2 (Fig. 6c, d). Furthermore, western blotting showed that inhibition of ROS level with NAC prevented H2O2-induced expressional upregulation of PARP-1, cytoplasmic formation of PAR polymer, and nuclear accumulation of AIF (Fig. 6e–g). Moreover, mitochondrial membrane potential was maintained at a higher level in the glioma cell pretreated with NAC when compared with that in the cells treated with H2O2 alone (Fig. 6h). Therefore, these results indicated that ROS contributed to H2O2-induced parthanatos in glioma cells.

Inhibition of ROS with NAC rescued H2O2-induced glioma cell death via attenuation of parthanatos. a, b MTT assay showed that NAC prevented the viability reduction at 24 h caused by H2O2 in SHG-44 and U251 cells. c, d Flow cytometry with Annexin V/PI double staining proved that H2O2-induced cell death in either SHG-44 or U251 glioma cells was inhibited obviously in the presence of NAC. e–g H2O2-induced cytoplasmic formation of PAR polymer, upregulation of PARP-1, and nuclear accumulation of AIF were all suppressed by NAC. h NAC inhibited H2O2-induced mitochondrial membrane potential decline at 24 h in SHG-44 glioma cells. Data were shown as mean ± SD from three independent experiments
Activation of JNK by ROS-Regulated Glioma Cell Parthanatos
Since JNK is a stress response protein kinase that could be activated by ROS [27], we investigated the role of JNK in H2O2-induced parthanatos. Western blotting analysis showed that 24 h exposure to H2O2 at each indicated concentration did not induce obvious changes in JNK level when compared with that in the control cells but triggered concentration-dependent upregulation of phosphorylated JNK (Fig. 7a). In contrast, administration of antioxidant NAC at 2 mmol/L significantly inhibited H2O2-induced JNK phosphorylation (Fig. 7b), indicating that ROS contributed to JNK activation in the glioma cells treated with H2O2.

Activation of JNK by ROS modulated H2O2-induced parthanatos. a Western blotting showed that H2O2-induced JNK phosphorylation in a concentration-dependent manner. b NAC prevented the phosphorylation of JNK caused by H2O2 in SHG-44 and U251 cells. c SP600125 suppressed H2O2-induced increase in phosphorylated JNK in SHG-44 and U251 cells. d MTT assay demonstrated SP600125 at indicated concentrations protected SHG-44 and U251 cells against H2O2 toxicity at 24 h (*p < 0.01 versus the group treated with H2O2 alone). e, f Flow cytometry with Annexin V/PI double staining showed that 20 μmol/L SP600125 reversed H2O2-induced cell death in SHG-44 and U251 glioma cells. g–i Western blotting revealed that 20 μmol/L SP600125 suppressed H2O2-induced cytoplasmic formation of PAR polymer, upregulation of PARP-1, and nuclear translocation of AIF in SHG-44 and U251 cells. Data were shown as mean ± SD from three independent experiments
Thus, JNK inhibitor SP600125 was used to test the role of JNK activation in H2O2-induced glioma cell parthanatos. As shown by western blotting analysis, prior administration of 20 μmol/L SP600125 effectively counteracted the upregulation of phosphorylated JNK caused by H2O2 at either 250 or 500 μmol/L (Fig. 7c). Moreover, MTT assay and flow cytometry with Annexin V/PI double staining revealed that SP600125 prevented the reduction of glioma cells’ viabilities in a concentration-dependent manner (Fig. 7d), and 40 μmol/L SP600125 rescued markedly the cell death resulting from 24 h exposure to H2O2 (Fig. 7e, f). Western blotting analysis showed that H2O2-induced cytoplasmic formation of PAR polymer, upregulation of PARP-1, and nuclear accumulations of AIF were all attenuated, when JNK activation was inhibited by 40 μmol/L SP600125 (Fig. 7g–i). To further confirm the role of JNK activation in H2O2-induced parthanatos, we knocked down the JNK level with siRNA and examined its effect on H2O2-induced changes in PARP-1, cytoplasmic PAR polymer, and nuclear AIF. As proved by western blotting, ablation of JNK effectively inhibited the increase of phosphorylated JNK caused by H2O2. Moreover, when compared with that in the cells transfected with scramble siRNA, the upregulation of PARP-1, cytoplasmic formation of PAR polymer, and nuclear accumulation of AIF caused by H2O2 were all obviously mitigated in the cells transfected with JNK siRNA (Fig. 8a–d).

JNK regulated the expression of PARP-1 and the production of PAR polymer in the glioma cells exposed to H2O2. a Western blotting analysis showed that knockdown of JNK with siRNA mitigated H2O2-indcued upregulation in the phosphorylated JNK. b Ablation of JNK with JNK siRNA mitigated H2O2-induced cytoplasmic formation of PAR polymer in SHG-44 and U251 cells. c, d Knockdown of JNK with siRNA inhibited H2O2-induced upregulation of PARP-1 and nuclear translocation of AIF in SHG-44 and U251 cells
Therefore, these results suggested that JNK activation contributed to H2O2-induced parthanatos in glioma cells.
JNK Increased Intracellular ROS Level via Regulation of Mitochondrial Generation of Superoxide
Given that intracellular ROS accounted for glioma cell parthanatos caused by exogenous H2O2, we examined the changes of intracellular ROS when JNK was inhibited by SP600125. As shown in Fig. 9a, b, the increased intracellular level of ROS in the glioma cells exposed for 12 h to H2O2 at either 250 or 500 μmol/L was attenuated markedly by pretreatment with JNK inhibitor SP600125 at 20 μmol/L (Fig. 5d, e).

JNK regulated the generation of mitochondrial superoxide. a, b Intracellular ROS level induced by 12 h exposure to exogenous H2O2 was significantly mitigated by pretreatment with 20 μmol/L SP600125 (*p < 0.01 versus the group treated with H2O2 alone). c Fluorescence microscopy with MitoSOX staining showed the red fluorescence in the glioma cells exposed for 12 h to 500 μmol/L H2O2 was markedly brighter than that in control group. d, f Statistical analysis of the fluorescence density proved that 20 μmol/L SP600125 inhibited mitochondrial generation of superoxide caused by H2O2 at 12 h (*p < 0.01 versus control group. # p < 0.01 versus H2O2 group)
Furthermore, MitoSOX, which is a specific probe for mitochondrial superoxide, was used to examine whether mitochondrion is the origin of intracellular ROS, because superoxide is an important member of ROS. Observation under fluorescence microscope revealed that the red fluorescence in the glioma cells treated for 12 h with 500 μmol/L H2O2 was obviously higher than that in control cells (Fig. 9c), indicating that exogenous H2O2 induced overgeneration of mitochondrial superoxide. Moreover, statistical analysis of the fluorescence density showed that superoxide was excessively generated in the mitochondria of the glioma cells treated with H2O2 at 500 μmol/L, not at 250 μmol/L (Fig. 9d, e). This indicated that mitochondrial generation of superoxide is a factor leading to the increase of intracellular ROS caused by exogenous H2O2. In contrast, we found that SP600125 significantly counteracted mitochondrial generation of superoxide caused by H2O2 exposure at 12 h (Fig. 9d, e). Thus, these results suggested that the increase of intracellular ROS in the glioma cells exposed to exogenous H2O2 was partially due to the activation of JNK, which promoted mitochondrial generation of superoxide.
Discussion
In this study, we demonstrated that exogenous H2O2 triggered PARP-1-dependent cell death (parthanatos) in glioma cells via induction of intracellular accumulation of ROS. The phosphorylation of JNK caused by intracellular ROS contributed to expressional upregulation and hyper-activation of PARP-1. Moreover, the increase of intracellular ROS caused by exogenous H2O2 was partially due to the activation of JNK, which promoted mitochondrial overgeneration of superoxide. Therefore, our results suggested that oxidative stress contributed to glioma cell parthanatos via phosphorylation of JNK, which improved intracellular ROS level via promotion of mitochondrial superoxide generation.
Previous reports have shown that exogenous H2O2 could lead to apoptosis and necrosis in various types of cancer cell lines, including gastric carcinoma, cervical cancer, colorectal cancer, and breast cancer [19–26]. Although it was found that H2O2 induced caspase-dependent apoptosis and autophagic cell death in glioma cells [23–26], the present study showed that parthanatos also accounted for the programmed glioma cell death caused by exogenous H2O2. As a new modality of programmed cell death characterized by hyper-activation of PARP-1 and overproduction of PAR polymer, parthanatos is energy-independent and does not lead to the formation of apoptotic body, which is different with apoptosis that is energy-dependent and forms apoptotic body [28]. However, parthanatos has some similarities with apoptosis including phosphatidylserine flipping onto the outer plasma membrane, dissipation of mitochondrial membrane potential, and chromatin condensation [28]. Flow cytometry analysis combined with Annexin V and PI double staining is an effective method to detect phosphatidylserine that flips onto the outer plasma membrane and intracellular accumulation of PI. In this study, the glioma cells with positive staining of Annexin V (Annexin V+/PI−, Annexin+/PI+) decreased significantly when cytoplasmic PAR polymer was attenuated by inhibition of PARP-1 with 3AB or siRNA (Figs. 3c, d and 4e, f), which was consistent with the characteristic of parthanatos [28]. Interestingly, previous studies showed that PAR polymer also plays a role in mediating cell death due to necrosis and apoptosis [29], and caspase activation could accompany the occurrence of parthanatos at late stage [28]. Additionally, Zhao et al. found that parthanatos and RIP-1-mediated necroptosis were both involved in remote lung injury in the rats receiving ischemic renal allografts [30], indicating that parthanatos could occur with other form of programmed cell death.
Hyper-activation of PARP-1 is the key step to initiate parthanatos, which accounts for the synthesis of PAR polymer by using NAD+ [4]. PAR polymer is an effector of parthanatos because it is cytotoxic, which was supported by the finding that exogenous delivery of purified PAR polymer resulted in cell death via induction of mitochondrial depolarization and AIF translocation into the nucleus [4, 31]. In this study, we found that H2O2 induced significant accumulation of PAR polymer in cytoplasm in a concentration- and incubation time-dependent manner. In contrast, pharmacological inhibition of PARP-1 with 3AB or genetic knockdown of its level with siRNA not only inhibited cytoplasmic accumulation of PAR polymer but also rescued glioma cell death. These results were in accordance with the criteria used for determination of parthanatos [9]. Although PAR polymer influences cellular destiny via causing mitochondrial depolarization as demonstrated by our results showing that inhibition of cytoplasmic accumulation of PAR polymer with 3AB reversed H2O2-induced decline in mitochondrial membrane potential, it also could covalently bind with cellular proteins to inhibit or activate their physiological functions. Andrabi et al. found that PARP-1-mediated inhibition of glycolysis partially resulted from PAR polymer binding to hexokinase to cause its dysfunction [32]. In this study, the smear bands detected by anti-PAR antibody on western blotting membrane suggested that PAR polymer might bind with cellular proteins in the glioma cells exposed to H2O2.
Intracellular accumulation of ROS is a typical feature of oxidative stress. Consistent with previous studies showing that exposure to exogenous H2O2 resulted in intracellular accumulation of ROS in cancer cells [19–22], we found as well that the intracellular ROS increased significantly in human glioma cells exposed to H2O2, which demonstrated that exogenous H2O2 caused oxidative stress in glioma cells. Despite that DNA damage is thought to be a major factor leading to the activation of PARP-1 [15], ROS has been found recently to play an important role in regulating PARP-1 hyper-activation. Akhiani et al. reported that ROS-generating myeloid cells and exogenous H2O2 triggered PARP-1-dependent accumulation of PAR polymer in human lymphocytes [16]. Mashimo et al. proved that exposure to H2O2 resulted in parthanatos in mouse embryonic fibroblasts [29]. We reported previously that inhibition of ROS level with antioxidant NAC prevented glioma cell parthanatos induced by deoxypodophyllotoxin [8]. Consistently, we found in this study that inhibition of ROS level with antioxidant NAC counteracted H2O2-triggered glioma cell death via inhibition of parthanatos, indicating that ROS is an initiator to glioma cell parthanatos.
In addition, we thought that the JNK pathway was involved in H2O2-induced glioma cell parthanatos on the basis that pharmacological inhibition of JNK with SP600125 or genetic ablation of its level with siRNA significantly mitigated the upregulation of PARP-1 and cytoplasmic formation of PAR polymer. JNK is a subfamily of MAPK and associated with cell proliferation, motility, metabolism, DNA repair, and death [33]. As a stress-activated protein kinase, JNK was found to be activated under the condition of oxidative stress and participate in modulation of H2O2-induced programmed death in cancer cells [33–35]. Consistently, our results in this study showed that the phosphorylated level of JNK in the glioma cells exposed to H2O2 was obviously mitigated when intracellular ROS level was counteracted by antioxidant NAC. Although the mechanism underlying JNK activation induced by ROS in glioma cells was not investigated in this study, previous reports have shown that ROS modulates JNK activation primarily through two pathways. One is via ROS-sensitive apoptosis signaling kinase 1 (ASK1) to phosphorylate JNK [36], and the other is to maintain the level of phosphorylated JNK via inhibition of MAPK phosphatases-1 (MKP-1) which is involved in the inactivation of JNK [37, 38]. Additionally, another member of the MAPK family, ERK, was reported to be associated with H2O2-induced parthanatos in human lymphocytes [16], despite that we found in this study that neither ERK1/2 inhibitor U0126 nor P38 inhibitor SB203580 could inhibit H2O2-induced parthanatos in glioma cells (data not shown).
Despite that how H2O2 led to the increase of intracellular ROS in glioma cells was not reported previously, we proved that inhibition of JNK with SP600125 attenuated the increase of both intracellular ROS and mitochondrial superoxide caused by H2O2. Particularly, accumulating evidences suggest that JNK activation plays an important role in regulation of intracellular ROS level. Hanawa et al. reported that phosphorylated JNK promoted intracellular ROS via affecting mitochondrial bioenergetics after translocation into mitochondria and inhibition of JNK phosphorylation with SP600125 that suppressed the generation of ROS [39]. Furthermore, Win et al. found that phosphorylated JNK impaired mitochondrial respiration via interplay with Sab(Sh3bp5) [40]. Additionally, Kim demonstrated that JNK inhibited the expression and activity of catalase which accounts for transformation of H2O2 into water [31]. Therefore, we thought that activated JNK enhanced the expression and the hyper-activation of PARP-1 via improvement of intracellular ROS level in glioma cells.
In conclusion, we demonstrated that oxidative stress featured by higher intracellular level of ROS initiated parthanatos in glioma cells via activation of JNK. The activated JNK contributed to glioma cell parthanatos through improvement of intracellular level of ROS.
Abbreviations
- PARP-1:
-
Poly(ADP-ribose) synthetase 1
- PAR:
-
Poly(ADP-ribose)
- NAD+:
-
Nicotinamide adenine dinucleotide
- MAPK:
-
Mitogen-activated protein kinase
- JNK:
-
c-Jun-N-terminal protein kinase
- ERK1/2:
-
Extracellular signal regulated kinases 1 and 2
- AIF:
-
Apoptosis inducing factor
- RIP-1:
-
Receptor interacting protein-1
- ROS:
-
Reactive oxygen species
- LDH:
-
Lactate dehydrogenase
- Nec-1:
-
Necrostatin-1
- 3AB:
-
3-Aminobenzamide
- NAC:
-
N-acetyl-l-cysteine
References
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359:492–507
Beck C, Robert I, Reina-San-Martin B, Schreiber V, Dantzer F (2014) Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp Cell Res 329:18–25
Fatokun AA, Dawson VL, Dawson TM (2014) Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol 171:2000–2016
Galia A, Calogero AE, Condorelli R, Fraggetta F, La Corte A, Ridolfo F, Bosco P, Castiglione R et al (2012) PARP-1 protein expression in glioblastoma multiforme. Eur J Histochem 56, e9
Kase M, Vardja M, Lipping A, Asser T, Jaal J (2011) Impact of PARP-1 and DNA-PK expression on survival in patients with glioblastoma multiforme. Radiother Oncol 101:127–131
Karpel-Massler G, Pareja F, Aimé P, Shu C, Chau L, Westhoff MA, Halatsch ME, Crary JF et al (2014) PARP inhibition restores extrinsic apoptotic sensitivity in glioblastoma. PLoS One 9, e114583
Tentori L, Ricci-Vitiani L, Muzi A, Ciccarone F, Pelacchi F, Calabrese R, Runci D, Pallini R et al (2014) Pharmacological inhibition of poly(ADP-ribose) polymerase-1 modulates resistance of human glioblastoma stem cells to temozolomide. BMC Cancer 14:151
Ma D, Lu B, Feng C, Wang C, Wang Y, Luo T, Feng J, Jia H et al (2016) Deoxypodophyllotoxin triggers parthanatos in glioma cells via induction of excessive ROS. Cancer Lett 371:194–204
Zhao N, Mao Y, Han G, Ju Q, Zhou L, Liu F, Xu Y, Zhao X (2015) YM155, a survivin suppressant, triggers PARP-dependent cell death (parthanatos) and inhibits esophageal squamous-cell carcinoma xenografts in mice. Oncotarget 6:18445–18459
Lee Y, Kang HC, Lee BD, Lee YI, Kim YP, Shin JH (2014) Poly (ADP-ribose) in the pathogenesis of Parkinson’s disease. BMB Rep 47:424–432
Mohammad G, Siddiquei MM, Abu El-Asrar AM (2013) Poly (ADP-ribose) polymerase mediates diabetes-induced retinal neuropathy. Mediat Inflamm 2013:510451
Yang Z, Li L, Chen L, Yuan W, Dong L, Zhang Y, Wu H, Wang C (2014) PARP-1 mediates LPS-induced HMGB1 release by macrophages through regulation of HMGB1 acetylation. J Immunol 193:6114–6123
Lu P, Kamboj A, Gibson SB, Anderson CM (2014) Poly(ADP-ribose) polymerase-1 causes mitochondrial damage and neuron death mediated by Bnip3. J Neurosci 34:15975–15987
Wu P, Zhu X, Jin W, Hao S, Liu Q (2015) Oxaliplatin triggers necrosis as well as apoptosis in gastric cancer SGC-7901 cells. Biochem Biophys Res Commun 460:183–190
Chiu LY, Ho FM, Shiah SG, Chang Y, Lin WW (2011) Oxidative stress initiates DNA damager MNNG-induced poly(ADP-ribose)polymerase-1-dependent parthanatos cell death. Biochem Pharmacol 81:459–470
Akhiani AA, Werlenius O, Aurelius J, Movitz C, Martner A, Hellstrand K, Thorén FB (2014) Role of the ERK pathway for oxidant-induced parthanatos in human lymphocytes. PLoS One 9, e89646
Zhang L, Wang H, Xu J, Zhu J, Ding K (2014) Inhibition of cathepsin S induces autophagy and apoptosis in human glioblastoma cell lines through ROS-mediated PI3K/AKT/mTOR/p70S6K and JNK signaling pathways. Toxicol Lett 228:248–259
Barbouti A, Doulias PT, Nousis L, Tenopoulou M, Galaris D (2002) DNA damage and apoptosis in hydrogen peroxide-exposed Jurkat cells: bolus addition versus continuous generation of H(2)O(2). Free Radic Biol Med 33:691–702
Mao Y, Song G, Cai Q, Liu M, Luo H, Shi M, Ouyang G, Bao S (2006) Hydrogen peroxide-induced apoptosis in human gastric carcinoma MGC803 cells. Cell Biol Int 30:332–337
Singh M, Sharma H, Singh N (2007) Hydrogen peroxide induces apoptosis in HeLa cells through mitochondrial pathway. Mitochondrion 7:367–373
Wang X, Wang J, Lin S, Geng Y, Wang J, Jiang B (2008) Sp1 is involved in H2O2-induced PUMA gene expression and apoptosis in colorectal cancer cells. J Exp Clin Cancer Res 27:44
McKeague AL, Wilson DJ, Nelson J (2008) Staurosporine-induced apoptosis and hydrogen peroxide-induced necrosis in two human breast cell lines. Br J Cancer 88:125–131
Datta K, Babbar P, Srivastava T, Sinha S, Chattopadhyay P (2002) p53 dependent apoptosis in glioma cell lines in response to hydrogen peroxide induced oxidative stress. Int J Biochem Cell Biol 34:148–157
Byun YJ, Kim SK, Kim YM, Chae GT, Jeong SW, Lee SB (2009) Hydrogen peroxide induces autophagic cell death in C6 glioma cells via BNIP3-mediated suppression of the mTOR pathway. Neurosci Lett 461:131–135
Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB (2008) Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ 15:171–182
Zhang H, Kong X, Kang J, Su J, Li Y, Zhong J, Sun L (2009) Oxidative stress induces parallel autophagy and mitochondria dysfunction in human glioma U251 cells. Toxicol Sci 110:376–388
Lennicke C, Rahn J, Lichtenfels R, Wessjohann LA, Seliger B (2015) Hydrogen peroxide-production, fate and role in redox signaling of tumor cells. Cell Commun Signal 13:39
Wang Y, Dawson VL, Dawson TM (2009) Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol 218:193–202
Mashimo M, Kato J, Moss J (2013) ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc Natl Acad Sci U S A 110:18964–18969
Zhao H, Ning J, Lemaire A, Koumpa FS, Sun JJ, Fung A, Gu J, Yi B et al (2015) Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int 87:738–748
Andrabi SA, Kim NS, Yu S, Wang H, Koh DW, Sasaki M, Klaus JA, Otsuka T et al (2006) Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A 103:18308–18313
Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagné JP, Poirier GG, Dawson VL et al (2014) Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A 28:10209–10214
Paul M, Hemshekhar M, Thushara RM, Sundaram MS, NaveenKumar SK, Naveen S, Devaraja S, Somyajit K et al (2015) Methotrexate promotes platelet apoptosis via JNK-mediated mitochondrial damage: alleviation by N-acetylcysteine and N-acetylcysteine amide. PLoS One 10, e0127558
Dixit D, Ghildiyal R, Anto NP, Sen E (2014) Chaetocin-induced ROS-mediated apoptosis involves ATM-YAP1 axis and JNK-dependent inhibition of glucose metabolism. Cell Death Dis 5, e1212
Zhang C, Yang L, Wang XB, Wang JS, Geng YD, Yang CS, Kong LY (2013) Calyxin Y induces hydrogen peroxide-dependent autophagy and apoptosis via JNK activation in human non-small cell lung cancer NCI-H460 cells. Cancer Lett 340:51–62
Palit S, Kar S, Sharma G, Das PK (2015) Hesperetin induces apoptosis in breast carcinoma by triggering accumulation of ROS and activation of ASK1/JNK pathway. J Cell Physiol 230:1729–1739
Nomura J, Busso N, Ives A, Tsujimoto S, Tamura M, So A, Yamanaka Y (2013) Febuxostat, an inhibitor of xanthine oxidase, suppresses lipopolysaccharide-induced MCP-1 production via MAPK phosphatase-1-mediated inactivation of JNK. PLoS One 8, e75527
Zhou JY, Liu Y, Wu GS (2006) The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death. Cancer Res 66:4888–4894
Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N (2008) Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem 283:13565–13577
Win S, Than TA, Le BH, García-Ruiz C, Fernandez-Checa JC, Kaplowitz N (2015) Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J Hepatol 62:1367–1374
Acknowledgments
This work was supported by the National Nature Science Foundation of China (81171234, 81372697, 11432016, and 11272134), the Changbaishan Scholar Project of Jilin province (2013026), the Scientific Research Foundation of Jilin province (20150414013GH, 20121809), and the Bethune project B of Jilin University (no.2012203).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interests
The authors declare that they have no conflict of interests.
Rights and permissions
About this article

Cite this article
Zheng, L., Wang, C., Luo, T. et al. JNK Activation Contributes to Oxidative Stress-Induced Parthanatos in Glioma Cells via Increase of Intracellular ROS Production. Mol Neurobiol 54, 3492–3505 (2017). https://doi.org/10.1007/s12035-016-9926-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-016-9926-y